Epidemiology of Hereditary Diseases in the Karachay-Cherkess Republic

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. The Structure of the Diversity of Hereditary Diseases in Populations of KCHR
2.2. Genetic Heterogeneity (Allelic, Locus) of Monogenic Hereditary Diseases in KCHR
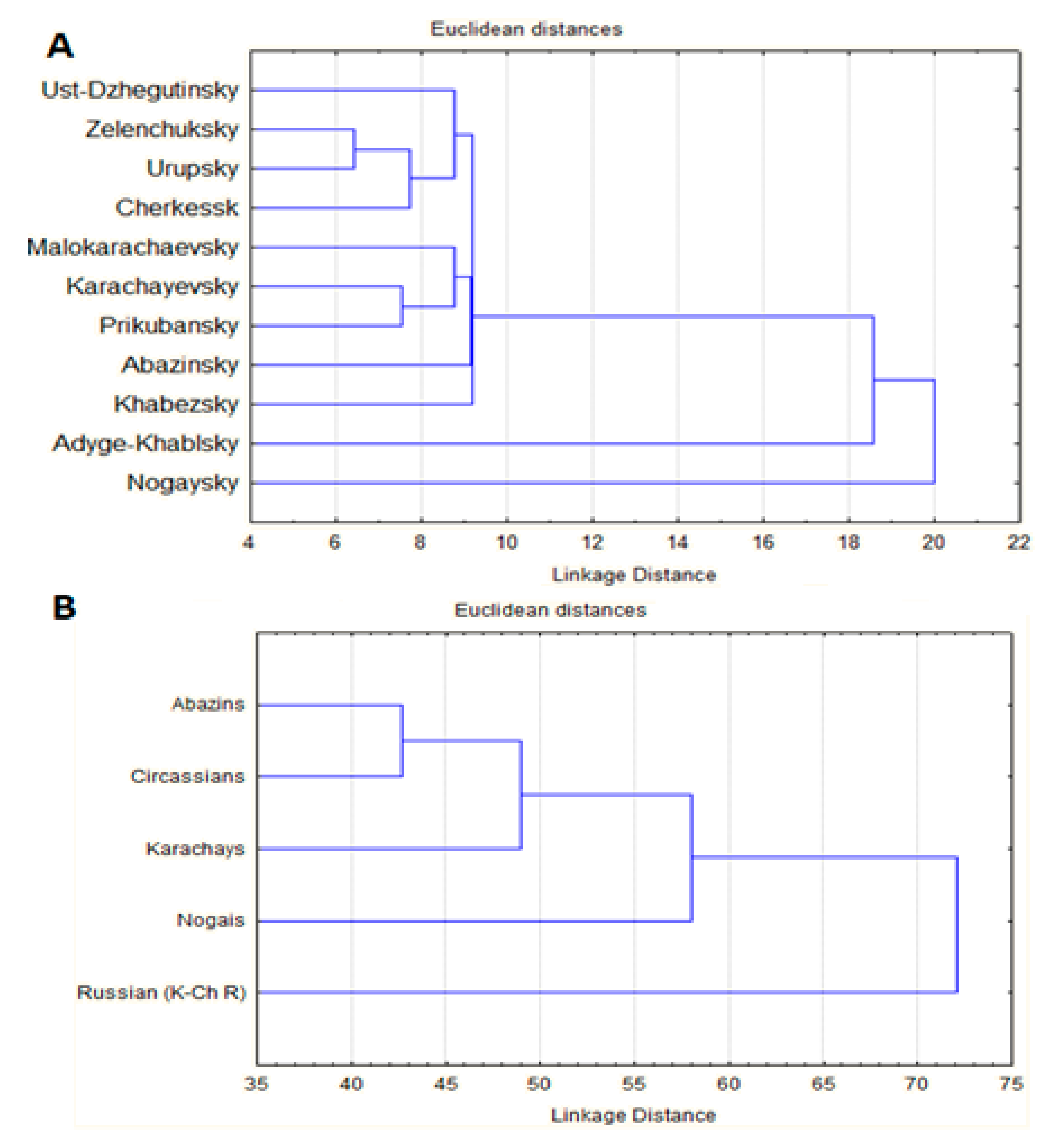
2.3. Analysis of Genetic Relationships for Prevalence of Hereditary Diseases between Districts and Main Ethnic Groups of KCHR
2.4. Values of Hereditary Disease Load in Populations and Ethnic Groups of KChR
2.5. The Study of the Possible Causes of the Differentiation of KCHR Populations of Load of HDs
3. Conclusions
4. Materials and Methods
4.1. Survey Population
4.2. Survey and Protocol
4.3. Molecular Genetic Analysis
4.4. Statistical Methods
4.5. Cluster Analysis
4.6. FST Analysis
4.7. Data Availability Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- United Nations Scientific Committee on the Effects of Atomic Radiation. UNSCEAR: Genetic and Somatic Effects of Ionizing Radiation; Report to the General Assembly with Annexes; United Nations: New York, NY, USA, 1986. [Google Scholar]
- United Nations Scientific Committee on the Effects of Atomic Radiation. UNSCEAR: Sources and Effects of Ionizing Radiation; Report to the General Assembly; United Nations: New York, NY, USA, 1977. [Google Scholar]
- United Nations Scientific Committee on the Effects of Atomic Radiation. UNSCEAR: Genetic and Somatic Effects of Ionizing Radiation; Report to the General Assembly with Annexes; United Nations: New York, NY, USA, 2010. [Google Scholar]
- Baird, P.A.; Anderson, T.W.; Newcombe, H.B.; Lowry, R.B. Genetic disorders in children and young adults: A population study. Am. J. Hum. Genet. 1988, 42, 677–693. [Google Scholar]
- Congenital Anomalies Surveillance in Canada: Results of a 2006–2007 Survey on Availability of Selected Data Variables in Canadian Provinces and Territories; Public Health Agency of Canada: Ottawa, ON, Canada, 2010.
