Oxidative Stress-Responsive MicroRNAs in Heart Injury
by
 , , and
, , and
Branislav Kura
1 ,
,
Barbara Szeiffova Bacova
1,
Barbora Kalocayova
1,
Matus Sykora
1,2 and
Jan Slezak
1,* 1
Centre of Experimental Medicine, Institute for Heart Research, Slovak Academy of Sciences, 841 04 Bratislava, Slovakia
2
Department of Animal Physiology and Ethology, Faculty of Natural Sciences, Comenius University, 842 15 Bratislava, Slovakia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(1), 358; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010358
Submission received: 30 October 2019
/
Revised: 31 December 2019
/
Accepted: 3 January 2020
/
Published: 5 January 2020
(This article belongs to the Special Issue Crosstalk between MicroRNA and Oxidative Stress in Physiology and Pathology 2.0)
Abstract
:Reactive oxygen species (ROS) are important molecules in the living organisms as a part of many signaling pathways. However, if overproduced, they also play a significant role in the development of cardiovascular diseases, such as arrhythmia, cardiomyopathy, ischemia/reperfusion injury (e.g., myocardial infarction and heart transplantation), and heart failure. As a result of oxidative stress action, apoptosis, hypertrophy, and fibrosis may occur. MicroRNAs (miRNAs) represent important endogenous nucleotides that regulate many biological processes, including those involved in heart damage caused by oxidative stress. Oxidative stress can alter the expression level of many miRNAs. These changes in miRNA expression occur mainly via modulation of nuclear factor erythroid 2-related factor 2 (Nrf2), sirtuins, calcineurin/nuclear factor of activated T cell (NFAT), or nuclear factor kappa B (NF-κB) pathways. Up until now, several circulating miRNAs have been reported to be potential biomarkers of ROS-related cardiac diseases, including myocardial infarction, hypertrophy, ischemia/reperfusion, and heart failure, such as miRNA-499, miRNA-199, miRNA-21, miRNA-144, miRNA-208a, miRNA-34a, etc. On the other hand, a lot of studies are aimed at using miRNAs for therapeutic purposes. This review points to the need for studying the role of redox-sensitive miRNAs, to identify more effective biomarkers and develop better therapeutic targets for oxidative-stress-related heart diseases.
1. Introduction
Despite advances in disease prevention, diagnosis, and treatment, cardiovascular diseases (CVDs) are still in the leading position as the cause of mortality and morbidity worldwide. It is estimated that by 2030, nearly 23.6 million people will die from CVDs, primarily from heart disease and stroke, per year [1,2]. As both reactive oxygen species (ROS) production and microRNA (miRNA) expression signature have been associated with the development of CVDs, it is important to understand the crosstalk between ROS and miRNAs [3,4].
ROS are constantly released during mitochondrial oxygen consumption for energy production. Any imbalance between ROS production and its scavenger system induces oxidative stress. Oxidative stress, a critical contributor to tissue damage, is well-known to be associated with various diseases [5]. It has been long recognized that an increase of ROS can modify the cell-signaling proteins and has functional consequences, which mediate pathological processes included in the development of CVDs related to hypoxia, cardiotoxicity, and ischemia-reperfusion [6]. It was reported that myocardial ROS levels were elevated in animal models of ischemia/reperfusion injury [7] and heart failure [8]. Recent data from T. Wongsurawat study reveal elevated level of ROS species at the proteomic and transcriptomic level in the vessel wall [9,10,11].
MiRNAs are integrated into a group of small, naturally occurring and noncoding RNAs (size 21–25 nucleotides), which modulate gene expression at the post-transcriptional level. MiRNAs play a role as regulators of gene expression through binding to complementary sequences on the 3′-untranslated region (3′-UTR) of their target mRNA, thus inhibiting mRNA translation or promoting mRNA degradation [12,13]. Many miRNA genes are transcribed by enzyme RNA polymerase II from intergenic, intronic, or polycistronic loci as a long primary miRNA transcript (pri-miRNA), which is then cleaved by the enzyme Drosha endoribonuclease to a 70-nt-long hairpin structure with 2-nt-30 overhangs (pre-miRNA). Pre-miRNA is thereafter exported to the cytoplasm and processed by a second endoribonuclease enzyme (Dicer), to form a 22-nucleotide-long miRNA:miRNA* duplex with partial complementarity. One strand of this duplex then combines with the Argonaute (AGO) protein into the RNA-induced silencing complex (RISC), while the passenger strand gets degraded. One mRNA can contain multiple binding sites for different miRNAs, thus creating a complicated network of miRNA–mRNA interactions. MiRNAs are distributed in tissue-specific patterns and are able to regulate the expression of approximately 30% of human genes [14,15,16].
