An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Nitrogen Availability Affects the Morphology, Physiology, and Growth Characteristics of Rice Leaves
2.2. Metabolite Profiles of Rice Leaves in Response to Nitrogen Availability
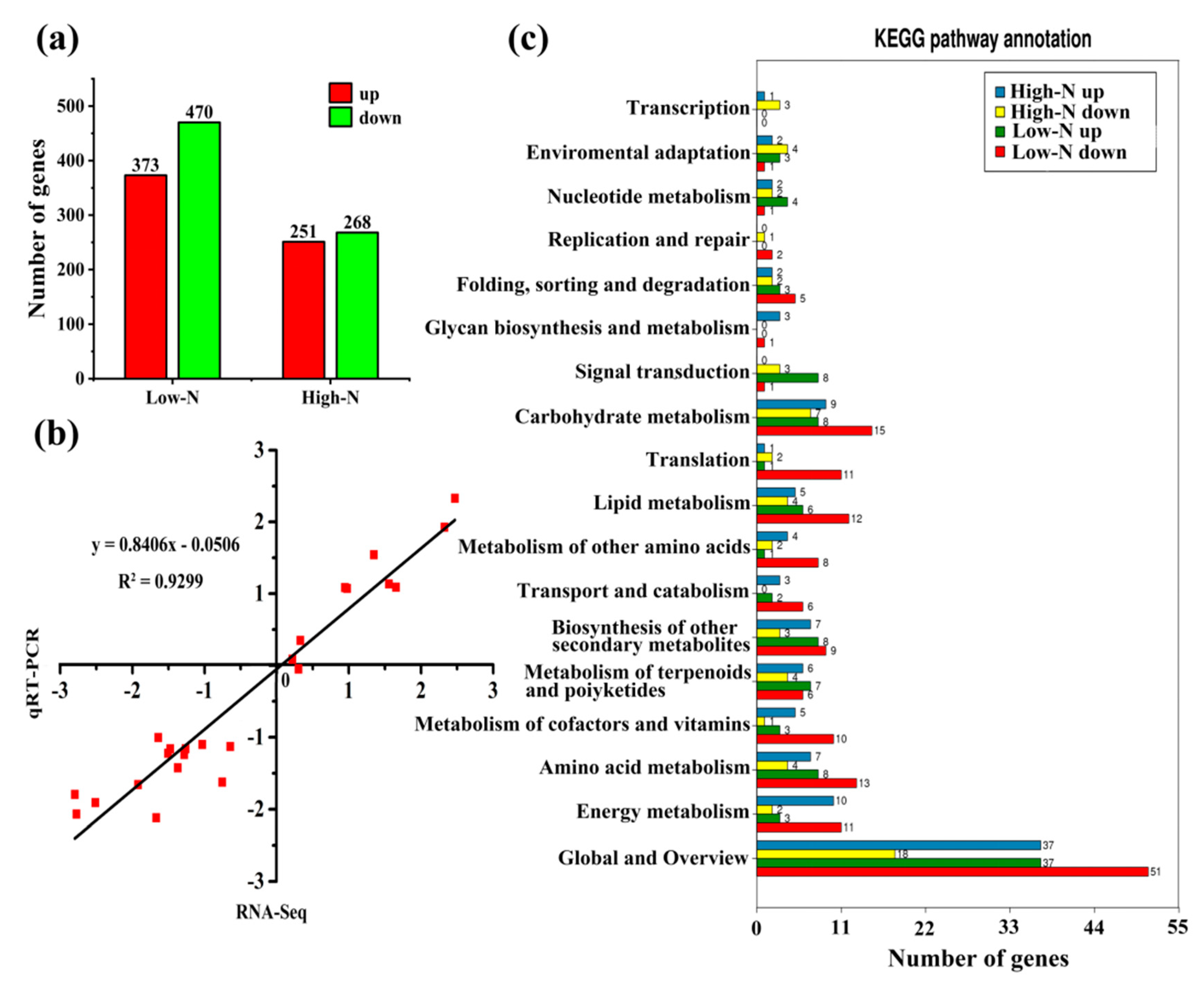
2.3. Transcriptome Profiles of Rice Leaves in Response to Nitrogen Availability
2.4. Nitrogen Assimilation and Amino Acid Metabolism
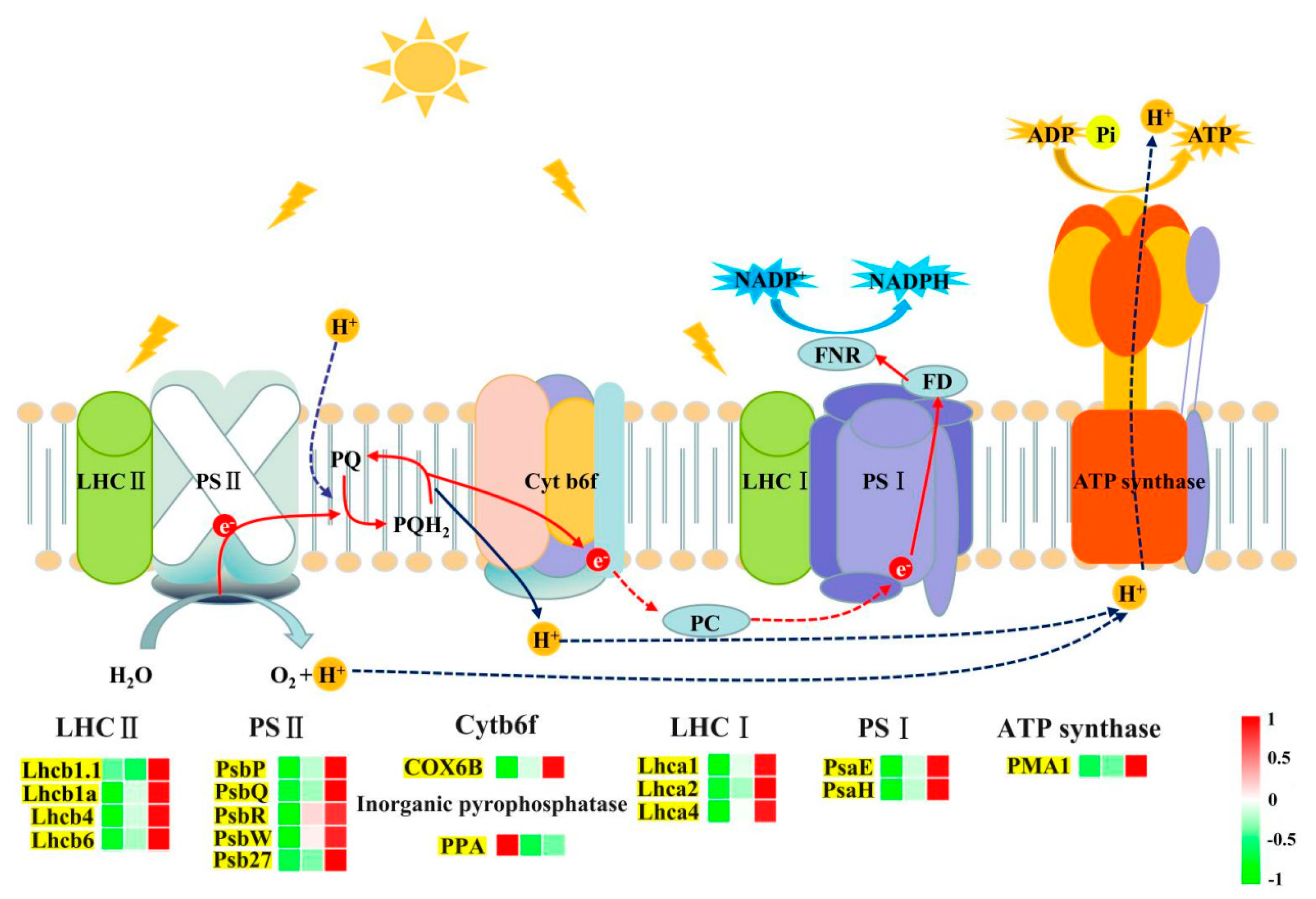
2.5. Carbon Metabolism
2.6. Regulation of Differential Gene Expression by Transcription Factors
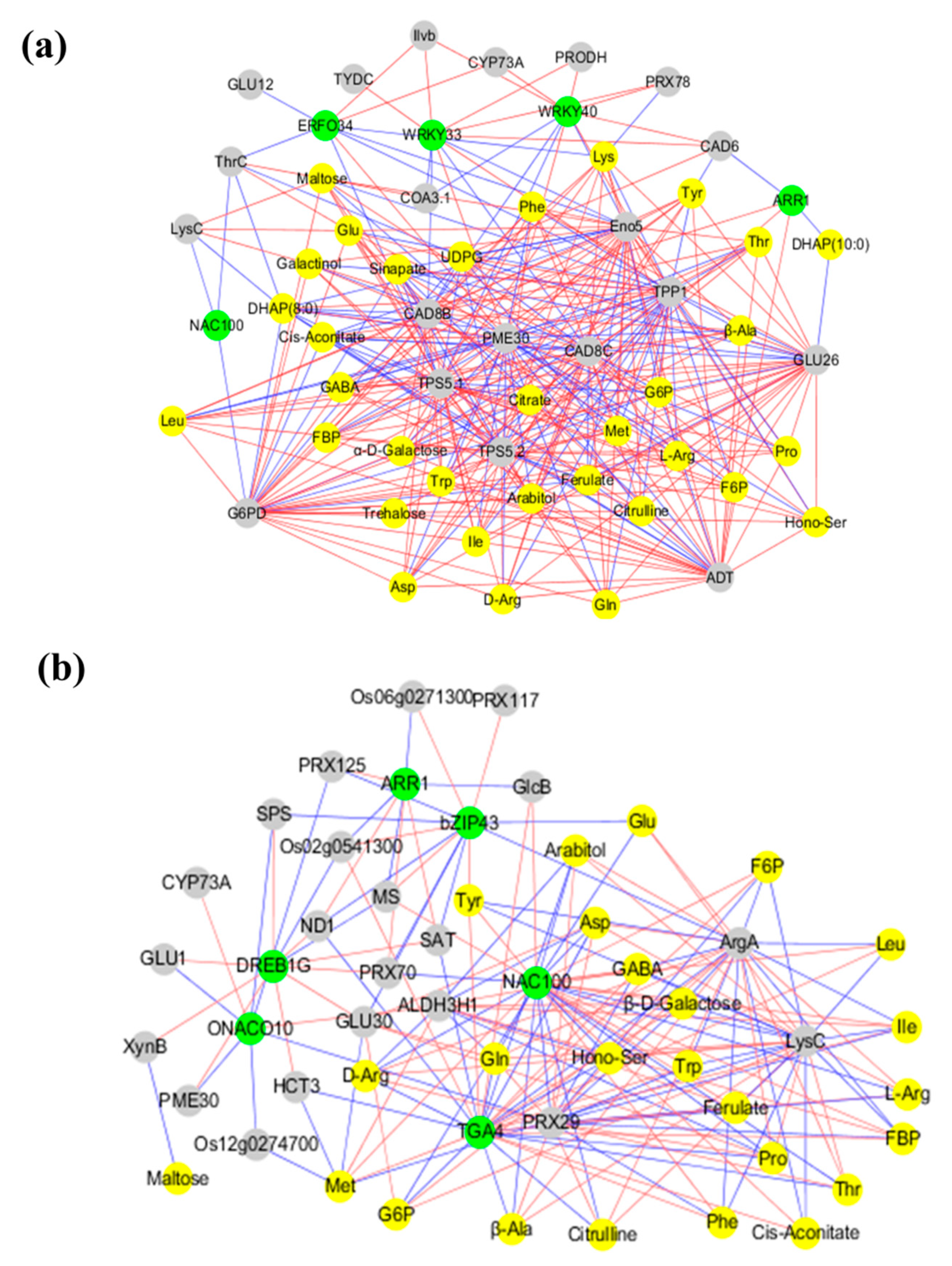
2.7. Co-Expression Networks Reveal a Differential Regulatory Network of Carbon and Nitrogen Metabolism under Low N and High N
3. Discussion
4. Materials and Methods
4.1. Plant Materials and N Treatment
4.2. Analysis of Leaf Characteristics
4.3. Metabolite Extraction and Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry (LC-ESI-MS/MS) Analysis
4.4. RNA Extraction and Transcriptome Sequencing Analysis
4.5. Co-Expression Network Analysis of Genes and Metabolites
4.6. Confirmation of Transcriptome Data Using qRT-PCR Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, H.; Hu, B.; Chu, C. Nitrogen use efficiency in crops: Lessons from Arabidopsis and rice. J. Exp. Bot. 2017, 68, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Curci, P.L.; Cigliano, R.A.; Zuluaga, D.L.; Janni, M.; Sanseverino, W.; Sonnante, G. Transcriptomic response of durum wheat to nitrogen starvation. Sci. Rep. 2017, 7, 1176. [Google Scholar] [CrossRef] [PubMed]
- Hakeem, K.R.; Ahmad, A.; Iqbal, M.; Gucel, S.; Ozturk, M. Nitrogen-efficient rice cultivars can reduce nitrate pollution. Environ. Sci. Pollut. Res. 2011, 18, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, R.A. Systems biology for enhanced plant nitrogen nutrition. Science 2012, 336, 1673–1675. [Google Scholar] [PubMed]
- Chikov, V.; Bakirova, G. Relationship between Carbon and Nitrogen Metabolisms in Photosynthesis. The Role of Photooxidation Processes. Photosynthetica 2000, 37, 519–527. [Google Scholar] [CrossRef]
- Nunes-Nesi, A.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar] [CrossRef] [PubMed]
- Sweetlove, L.J.; Beard, K.F.; Nunesnesi, A.; Fernie, A.R.; Ratcliffe, R.G. Not just a circle: Flux modes in the plant TCA cycle. Trends Plant Sci. 2010, 15, 462–470. [Google Scholar] [CrossRef]