- Norio, R. Diseases of Finland and Scandinavia. In Biocultural Aspects of Disease; Academic Press: New York, NY, USA, 1981; pp. 359–415. [Google Scholar]
- Goodman, R.M. Genetic Disorders among the Jewish People; The Gohn Hopkins Univ. Press: Baltimor, MA, USA, 1979. [Google Scholar]
- McKusick, V.A. Medical genetic studies of the Amish, with comparison to other populations. In Population Structure and Genetic Disorders; Academic Press: London, UK, 1980; pp. 291–300. [Google Scholar]
- De Braekeleer, M.; Dao, T. Hereditary Disorders in the French Canadian Population of Quebec. II. Contribution of Perche. Hum. Biol. 1994, 66, 225–249. [Google Scholar] [PubMed]
- De Braekeleer, M.; Dao, T. Hereditary Disorders in the French Canadian Population of Quebec. I. In Search of Founders. Hum. Biol. 1994, 66, 205–223. [Google Scholar] [PubMed]
- Carter, C.O. Monogenic disorders. J. Med. Genet. 1977, 14, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsiev, A. Atlas of the Ethno-Political History of the Caucasus (Historical Atlas); Yale University Press: New Haven, CT, USA, 2014. [Google Scholar]
- Cavalli-Sforza, L.L.; Bodmer, W.F. The Genetics of Human Populations; Dover Publication: Minneola, NY, USA, 1999. [Google Scholar]
- Morton, N.E. Genetic tests under incomplete ascertainment. Am. J. Hum. Genet. 1959, 11, 1–16. [Google Scholar]
- Zinchenko, R.A.; Elchinova, G.I.; Nurbaev, S.D.; Ginter, E.K. Diversity of autosomal dominant diseases in populations of Russia. Russ. J. Genet. 2001, 37, 290–301. [Google Scholar] [CrossRef]
- Zinchenko, R.A.; Elchinova, G.I.; Gavrilina, S.G.; Ginter, E.K. Analysis of diversity of autosomal recessive diseases in populations of Russia. Russ. J. Genet. 2001, 37, 1312–1322. [Google Scholar] [CrossRef]
- Zinchenko, R.A.; Kadyshev, V.V.; El’chinova, G.I.; Marakhonov, A.V.; Galkina, V.A.; Dadali, E.L.; Khlebnikova, O.V.; Mikhailova, L.K.; Petrova, N.V.; Petrina, N.E.; et al. Study of the genetic load and diversity of hereditary diseases in the Russian population of the Karachay-Cherkess Republic. Int. J. Mol. Epidemiol. Genet. 2018, 9, 34–42. [Google Scholar]
- «Prevalence of Rare Diseases: Bibliographic Data», Orphanet Report Series, Rare Diseases Collection, January 2019, Number 2: Diseases Listed by Decreasing Prevalence, Incidence or Number of Published Cases. Available online: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_decreasing_prevalence_or_cases.pdf (accessed on 12 January 2019).