Numerous studies have shown that miRNAs have essential roles in cardiovascular development, pathology, regeneration, and repair and could be used for the diagnosis and prevention of cardiovascular diseases, such as hypertrophy, myocardial infarction, contractility defects, arrhythmias, etc. [3,17,18,19,20,21]. In response to increased ROS or stress stimuli, CVDs are obviously initiated and progressed by apoptosis, autophagy, necrosis, and fibrosis, as well as proliferation and migration of cardiomyocytes and endothelial cells, cardiac fibroblasts, and vascular smooth muscle cells. It was documented that miRNAs are involved in these processes [3,18,22]. Moreover, some miRNAs have been assigned as regulators of oxidative stress in the cardiovascular system by targeting ROS generators, antioxidant signaling pathways, and selected antioxidant effectors [23].
In this review, we tried to highlight recent findings about the association of miRNAs with the development of CVDs, including atherosclerosis, myocardial infarction, cardiac hypertrophy, or heart failure. There is evidence of interactions between cardiac miRNAs and ROS, but further studies need to be provided to reveal the molecular mechanisms of miRNAs regulating CVD diseases under ROS-related stress conditions.
2. Oxidative Stress and Cardiovascular System
The common pathological feature of most cardiac and vascular diseases is an imbalance of biological system in oxidation and antioxidation or between the generation and detoxification of ROS, which is generally called oxidative stress [24,25].
ROS are small reactive molecules implicated in the regulation of various cell functions and biological processes [26]. They are defined as molecules containing at least one atom of oxygen with higher reactivity than molecular oxygen, like superoxide anion (O2−), hydrogen peroxide (H2O2), hydroxyl radical (OH▪), peroxynitrite (ONOO−), hypochlorous acid (HOCl), and others [27]. In smaller or moderate concentrations, ROS can serve as signaling molecules, but an uncontrolled higher level of ROS leads to free-radical damage associated with the structural and functional alterations of proteins, lipids, and deoxyribonucleic acid [28,29].
The generation of ROS could be divided into two categories. Firstly, they are produced mainly by mitochondrial oxidative metabolism as a by-product or waste product, and secondly, as a cellular response to stress, xenobiotics, cytokines, and bacterial invasion, where ROS are formed intentionally as part of a signal transduction pathway [6,30]. Most relevant enzymatic sources of ROS are the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), xanthine oxidase, uncoupled nitric oxide (NO) synthase, and mitochondria [24]. To the other incentives belong tumor necrosis factor-alpha (TNF-α), epidermal growth factor, Interleukin-1beta (IL-1β), hypoxia, and irradiation [6].
To ensure homeostasis in these processes, proteins with an antioxidant activity that protect an aerobic organism from increased toxicity of ROS have been found [31]. Major antioxidant proteins are superoxide dismutases (SODs), which catalyze conversion of superoxide into oxygen and hydrogen peroxide [32]. Hydrogen peroxide produced by SOD is then subsequently transformed into water and oxygen by other antioxidant proteins, glutathione peroxidases, catalases, and thioredoxins [33]. Taken together, to keep redox equilibrium is very difficult due to a variety of mechanisms, and, in the case of the antioxidant cell defense system that is suppressed by oxidative stress, many diseases can occur, especially in the cardiovascular system, where oxygen delivery to myocardium is approximately 1.6–1.8 times higher than in other tissues [34].
Increased levels of ROS are unfavorably implicated in the myocardial calcium handling, cardiac remodeling induced by hypertrophic signaling, apoptosis, and necrosis. Oxidative stress similarly has a negative effect on blood vessels, their function, angiogenesis, apoptosis, vascular tone, and genomic stability [24]. This suggests that cardiovascular risk factors with elevated ROS levels are interconnected with endothelial dysfunction. Dysregulated generation of ROS also contributes to the pathogenesis of atherosclerosis, heart failure, cardiomyopathy, and cardiac hypertrophy [35] (Figure 1).