- Zheng, Z.J. Carbon and nitrogen nutrient balance signaling in plants. Plant Signal. Behav. 2009, 4, 584–591. [Google Scholar]
- Bao, A.; Zhao, Z.; Ding, G.; Shi, L.; Xu, F.; Cai, H. Accumulated Expression Level of Cytosolic Glutamine Synthetase 1 Gene (OsGS1;1 or OsGS1;2) Alter Plant Development and the Carbon-Nitrogen Metabolic Status in Rice. PLoS ONE 2014, 9, e95581. [Google Scholar] [CrossRef]
- Bao, A.; Zhao, Z.; Ding, G.; Shi, L.; Xu, F.; Cai, H. The Stable Level of Glutamine synthetase 2 Plays an Important Role in Rice Growth and in Carbon-Nitrogen Metabolic Balance. Int. J. Mol. Sci. 2015, 16, 12713–12736. [Google Scholar] [CrossRef] [PubMed]
- Wolf-Rüdiger, S.; Mark, S. Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol. 2004, 136, 2483–2499. [Google Scholar]
- Bi, Y.M.; Wang, R.L.; Tong, Z.; Rothstein, S.J. Global transcription profiling reveals differential responses to chronic nitrogen stress and putative nitrogen regulatory components in Arabidopsis. BMC Genom. 2007, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Krapp, A.; Berthomã, R.; Orsel, M.; Mercey-Boutet, S.; Yu, A.; Castaings, L.; Elftieh, S.; Major, H.; Renou, J.P.; Daniel-Vedele, F. Arabidopsis roots and shoots show distinct temporal adaptation patterns toward nitrogen starvation. Plant Physiol. 2011, 157, 1255–1282. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Noctor, G.; Hodges, M. Respiration and nitrogen assimilation: Targeting mitochondria-associated metabolism as a means to enhance nitrogen use efficiency. J. Exp. Bot. 2011, 62, 1467–1482. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, P.P.; Bligny, R.; Gout, E.; Mahé, A.; Nogués, S.; Hodges, M.; Tcherkez, G.G. In folio isotopic tracing demonstrates that nitrogen assimilation into glutamate is mostly independent from current CO2 assimilation in illuminated leaves of Brassica napus. New Phytol. 2010, 185, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Nian, J.; Xie, Q.; Feng, J.; Zhang, F.; Jing, H.; Zhang, J.; Dong, G.; Liang, Y.; Peng, J. Rice Ferredoxin-Dependent Glutamate Synthase Regulates Nitrogen–Carbon Metabolomes and Is Genetically Differentiated between japonica and indica Subspecies. Mol. Plant 2016, 9, 1520–1534. [Google Scholar] [CrossRef]