- Gundorova, P.; Stepanova, A.A.; Makaov, A.K.; Zinchenko, R.A.; Abaykhanova, Z.M.; Polyakov, A.V. Mutation spectrum of the PAH gene in phenylketonuria patients in the Karachay-Cherkess Republic (Russia). Russ. J. Genet. 2016, 52, 1282–1290. [Google Scholar] [CrossRef]
- Gundorova, P.; Zinchenko, R.A.; Kuznetsova, I.A.; Bliznetz, E.A.; Stepanova, A.A.; Polyakov, A.V. Molecular-genetic causes for the high frequency of phenylketonuria in the population from the North Caucasus. PLoS ONE 2018, 13, e0201489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, N.V.; Kashirskaya, N.Y.; Vasilyeva, T.A.; Timkovskaya, E.E.; Voronkova, A.Y.; Shabalova, L.A.; Kondratyeva, E.I.; Sherman, V.D.; Novoselova, O.G.; Kapranov, N.I.; et al. High prevalence of W1282x mutation in cystic fibrosis patients from Karachay-Cherkessia. J. Cyst. Fibros. 2016, 15, e28–e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marakhonov, A.V.; Konovalov, F.A.; Makaov, A.K.; Vasilyeva, T.A.; Kadyshev, V.V.; Galkina, V.A.; Dadali, E.L.; Kutsev, S.I.; Zinchenko, R.A. Primary microcephaly case from the Karachay-Cherkess Republic poses an additional support for microcephaly and Seckel syndrome spectrum disorders. BMC Med. Genom. 2018, 11, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elizabeth, T.; Amin, M.; Lyudmila, M.; Vasilyeva, T.A.; Marakhonov, A.V.; Galkina, V.A.; Rena, Z. Metatropic dysplasia: Clinical and molecular diagnostics, genetic counseling. Med. News North Caucasus 2016, 11, 173–176. [Google Scholar] [CrossRef]
- Crow, J.F. Surnames as biological marker. Hum. Biol. 1983, 55, 383–397. [Google Scholar]
- Wu, D.D.; Zhang, Y.P. Different level of population differentiation among human genes. BMC Evol. Biol. 2011, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Zinchenko, R.A.; El’chinova, G.I.; Ginter, E.K. Factors determining the spread of hereditary diseases in Russian populations. Med. Genet. 2009, 12, 7–23. (In Russia) [Google Scholar]
- Ginter, E.K.; Zinchenko, R.A. Epidemiology of Hereditary Diseases in European Sector of Russia. In Genomics and Health in The Developing World; Kumar, D., Ed.; Oxford University Press: New York, NY, USA, 2012; pp. 1281–1314. [Google Scholar]
- Full Collection of Legislation of the USSR. Available online: http://www.ussrdoc.com/ (accessed on 12 January 2019).
- Tishkov, V.A. (Ed.) The Russian Caucasus: Book for Politicians; Rosinformagrotech: Moscow, Russia, 2007. [Google Scholar]
- Elchinova, G.I.; Makaov AKh Bikanov, R.A.; Gavrilina, S.G.; Petrin, A.N.; Marakhonov, A.V.; Zinchenko, R.A. Analysis of the age and sex structure of the population of Karachay-Cherkessia Republic. Modern Probl. Sci. Educ. 2017, 2, 52. [Google Scholar] [CrossRef] [Green Version]
- Emery, A.E.H.; Elliott, D.; Moores, M.; Smith, C. A genetic register system (RAPID). J. Med. Genet. 1974, 11, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.A. Statistical Methods for Research Workers, 7th ed.; Electronic Resource; Edinburgh Oliver and Boyd—Biological Monographs and Manuals: Edinburgh, UK, 1938; 356p. [Google Scholar]
- Morton, N.E.; Rao, D.C. Methods in Genetic Epidemiology; Karger: Basel, Germany, 1983; p. 64. [Google Scholar]
- Morton, N.E.; Yee, S.; Harris, D.E.; Lew, R. Bioassay of kinship. Theor. Popul. Biol. 1971, 2, 507–524. [Google Scholar] [CrossRef]
- Elchinova, G.I.; Makaov, A.K.; Zinchenko, R.A. Analysis of surname landscape Karachay-Cherkessia. Mod. Probl. Sci. Educ. 2015, 5, 687. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Diagnosis | Number of Diseases (%) | Number of Patients (%) | Prevalence (per 100,000 People) | |||
|---|---|---|---|---|---|---|
| KChR | AvR | KChR | AvR | KChR | AvR | |