It was observed that activation of neuroendocrine pathways, namely sympathetic and the renin-angiotensin-aldosterone system in patients with failing myocardium, was associated with oxidative stress [36,37]. In experimental and human studies dealing with heart failure, the elevated activity of NOX has been consistently observed [37,38]. For example, the implication of NOX isoforms in the development of left ventricular hypertrophy (LVH) was demonstrated in NOX2 knockout mice, where infusion of Angiotensin II resulted in less incidence of LVH compared to control wild-type mice infused with the same concentration of Angiotensin II induced LVH [39]. A similar conclusion was drawn from neonatal rat ventricular cardiomyocytes where activation of NOX2 was associated with angiotensin II-induced cardiac hypertrophy [40]. Besides that, inactivation of NOX2 leads to the reduction of infarct size in a model of myocardial infarction [41]. Activation of NOX4 also has an impact on heart failure [42]. This implication was shown in experimental study with transgenic mice with cardiac-specific overexpression of NOX4, where incidence of fibrosis, apoptosis, and enlargement of cardiomyocytes were found [43]. Besides, upregulated expression of NOX4 was associated with overexpression of lysocardiolipin acyltransferase-1, which was implicated in the catalysis of cardiolipin synthesis [44]. Upregulated lysocardiolipin acyltransferase-1 by oxidative stress damages phospholipid cardiolipin, a component of the inner mitochondrial membrane leading to increased levels of O2−, ONOO−, and NO radicals, which results in the decline of ATP production and disorganization of the dimeric ADP/ATP carrier functional capacity [45,46]. Unlike this, in mice with cardiac-specific deletion of NOX4 with applied transverse aortic constriction, the pathological changes were less pronounced [43]. Expression and activity of xanthine oxidase were also increased in patients with heart failure, whereas inhibition of this enzyme ameliorated heart contractility, as well as endothelial dysfunction [47].
Hypertension, hypercholesterolemia, hyperglycemia, and atherosclerosis are also typical with the activation of ROS enzyme sources [24]. Elevated ROS levels, activation of NOX [48], impairment of NO/cGMP signaling, and subsequently reduced acetylcholine-mediated vasodilation in hypertension models were induced by Angiotensin II stimulation [49]. Moreover, a decline in superoxide dismutase and glutathione peroxidase activity were inversely correlated with blood pressure in untreated hypertensive patients [50]. The process of atherosclerosis is closely linked with lack of NO production or its accelerated scavenging [51]. Generated ONOO− induces transformation of smooth muscle cells into foam cells, as well as release of matrix metalloproteinases, which degrade atheromatous plaque and basement membrane of the endothelial cells leading to physical disruption of the plaques [26].
Another situation of oxidative-stress-related cardiac damage is heart transplantations, where the donor heart, graft, is exposed to cold ischemia-reperfusion injury associated with increased ROS. This leads to graft dysfunction, like allograft rejection, delayed graft function, or primary nonfunction, as well as to endothelial and parenchymal cell injury [52,53]. To increase success of the heart transplantation, experimental studies have focused on the enrichment of antioxidative substrates in cardioplegia solution and pump prime solution [54,55,56,57,58,59], suggesting the importance of continuous research into antioxidants.
3. Oxidative Stress and MiRNA
Many previous studies have demonstrated that multiple molecular mechanisms and signaling pathways can regulate oxidative stress [60]. Growing evidence has confirmed that miRNAs can be considered as potential targets and modulators of oxidative-stress-related pathways [61]. By analysis of miRNA expression signature implicated in oxidative stress-related pathways, several miRNAs were identified and termed as oxidative stress-responsive miRNAs [62]. The increasing number of studies shows that intracellular ROS can either inhibit or induce miRNA expression level, which results in subsequent biological effects through regulation of their direct target genes (Figure 2) [63]. Among them, several pathways (Nrf2—nuclear factor erythroid 2-related factor 2, SIRT1—sirtuin 1, and NF-κB—nuclear factor kappa B) have been the most intensively studied in connection with oxidative stress and miRNA. These will be further described in this section.
3.1. Nrf2 Pathway
Nrf2 plays a part in the cellular antioxidant defense system by upregulating the expression levels of antioxidant enzymes [64], such as glutathione S-transferase (GST), NAD(P)H:quinone oxidoreductase (NQO) 1, SOD1, and heme oxygenase (HO) 1, through binding to antioxidant response elements (AREs) in their promoters [23]. Under physiological conditions, Nrf2 is bound to its inhibitory protein, Kelch-like ECH-associated protein 1 (Keap1), which limits its transcriptional activity in the nucleus. Oxidative stress causes Nrf2 to dissociate from Keap1, which results in its binding to ARE and the transcription of downstream target genes [65].
Some miRNAs were reported to target Nrf2 directly. Zhu et al. revealed that miRNA-153 promotes oxidative stress by negatively regulating Nrf2 in an in vitro model of Parkinson’s disease [66]. The study of Sangokoya et al. shows that increased expression of miRNA-144 is associated with reduced Nrf2 levels in homozygous sickle cell disease (HbSS) reticulocytes and with decreased glutathione regeneration and attenuated antioxidant capacity in HbSS erythrocytes [67]. Another study found that the downregulation of miRNA-93 elevates Nrf2 expression and alleviates reactive oxygen species and cell apoptosis in diabetic retinopathy [68].