- Coruzzi, G.M.; Zhou, L. Carbon and nitrogen sensing and signaling in plants: Emerging ‘matrix effects’. Curr. Opin. Plant Biol. 2001, 4, 247–253. [Google Scholar] [CrossRef]
- Martin, T.; Oswald, O.; Graham, I.A. Arabidopsis seedling growth, storage lipid mobilization, and photosynthetic gene expression are regulated by carbon: Nitrogen availability. Plant Physiol. 2002, 128, 472–481. [Google Scholar] [CrossRef]
- Krapp, A.; Traong, H.N. Regulation of C/N Interaction in Model Plant Species. J. Crop Improv. 2005, 15, 127–173. [Google Scholar] [CrossRef]
- Peng, S.; Buresh, R.J.; Huang, J.; Yang, J.; Zou, Y.; Zhong, X.; Wang, G.; Zhang, F. Strategies for overcoming low agronomic nitrogen use efficiency in irrigated rice systems in China. Field Crop. Res. 2006, 96, 37–47. [Google Scholar] [CrossRef]
- Kusano, M.; Fukushima, A.; Redestig, H.; Saito, K. Metabolomic approaches toward understanding nitrogen metabolism in plants. J. Exp. Bot. 2011, 62, 1439–1453. [Google Scholar] [CrossRef]
- Glaubitz, U.; Li, X.; Schaedel, S.; Erban, A.; Sulpice, R.; Kopka, J.; Hincha, D.K.; Zuther, E. Integrated analysis of rice transcriptomic and metabolomic responses to elevated night temperatures identifies sensitivity- and tolerance-related profiles: Integrated profiling analysis of rice under HNT. Plant Cell Environ. 2017, 40, 121–137. [Google Scholar] [CrossRef]
- Lou, Q.; Liu, Y.; Qi, Y.; Jiao, S.; Tian, F.; Jiang, L.; Wang, Y. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J. Exp. Bot. 2014, 65, 3157–3164. [Google Scholar] [CrossRef]
- Cho, K.; Cho, K.S.; Sohn, H.B.; Ha, I.J.; Hong, S.Y.; Lee, H.; Kim, Y.M.; Nam, M.H. Network analysis of the metabolome and transcriptome reveals novel regulation of potato pigmentation. J. Exp. Bot. 2016, 67, 1519. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, J.; Li, S.S.; Zhang, H.J.; Feng, C.Y.; Yin, D.D.; Wu, R.Y.; Wang, L.S. Transcriptome sequencing and metabolite analysis for revealing the blue flower formation in waterlily. BMC Genom. 2016, 17, 897. [Google Scholar] [CrossRef]
- Liu, G.F.; Han, Z.X.; Feng, L.; Gao, L.P.; Gao, M.J.; Gruber, M.Y.; Zhang, Z.L.; Xia, T.; Wan, X.C.; Wei, S. Metabolic Flux Redirection and Transcriptomic Reprogramming in the Albino Tea Cultivar ‘Yu-Jin-Xiang’ with an Emphasis on Catechin Production. Sci. Rep. 2017, 7, 45062. [Google Scholar] [CrossRef]