| Neurological diseases | 54 (23.48%) | 87 (17.51%) | 453 (24.39%) | 1844 (23.59%) | 110.39 | 66.50 |
| Ophthalmic diseases | 42 (18.26%) | 66 (13.28%) | 204 (10.99%) | 1217 (23.59%) | 49.71 | 43.89 |
| Genodermatoses | 14 (6.09%) | 36 (7.24%) | 169 (9.10%) | 1229 (15.57%) | 41.18 | 44.32 |
| Skeletal diseases | 29 (12.61%) | 77 (15.72%) | 115 (6.19%) | 1000 (15.72%) | 28.02 | 36.06 |
| Hereditary syndromes | 62 (26.95%) | 191 (38.43%) | 435 (23.43%) | 1483 (12.79%) | 106.00 | 53.48 |
| Other hereditary diseases (diseases of metabolism, blood, etc.) | 29 (12.61%) | 40 (8.05%) | 481 (25.90%) | 1045 (13.37%) | 117.21 | 37.68 |
| Total | 230 | 497 | 1857 | 7818 | 452.52 | 281.92 |
| District/City | Population | Load per 1000 People/Men * | |||
|---|---|---|---|---|---|
| AD | AR | XL * | Total | ||
| Load in Administrative Units (Districts, City) | |||||
| Ust-Dzhegutinsky District | 43,396 | 2.30 ± 0.230 | 2.00 ± 0.215 | 0.74 ± 0.184 | 5.05 ± 0.328 |
| Karachaevsky District | 35,500 | 2.54 ± 0.267 | 2.06 ± 0.240 | 0.9 ± 0.225 | 5.49 ± 0.376 |
| Malokarachaevsky District | 36,594 | 3.96 ± 0.328 | 2.62 ± 0.267 | 0.66 ± 0.189 | 7.24 ± 0.433 |
| Cherkessk City | 138,900 | 1.03 ± 0.086 | 0.95 ± 0.083 | 0.36 ± 0.072 | 2.34 ± 0.125 |
| Prikubansky District | 27,557 | 2.58 ± 0.305 | 1.81 ± 0.256 | 1.16 ± 0.290 | 5.55 ± 0.424 |
| Urupsky District | 18,074 | 1.88 ± 0.322 | 1.27 ± 0.265 | 0.89 ± 0.312 | 4.04 ± 0.445 |
| Zelenchuksky District | 43,588 | 1.88 ± 0.208 | 1.65 ± 0.195 | 1.06 ± 0.220 | 4.59 ± 0.305 |
| Abazinsky District | 14,631 | 2.87 ± 0.442 | 1.71 ± 0.341 | 1.78 ± 0.492 | 6.36 ± 0.610 |
| Khabezsky District | 25,474 | 4.71 ± 0.429 | 2.67 ± 0.323 | 1.26 ± 0.314 | 8.64 ± 0.558 |
| Adyge-Khablsky District | 11,178 | 3.58 ± 0.565 | 3.4 ± 0.551 | 1.97 ± 0.593 | 8.95 ± 0.841 |
| Nogaysky District | 15,475 | 5.82 ± 0.611 | 2.58 ± 0.408 | 1.42 ± 0.428 | 9.82± 0.764 |
| Load in the Ethnic Groups | |||||
| Karachays | 162,444 | 2.68 ± 0.128 | 2.00 ± 0.110 | 0.86 ± 0.103 | 5.55 ± 0.177 |
| Russian | 134,756 | 1.43 ± 0.103 | 1.13 ± 0.091 | 0.59 ± 0.049 | 3.15 ± 0.145 |
| Circassians | 50,817 | 2.83 ± 0.236 | 1.69 ± 0.182 | 1.02 ± 0.201 | 5.55 ± 0.314 |
| Abazins | 33,264 | 2.35 ± 0.263 | 1.60 ± 0.229 | 1.27 ± 0.275 | 5.22 ± 0.375 |
| Nogais | 14,741 | 5.22 ± 0.594 | 2.10 ± 0.377 | 0.14 ± 0.136 | 7.51 ± 0.706 |
| Other ethnos | 14,345 | 2.37 ± 0.406 | 2.30 ± 0.3400 | 1.39 ± 0.441 | 6.06 ± 0.610 |
| Total/average | 410,367 | 2.33 ± 0.075 | 1.72 ± 0.065 | 0.81 ± 0.063 | 4.86 ± 0.104 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zinchenko, R.A.; Makaov, A.K.; Marakhonov, A.V.; Galkina, V.A.; Kadyshev, V.V.; El’chinova, G.I.; Dadali, E.L.; Mikhailova, L.K.; Petrova, N.V.; Petrina, N.E.; et al. Epidemiology of Hereditary Diseases in the Karachay-Cherkess Republic. Int. J. Mol. Sci. 2020, 21, 325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010325
Zinchenko RA, Makaov AK, Marakhonov AV, Galkina VA, Kadyshev VV, El’chinova GI, Dadali EL, Mikhailova LK, Petrova NV, Petrina NE, et al. Epidemiology of Hereditary Diseases in the Karachay-Cherkess Republic. International Journal of Molecular Sciences. 2020; 21(1):325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010325
Chicago/Turabian StyleZinchenko, Rena A., Amin Kh. Makaov, Andrey V. Marakhonov, Varvara A. Galkina, Vitaly V. Kadyshev, Galina I. El’chinova, Elena L. Dadali, Lyudmila K. Mikhailova, Nika V. Petrova, Nina E. Petrina, and et al. 2020. "Epidemiology of Hereditary Diseases in the Karachay-Cherkess Republic" International Journal of Molecular Sciences 21, no. 1: 325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010325