Besides directly targeting Nrf2, miRNAs may also target its regulators. Cheng et al. demonstrated that miRNA-141 attenuates UV-induced oxidative stress via targeting Keap1 to activate Nrf2 signaling in human retinal pigment epithelium cells and retinal ganglion cells [69]. MiRNA-7 represses Keap1 expression in human neuroblastoma cells, decreases the intracellular hydroperoxide level, and increases the level of the reduced form of glutathione [70]. Zhang et al. demonstrated that miRNA-455-3p activated the Nrf2/ARE signal pathway through suppressing Keap1, thereby suppressing oxidative stress and promoting osteoblasts growth [71]. Other authors showed that miRNA-200a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis [72].
3.2. SIRT1 Pathway
The increasing number of studies confirms that SIRT1 is an important component of cellular responses to oxidative stress [73,74]. SIRT1 is a target of various redox-sensitive pathways [23]. To induce an antioxidant response, activated SIRT1 deacetylates multiple targets, including endothelial nitric oxide synthase (eNOS), peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α), p53, Forkhead box O transcription factors (FoxO), Nrf2, and NF-κB [65]. In the cell, the deacetylation of FoxO1 by SIRT1 increases transcriptional activity and upregulates downstream antioxidants such as SOD2 and catalase [75].
Several miRNAs have been reported to influence oxidative stress by directly targeting SIRT1. Downregulation of SIRT1 by miRNA-34a promoted vascular smooth muscle cells senescence and inflammation in aged mouse aortas [76]. Similarly, miRNA-217 was identified as an endogenous SIRT1 inhibitor, which promotes endothelial senescence. MiRNA-217 was expressed in human atherosclerotic lesions and was negatively correlated with SIRT1 expression and with FoxO1 acetylation status [77]. Zhao el al. showed that miRNA-128-3p aggravated the doxorubicin-induced liver injury by promoting oxidative stress via targeting SIRT1 [78]. The study of Zhu et al. demonstrates a pro-apoptotic role of miRNA-195 in cardiomyocytes and identifies SIRT1 as a direct target of miRNA-195. The effect of miRNA-195 on apoptosis is mediated through the downregulation of SIRT1, Bcl-2 (B-cell lymphoma 2), and ROS production [79]. D′Adamo et al. identified miRNA-9 as a post-transcriptional regulator of SIRT1. MiRNA-9 and SIRT1 levels showed opposite changes in chondrocytes, following H2O2 treatment [80].
3.3. NF-κB Pathway
Excessive levels of ROS would also activate NF-κB pathway that is a redox-sensitive pathway [81]. NF-κB is present in all kinds of cells controlling the transcription of a wide variety of genes, including pro-apoptotic and pro-survival genes, pro-inflammatory cytokines, antioxidant and pro-oxidant enzymes, and many others [82]. The mammalian NF-κB family is composed of five members: p65 (RelA), RelB, c-Rel, NF-κB1 (p50 and its precursor p105), and NF-κB2 (p52 and its precursor p100), which can form homodimers and heterodimers among themselves. The NF-κB proteins are normally sequestered in the cytoplasm by a family of inhibitory proteins, including IκB family members. The most common activation reactions of NF-κB are represented by phosphorylation and activation of the IκB kinase complex [83].
One of the most important ways in which NF-κB activity influences ROS levels is via increased expression of antioxidant proteins such as SOD, glutathione peroxidase, or heme oxygenase. Since NF-κB is important in inflammation, some enzymes that promote the production of ROS (e.g., NOX2, inducible nitric oxide synthase (iNOS), cyclooxygenase (COX) 2, or cytochrome P450 enzymes) are also regulated as its targets, especially in cells of the immune system [82]. Abnormal NF-κB activity is frequently associated with an abnormal level of miRNAs, which is found to play critical roles in disease progression [84].
Downregulation of miRNA-155 ameliorates high-glucose-induced endothelial injury by inhibiting NF-κB activation and promoting HO-1 and NO production [71]. Gu revealed that miRNA-124 prevents H2O2-induced oxidative stress and apoptosis in human lens epithelial cells by suppressing the activation of the NF-κB pathway [85]. Results of Wei et al. indicated that NF-κB positively regulated miRNA-21 expression under oxidative stress, and programmed cell death protein 4 (PDCD4) was a direct target for miRNA-21 [86]. In another study, Xie et al. studied the role of miRNA-146a in the brain of chronic type 2 diabetes mellitus (cT2DM) rats. Increased inflammation and oxidative stress were associated with brain impairment in cT2DM rats, which were negatively correlated with miR-146a expression. The expressions of NF-κB p65 and its specific modulators were elevated in the brain of cT2DM rats, which might be inhibited by miR-146a [87].