- Taiz, L.; Zeiger, E. Plants Physiology, 5th ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 2010; p. 126. [Google Scholar]
- Cai, H.; Lu, Y.; Xie, W.; Zhu, T.; Lian, X. Transcriptome response to nitrogen starvation in rice. J. Biosci. 2012, 37, 731–747. [Google Scholar] [CrossRef]
- Yang, W.; Yoon, J.; Choi, H.; Fan, Y.; Chen, R.; An, G. Transcriptome analysis of nitrogen-starvation-responsive genes in rice. BMC Plant Biol. 2015, 15, 31. [Google Scholar] [CrossRef]
- Subodh, S.; Amitha, S.V.; Saurabh, C.; Punit, T.; Sureshkumar, V.; Manju, R.; Pranab, M. Transcriptome Analysis of Two Rice Varieties Contrasting for Nitrogen Use Efficiency under Chronic N Starvation Reveals Differences in Chloroplast and Starch Metabolism-Related Genes. Genes 2018, 9, 206. [Google Scholar]
- Tovar-Mendez, A.; Miernyk, J.A.; Randall, D.D. Regulation of pyruvate dehydrogenase complex activity in plant cells. Eur. J. Biochem. 2003, 270, 1043–1049. [Google Scholar] [CrossRef]
- Fan, J.; Kang, H.B.; Shan, C.; Elf, S.; Lin, R.; Xie, J.; Gu, T.L.; Aguiar, M.; Lonning, S.; Chung, T.W.; et al. Tyr-301 Phosphorylation Inhibits Pyruvate Dehydrogenase by Blocking Substrate Binding and Promotes the Warburg Effect. J. Biol. Chem. 2014, 289, 26533–26541. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Fan, X.; Miller, A.J. Plant Nitrogen Assimilation and Use Efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef] [PubMed]
- Black, B.L.; Fuchigami, L.H.; Coleman, G.D. Partitioning of nitrate assimilation among leaves, stems and roots of poplar. Tree Physiol. 2002, 22, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Bi, Y.M.; Rothstein, S.J. Understanding plant response to nitrogen limitation for the improvement of crop nitrogen use efficiency. J. Exp. Bot. 2011, 62, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Hoff, T.; Truong, H.N.; Caboche, M. Use of mutants and transgenic plants to study nitrate assimilation. Plant Cell Environ. 1994, 17, 489–506. [Google Scholar] [CrossRef]
- Vincentz, M.; Moureaux, T.; Leydecker, M.T.; Vaucheret, H.; Caboche, M. Regulation of nitrate and nitrite reductase expression in Nicotiana plumbaginifolia leaves by nitrogen and carbon metabolites. Plant J. 2010, 3, 315–324. [Google Scholar] [CrossRef]