Several other genes and related pathways have been described in the literature to be involved in the regulation of oxidative stress by miRNAs. Examples of these are MAPK (mitogen-activated protein kinase) signaling pathway, TGF-beta (transforming growth factor beta) signaling pathway, cell adhesion molecules (CAMs), calcium signaling pathway, VEGF (vascular endothelial growth factor) signaling pathway, etc. [61].
4. MiRNA in Oxidative-Stress-Induced Heart Diseases
Oxidative stress plays a crucial role in many cardiovascular diseases, like hypoxia, ischemia/reperfusion injury, or heart failure [88]. As mentioned before, intracellular ROS are formed in normal conditions as the result of normal mitochondrial respiration, but ROS are also produced during reperfusion in hypoxic tissue and in association with infection and inflammation, leading to pathological conditions of the heart [88,89]. One of the effects of ROS accumulation in cardiomyocytes is a different expression of noncoding RNAs (ncRNAs), subsequently contributing to cell apoptosis and heart pathology. Among these ncRNAs, miRNAs are the most intensively studied, as they have a huge impact on heart condition by inhibiting protein translation or target mRNA degradation [16,90,91].
4.1. Cardiac Hypertrophy
Oxidative stress generates many complex cellular changes in the heart which force it to adaptation in the form of cardiac hypertrophy of ventricles. These adaptations may provide initial salutary compensation to the arisen stress, sustained hypertrophic stimulation becomes maladaptive, worsening morbidity, and mortality risks because of congestive heart failure and sudden cardiac death [92]. During hypertrophy in different experiments with animal models or in clinical trials, changes in miRNA expression were observed—mainly miRNA-1 and -133. Zhao et al. demonstrated that their lower levels indicate a significant cardiac injury [93]. MiRNA-1 is connected with cardiomyocyte growth and hypertrophy most probably through the calcineurin/nuclear factor of activated T cell (NFAT) signaling pathway inhibition [94,95]. As a previous miRNA-1 case, also patients and animals with cardiac hypertrophy have lower levels of miRNA-133, probably by regulating antihypertrophic genes like guanosine diphosphate–guanosine triphosphate (GDP–GTP) exchange protein, or signal transduction kinase cell division control protein 42 (Cdc42) [96,97].
MiRNA-208 belongs to the cardiac-specific miRNAs, and its regulation is important in the processes of cardiac remodeling. Several studies demonstrated that cardiac hypertrophy is caused by switching of adult alpha myosin heavy chain (α-MHC, known as Myh6) to fetal beta myosin heavy chain (β-MHC, known as Myh7) gene expression. In experimental studies, deletion of miRNA-208a, which is encoded in the intron of the Myh6 gene, leads to the decreased expression of the Myh7 gene in response to stress and to hypertrophy [88,98]. These results were confirmed in the study of Rawal et al., where inhibition of miRNA-208a hampers the activation of β-MHC and hypertrophic response [99].
Important miRNAs involved in the cardiac hypertrophy are also miRNA-22 (influence phosphatidylinositol-3-kinase (PI3K)-protein kinase B (AKT)) [100], miRNA-212/132 family (active through antihypertrophic FoxO3 transcription factor), or miRNA-199 (miRNA-199a targets the pro-autophagic and antihypertrophic factor glycogen synthase kinase 3β; miRNA-199b acts through targeting tyrosine phosphorylation regulated kinase 1A (Dyrk1a) gene, involved in the phosphorylation of NFAT factors) [101,102,103]. Other studies dealing with miRNAs associated with cardiac hypertrophy observed changed expression of miRNA-21, -18b, -195, -199, -29, -22, or -23 levels [104,105,106,107].
4.2. Ischemia/Reperfusion Injury
Cardiac ischemia/reperfusion (I/R) injury involves the damage caused by reduced coronary blood flow, causing depletion of ATP, reduced partial pressure of oxygen, and production of toxins. Reperfusion leads to further damage through generation of ROS and a proton gradient across both the sarcolemma and the inner mitochondrial membrane [108]. Many miRNAs are involved in these processes, either as a result of damage caused by ROS generation or directly responsive to ROS.
One of the most promising miRNAs for potential use as a diagnostic or therapeutic target is miRNA-24-3p. In a very recent study of Xiao et al., decrease expression of miRNA-24-3p during induced ischemia/reperfusion injury in mouse hearts and decreasing levels of apoptosis of cardiomyocytes caused by ROS during ischemia/reperfusion injury after application of miRNA mimics were confirmed [109]. In addition to these observations, the authors identified the Keap1-Nrf2 pathway as one of the possible targets of miRNA-24-3p [109].
Lusha et al. showed that miRNA-144, which is primarily connected with the regulation of apoptosis in human cancer diseases, is another miRNA with changed expression levels in the I/R model through regulation of FoxO1. The authors observed in their study reduced infarct size and apoptosis in cardiomyocytes during the upregulation of miRNA-144 and increased sensitivity to I/R in the situation with depleted miRNA-144 [110]. FoxO1 protein is an important transcription factor which mediates apoptosis by activating iNOS expression in cardiomyocytes [111], and it can be regulated by sirtuin 1 in the cardiovascular system [112].
In another study, Fang and Yeh explored the role of miRNAs in cardiomyocyte apoptosis induced by ischemia. They found an increased level of miRNA-302 expression induced by hypoxia/reoxygenation injury. This aggravated cardiomyocyte apoptosis probably by inhibiting antiapoptotic protein myeloid cell leukemia 1 (Mcl-1) expression, thereby activating pro-apoptotic molecules. Based on this data, the authors suggested that elevated miRNA-302 levels can be detrimental to cells, but decreased levels are beneficial and can lead to effective therapeutic intervention [113]. Several publications reveal profound effects of myocardial ischemia on miRNA transcript in the vessel wall and vascular smooth muscle cells, in particular [114,115,116].
MiRNA-23a promotes cardiomyocyte apoptosis and myocardial infarction induced by I/R through directly suppressing the expression of manganese SOD, an important antioxidant for scavenging of superoxide [117]. This is one of the endogenous enzymes that protects cells from oxidative stress. Wang et al. observed that miRNA-1 worsens cardiac oxidative stress by post-transcriptional modification of the antioxidant network in the I/R injury C57BL/6 mice model. They found that miRNA-1 reduced the protein levels of antioxidant enzymes glutamate cysteine ligase (Gclc), SOD1, and glucose-6-phosphate dehydrogenase (G6PD) under oxidative stress conditions [118]. Other miRNAs, such as miRNA-130a [119] and miRNA-98 [120], are investigated to be associated with ROS-related cardiomyocyte apoptosis. MiRNA-208a promoted apoptosis and oxidative stress in the I/R injury rat model by regulation of protein tyrosine phosphatase receptor type G and protein tyrosine phosphatase, non-receptor type 4 [121].
Another situation of ischemia/reperfusion injury is heart transplantations. During the transplantation process in the final step, where heart graft is connected to the circulatory system of the recipient and when reperfusion of the graft is started, there is an excessive production of free radicals, what could lead to graft failures and lower long-term survival rate of the patients [122]. In several works, more attention is focused on miRNA′s changed expression after transplantation, making miRNAs as an ideal candidate for biomarkers of transplant rejection [122,123,124]. Zhou et al. found upregulation of miRNA-711, -2137, -705, -5130, -346, -714, and -744 and downregulation of miRNA-210, -490, -491, -425, -423-3p, and -532-3p in experiments on C56BL/6 mouse animal models after heart transplantation in I/R injured hearts [122].
MiRNAs were measured in the samples from endomyocardial biopsies (EMB) and blood serum in 30 patients with rejecting heart allograft and 30 patients without allograft rejection. MiRNA analyzes revealed changed expression of miRNA-10a, -31, -92a, and -155 in EMB, as well as in blood serum [125]. These miRNAs are strongly associated with inflammatory processes, as they influenced NF-κB, TNF-α, interleukins -6, -8, and -1, monocyte chemoattractant protein-1, eNOS, or vascular cell adhesion protein [126,127,128]. Wei et al. also suggested that miRNA-183, -182, and -96 have an important function in the regulation of graft rejection probably through regulation of FoxO1 expression and could be new potential noninvasive biomarkers of allograft rejection in heart transplantation [129].
All these results suggested the potential role of miRNAs in the regulation and adaptation of transplanted allografts and their potential use as biomarkers in grafts rejection.
4.3. Coronary Artery Diseases (CAD)
CAD is an atherosclerotic disease which is inflammatory in nature. Atherosclerosis starts due to the accumulation of lipoproteins in the intima of the coronary vessels. Oxidized or modified low-density lipoprotein then attract leukocytes into the intima of the coronary vessels, which can be scavenged by macrophages, leading to the formation of foamy cells. The atherosclerotic plaque starts developing. Cell death or apoptosis occurs commonly in the atherosclerotic lesions. The modified lipoproteins propagate inflammatory responses. As a result, obstruction of blood flow occurs, and this leads to a mismatch between myocardial oxygen demand and supply [130].
Endothelial cell apoptosis under oxidative stress plays a critical role in the initiation and progression of atherosclerosis [131,132,133,134,135,136,137,138,139,140,141]. Li et al. observed that overexpression of miRNA-210 caused inhibition of apoptosis and reduction of ROS level in human umbilical vein endothelial cells (HUVECs) treated with H2O2 and also downregulation of caspase levels. This study leads to the conclusion that miRNA-210 could have a role in the protection against oxidative-stress-induced apoptosis in HUVECs [131].