- Cong, F.; Diehl, B.G.; Hill, J.L.; Brown, N.R.; MingTien, M. Covalent bond formation between amino acids and lignin: Cross-coupling between proteins and lignin. Phytochemistry 2013, 96, 449–456. [Google Scholar] [CrossRef]
- Zhang, X.; Misra, A.; Nargund, S.; Coleman, G.D.; Sriram, G. Concurrent isotope-assisted metabolic flux analysis and transcriptome profiling reveal responses of poplar cells to altered nitrogen and carbon supply. Plant J. 2018, 93, 472–488. [Google Scholar] [CrossRef]
- Ray, D.; Sheshshayee, M.S.; Mukhopadhyay, K.; Bindumadhava, H.; Prasad, T.G.; Kumar, M.U. High Nitrogen Use Efficiency in Rice Genotypes is Associated with Higher Net Photosynthetic Rate at Lower Rubisco Content. Biol. Plant. 2003, 46, 251–256. [Google Scholar] [CrossRef]
- Bown, H.E.; Watt, M.S.; Mason, E.G.; Clinton, P.E.; Whitehead, D. The influence of nitrogen and phosphorus supply and genotype on mesophyll conductance limitations to photosynthesis in Pinus radiata. Tree Physiol. 2009, 29, 1143–1151. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, J.; Li, H.; Shi, W.; Polle, A.; Lu, M.; Sun, X.; Luo, Z.B. Global poplar root and leaf transcriptomes reveal links between growth and stress responses under nitrogen starvation and excess. Tree Physiol. 2015, tpv091. [Google Scholar] [CrossRef]
- Schlüter, U.; Mascher, M.; Colmsee, C.; Scholz, U.; Bräutigam, A.; Fahnenstich, H.; Sonnewald, U. Maize source leaf adaptation to nitrogen deficiency effects not only N and C metabolism but also control of P homeostasis. Plant Physiol. 2012, 160, 1384–1406. [Google Scholar]
- Juergens, M.T.; Deshpande, R.R.; Lucker, B.F.; Park, J.J.; Wang, H.; Gargouri, M.; Holguin, F.O.; Disbrow, B.; Schaub, T.; Skepper, J.N.; et al. The regulation of photosynthetic structure and function during nitrogen deprivation in Chlamydomonas reinhardtii. Plant Physiol. 2015, 167, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, X.; Ren, B.; Shen, Q.; Guo, S. Why nitrogen use efficiency decreases under high nitrogen supply in rice (Oryza sativa L.) seedlings. J. Plant Growth Regul. 2012, 31, 47–52. [Google Scholar] [CrossRef]
- Couee, I. Involvement of soluble sugars in reactive oxygen species balance and responses to oxidative stress in plants. J. Exp. Bot. 2006, 57, 449–459. [Google Scholar] [CrossRef]