MiRNA-24 is highly expressed in the vessel wall and changes of its expression are connected with dysfunction and injury of vascular endothelial cells [132]. It is believed that miRNA-24 participates in many pathophysiological processes including I/R injury or vascular oxidative stress [134,135,136,137]. In an experimental study by Zhang et al., miRNA-24 was upregulated, and it has a supportive effect on vascular endothelium repair by attenuating oxidative-stress-induced damage of endothelial cells. In this case, miRNA-24 regulated the Nrf2/HO-1 signaling pathway indirectly by regulating of Keap1 after influencing of O-linked β-N-acetylglucosamine transferase gene (Ogt). On the other hand, authors confirmed that miRNA-24 affects the expression of SOD, malondialdehyde, and glutathione peroxidase [137].
MiRNA-92a overexpression impairs endothelial function and suppresses HO-1 expression in endothelial cells. Inhibition of miRNA-92a attenuates oxidative stress and improves endothelial function through enhancing HO-1 expression and activity in diabetic mouse aortas [138]. Yamac et al. observed markedly lowered expression of miRNA-199a in patients with coronary artery disease [139]. In parallel, they also showed the induction of cardioprotective protein SIRT1, a potential target of miRNA-199. Significantly increased expression of miRNA-146a was observed in patients with acute coronary syndrome. This miRNA is connected with the inflammatory pathway by regulating NF-κB [140]. According to O´Sullivan et al., miRNA-93-5p belongs to one of the strongest predictors of coronary artery disease when its expression was significantly upregulated in the patients with CAD, most likely through modulation of ATP-binding cassette A1 (ABCA1) gene [141]. ABCA1 plays an important role in cholesterol homeostasis and atherogenesis, and it could be reduced by oxidative stress [133].
4.4. Heart Failure
A complex syndrome resulting from structural or functional cardiac disorders, leading to disability of ventricle to fill or eject blood, is called heart failure [142,143,144], and it is considered to be one of the leading cause of morbidity and mortality worldwide [142,145,146,147,148]. Development of heart failure depends on many circumstances in the organism, but one of the key pathophysiological pathways for it is oxidative stress [149,150,151]. According to a lot of animal and human studies, multiple miRNAs are changed in models of heart failure, including miRNA-199b, -195, -100, -133, -24, and -208 [94,152,153,154,155,156]. Upregulated levels of miRNA-199b were measured during heart failure and appeared to target the calcineurin/NFAT pathway. It was proved that this calcineurin/NFAT pathway is activated after oxidative stress stimuli [157,158]. In the in vivo experiments, inhibition of miRNA-199b caused normalization of the expression of Dyrk1a, a reduction of nuclear NFAT activity, and inhibition of hypertrophy and fibrosis in mouse models of heart failure [101,159]. Changed expressions of miRNA-1, -214, -29b, -342, -7, -107, -126, -125, -122, -423-5p, -320a, -650, -1228, -662, -583, -3175, -21, -22, and -92b have been shown in other studies of heart failure [106,152,156,160,161,162,163].
A brief review of selected miRNAs included in the cardiovascular diseases caused by oxidative stress is provided in Table 1.
5. Future Perspectives of Using MiRNA in Disease Diagnosis and Treatment
Since 2001, miRNAs have been recognized as biomarkers and possible therapeutic targets for the diagnosis and treatment of diseases [165]. One of the biggest advantages for using miRNAs as biomarkers is their stability under many different conditions. MiRNAs can be stored at room temperature, frozen, or thawed [166]. Bioavailability of miRNA is another great advantage. MiRNAs can be isolated from various biological materials, like from peripheral blood, fresh and frozen tissues, or formalin-fixed, paraffin-wax-embedded samples, but also from saliva, epithelium of the skin, or hair [167,168]. Difficulties in the use of therapeutically altering miRNAs lie in their non-specificity— single miRNA can target many genes and influence more than one gene expression, so they could affect also other pathways in the organisms [169]. MiRNAs impose a relatively modest effect on their target, reflecting that individual mRNAs are targeted by multiple miRNAs, while the cellular proteome might be able to compensate the absence of a single miRNA [170].