- Lancien, M. Simultaneous Expression of NAD-Dependent Isocitrate Dehydrogenase and Other Krebs Cycle Genes after Nitrate Resupply to Short-Term Nitrogen-Starved Tobacco. Plant Physiol. 1999, 120, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, S.; Akiyama, A.; Kisaka, H.; Uchimiya, H.; Miwa, T. Metabolic engineering with Dof1 transcription factor in plants: Improved nitrogen assimilation and growth under low-nitrogen conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 7833–7838. [Google Scholar] [CrossRef] [PubMed]
- Castaings, L.; Camargo, A.; Pocholle, D.; Gaudon, V.; Texier, Y.; Boutet-Mercey, S.; Taconnat, L.; Renou, J.P.; Daniel-Vedele, F.; Fernandez, E.; et al. The nodule inception-like protein 7 modulates nitrate sensing and metabolism in Arabidopsis. Plant J. 2009, 57, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Kurai, T.; Wakayama, M.; Abiko, T.; Yanagisawa, S.; Aoki, N.; Ohsugi, R. Introduction of the ZmDof1 gene into rice enhances carbon and nitrogen assimilation under low-nitrogen conditions. Plant Biotechnol. J. 2011, 9, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Bi, Y.M.; Zhu, T.; Rothstein, S.J. Genome-wide analysis of Arabidopsis responsive transcriptome to nitrogen limitation and its regulation by the ubiquitin ligase gene NLA. Plant Mol. Biol. 2007, 65, 775–797. [Google Scholar] [CrossRef]
- Imamura, S.; Kanesaki, Y.; Ohnuma, M.; Inouye, T.; Sekine, Y.; Fujiwara, T.; Kuroiwa, T.; Tanaka, K. R2R3-type MYB transcription factor, CmMYB1, is a central nitrogen assimilation regulator in Cyanidioschyzon merolae. Proc. Natl. Acad. Sci. USA 2009, 106, 12548–12553. [Google Scholar] [CrossRef]
- Ogawa, T.; Oikawa, S.; Hirose, T. Nitrogen-utilization efficiency in rice, an analysis at leaf, shoot, and whole-plant level: International journal of plant nutrition, plant chemistry, soil microbiology and soil-born plant diseases. Plant Soil 2016, 404, 321–344. [Google Scholar] [CrossRef]
- Ju, C.; Buresh, R.J.; Wang, Z.; Zhang, H.; Liu, L.; Yang, I.; Zhang, J. Root and shoot traits for rice varieties with higher grain yield and higher nitrogen use efficiency at lower nitrogen rates application. Field Crop. Res. 2015, 175, 47–55. [Google Scholar] [CrossRef]
- Cao, H.H.; Zhang, M.; Zhao, H.; Zhang, Y.; Wang, X.X.; Guo, S.S.; Zhang, Z.F.; Liu, T.X. Deciphering the Mechanism of β-Aminobutyric Acid-Induced Resistance in Wheat to the Grain Aphid, Sitobion avenae. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Maness, N. Extraction and analysis of soluble carbohydrates. Methods Mol. Biol. Plant Stress Toler. 2010, 341–370. [Google Scholar]
- Xu, J.; Zhang, Y.; Qi, D.; Huo, H.; Dong, X.; Tian, L.; Zhang, X.; Liu, C.; Cao, Y. Postharvest metabolomic changes in Pyrus ussuriensis Maxim. wild accession ‘Zaoshu Shanli’. J. Sep. Sci. 2018, 41, 4001–4013. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Saccenti, E.; Hoefsloot, H.C.J.; Smilde, A.K.; Westerhuis, J.K.; Hendriks, M.M.W.B. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2014, 10, 361–374. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; John, L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562. [Google Scholar] [CrossRef]
- Cosgrove, D.J. Plant cell wall extensibility: Connecting plant cell growth with cell wall structure, mechanics, and the action of wall-modifying enzymes. J. Exp. Bot. 2015, 67, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Zhou, X.; Chen, X.; Li, Q.; Tan, H.; Dong, X.; Xiao, Y.; Chen, L.; Chen, W. Dynamic metabolic and transcriptomic profiling of methyl jasmonate-treated hairy roots reveals synthetic characters and regulators of lignan biosynthesis in Isatis indigotica Fort. Plant Biotechnol. J. 2016. [Google Scholar] [CrossRef]
- Pujan, M.A.; Han, J.D.J.; Starita, L.M.; Stevens, K.N.; Tewari, M.; Ahn, J.S.; Rennert, G.; Moreno, V.; Kirchhoff, T.; Gold, B. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 2007, 39, 1338. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Low N | Control N | High N | FP Value |
|---|---|---|---|---|
| Leaf biomass (g) | 1.26 ± 0.11b | 1.87 ± 0.08a | 1.81 ± 0.08a | 42.22 *** |
| Leaf area (cm2) | 238.36 ± 20.17b | 352.20 ± 15.25a | 341.51 ± 14.24a | 53.72 *** |
| Chlorophyll a (Chl a, mg·g−1) | 0.82 ± 0.03c | 1.58 ± 0.08b | 1.73 ± 0.01a | 324.32 *** |
| Chlorophyll b (Chl b, mg·g−1) | 0.37 ± 0.01c | 0.71 ± 0.04b | 0.80 ± 0.01a | 301.64 *** |
| Intercellular CO2 concentration (Ci, μmol·mol−1) | 285.33 ± 16.07a | 292.67 ± 4.51a | 292.67 ± 2.31a | 0.57 ns |
| Photosynthetic rate (Pn, μmol m−2·s−1) | 18.13 ± 0.59b | 21.73 ± 1.00a | 21.97 ± 0.47a | 26.47 ** |
| Stomatal conductance (gs, mmol m−2·s−1) | 600.00 ± 14.42c | 731.33 ± 36.69b | 814.67 ± 52.44a | 24.49 ** |
| N content | 3.19 ± 0.10c | 4.55 ± 0.08b | 5.29 ± 0.03a | 574.51 *** |
| C content | 37.50 ± 0.30b | 41.05 ± 0.52a | 41.46 ± 0.70a | 50.36 *** |
| Carbon/Nitrogen (C/N) | 11.76 ± 0.35a | 9.01 ± 0.10b | 7.84 ± 0.09c | 261.47 *** |
| Soluble sugar (mg·mg−1) | 0.10 ± 0.01b | 0.10 ± 0.01b | 0.13 ± 0.01a | 25.4 ** |
| Free amino acids (μmol·mg−1) | 3.56 ± 0.05b | 4.07 ± 0.15a | 3.51 ± 0.30b | 7.35 * |
| Total protein (μg·mg−1) | 1.78 ± 0.02b | 1.83 ± 0.10b | 2.14 ± 0.08a | 19.29 ** |
| Nitrogen use efficiency (NUE, g·g−1) | 47.45 ± 2.13a | 32.34 ± 1.11b | 29.23 ± 1.02c | 125.14 *** |
| Photosynthetic nitrogen use efficiency (PUNE, μmol g−1·s−1) | 10.73 ± 0.51a | 9.0 ± 0.30b | 7.83 ± 0.19c | 49.27 *** |
| TF Family | Low N | High N | ||
|---|---|---|---|---|
| Up | Down | Up | Down | |
| bHLH | 6 | 1 | 1 | 1 |
| bZIP | 1 | 1 | 0 | 2 |
| C2H2 | 4 | 0 | 0 | 0 |
| CO-like | 1 | 0 | 1 | 0 |
| DBB | 1 | 1 | 0 | 0 |
| E2F/DP | 0 | 1 | 1 | 0 |
| EIL | 0 | 0 | 0 | 1 |
| ERF | 4 | 1 | 1 | 0 |
| G2-like | 0 | 1 | 1 | 0 |
| GRAS | 1 | 0 | 1 | 0 |
| HD-ZIP | 1 | 1 | 0 | 0 |
| HSF | 1 | 0 | 0 | 0 |
| LSD | 1 | 0 | 0 | 0 |
| M-type_MADS | 0 | 0 | 1 | 0 |
| MYB | 1 | 0 | 0 | 0 |
| MYB_related | 3 | 1 | 0 | 6 |
| NAC | 5 | 1 | 0 | 3 |
| NF-YA | 1 | 0 | 0 | 0 |
| NF-YC | 0 | 1 | 1 | 0 |
| Nin-like | 0 | 0 | 1 | 1 |
| Whirly | 0 | 1 | 0 | 0 |
| WRKY | 6 | 0 | 0 | 0 |
| Total | 38 | 10 | 8 | 15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Li, Z.; Zhao, Y.; Peng, C.; Zhao, C. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability. Int. J. Mol. Sci. 2019, 20, 2349. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092349
Xin W, Zhang L, Zhang W, Gao J, Yi J, Zhen X, Li Z, Zhao Y, Peng C, Zhao C. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability. International Journal of Molecular Sciences. 2019; 20(9):2349. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092349
Chicago/Turabian StyleXin, Wei, Lina Zhang, Wenzhong Zhang, Jiping Gao, Jun Yi, Xiaoxi Zhen, Ziang Li, Ying Zhao, Chengcheng Peng, and Chen Zhao. 2019. "An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability" International Journal of Molecular Sciences 20, no. 9: 2349. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092349