Manipulation of RNA using miRNA mimics and antagomirs holds significant therapeutic potential for treating a variety of diseases. With recent technological advances, identification and validation of potential therapeutic miRNA targets are readily available [165]. Treatment of diseases by modulation of selected miRNAs in the organisms is based on two approaches. First, miRNA mimics is an approach for gene silencing due to generating synthetized artificial double-stranded miRNA-like RNA fragments. These molecules are able to bind to target mRNA and suppressed genes [171]. The second approach uses antagomirs, chemically designed oligonucleotides. These oligonucleotides specifically inhibit target miRNA by binding to them, which leads to reduction of RISC activation and to upregulation of genes [172,173]. MiRNAs could be modulated also by miRNA sponges (target mimicry), masking, and erasers. MiRNA sponges contain a binding site for the miRNA family, leading to the blocking of the activity of miRNAs [174,175]. Masking is based on the occupation of the binding site on target mRNA by oligonucleotides [176]. Erasers are oligonucleotides complementary to specific miRNA, leading to inhibition of its function [177]. However, delivery of anti-miRNAs and miRNAs in vivo may prove to be challenging [165].
6. Conclusions
Oxidative stress is one of the important contributing factors in cardiovascular disease genesis and development. Excessive ROS production has a significant impact on the pathogenesis of cardiovascular diseases related to atherosclerosis, cardiomyopathy, ischemia/reperfusion, and heart failure. Published literature highlights the increasing importance of studying the role of redox-sensitive miRNAs to identify more effective biomarkers and develop better therapeutic targets for oxidative-stress-related diseases. It is necessary to define the roles of individual miRNAs and their important targets, to determine their potential for possible diagnosis/treatment of cardiovascular disorders. Although a number of targets of oxidative-stress-responsive miRNAs have been identified, e.g., Nrf2, SIRT1, and NF-κB, future studies are still needed to determine further potential targets and their links to cardiovascular disease. MiRNA may be a promising novel tool and means in the clinical diagnosis, prognostic evaluation, and even therapeutic intervention of oxidative-stress-related CVD. The knowledge of the crosstalk between miRNAs, ROS, and cardiovascular diseases can contribute to new therapeutic approaches based on the suppression of ROS effects, with the potential to ameliorate or prevent the progression of cardiovascular diseases. However, several studies are still required to validate the present findings before the application of miRNA in clinical practice.
Author Contributions
J.S. supervised the writing project of the manuscript; B.K. (Branislav Kura), B.K. (Barbora Kalocayova), and B.S.B. prepared the manuscript and wrote the draft together; M.S. prepared the figures. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant from the Slovak Research and Development Agency of Ministry of Education, Science, Research, and Sport of the Slovak Republic (APVV-15-0376) and grants from the Scientific Grant Agency of the Ministry of Education, Science, Research, and Sport of the Slovak Republic and the Slovak Academy of Sciences (VEGA 2/0021/15, VEGA 2/0063/18, VEGA 2/0158/19, VEGA 2/0166/17, VEGA 2/0002/20) and grant from European Union Structural funds (ITMS 26230120009).
Acknowledgments
The authors thanks all of the individuals who participated in the investigations.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- WHO. About Cardiovascular Diseases. Available online: https://www.who.int/cardiovascular_diseases/about_cvd/en/ (accessed on 30 October 2019).
- Li, M.; Duan, L.; Li, Y.; Liu, B. Long noncoding RNA/circular noncoding RNA–miRNA–mRNA axes in cardiovascular diseases. Life Sci. 2019, 233, 116440. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Hirakawa, Y.; Nangaku, M. The role of oxidative stress and hypoxia in renal disease. Kidney Res. Clin. Pract. 2019, 38, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugamura, K.; Keaney, J.F. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, D.B.; Siwik, D.A.; Xiao, L.; Pimentel, D.R.; Singh, K.; Colucci, W.S. Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell. Cardiol. 2002, 34, 379–388. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Woo, C.C.; Giannakakis, A.; Lin, X.Y.; Cheow, E.S.H.; Lee, C.N.; Richards, M.; Sze, S.K.; Nookaew, I.; Kuznetsov, V.A.; et al. Transcriptome alterations of vascular smooth muscle cells in aortic wall of myocardial infarction patients. Data Br. 2018, 17, 1112–1135. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Woo, C.C.; Giannakakis, A.; Lin, X.Y.; Cheow, E.S.H.; Lee, C.N.; Richards, M.; Sze, S.K.; Nookaew, I.; Kuznetsov, V.A.; et al. Distinctive molecular signature and activated signaling pathways in aortic smooth muscle cells of patients with myocardial infarction. Atherosclerosis 2018, 271, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Derda, A.A.; Woo, C.C.; Wongsurawat, T.; Richards, M.; Lee, C.N.; Kofidis, T.; Kuznetsov, V.A.; Sorokin, V.A. Gene expression profile analysis of aortic vascular smooth muscle cells reveals upregulation of cadherin genes in myocardial infarction patients. Physiol. Genom. 2018, 50, 648–657. [Google Scholar] [CrossRef]
- Date, R.A. Bradyrhizobium effectiveness responses in Stylosanthes hamata and S. seabrana. Trop. Grassl. 2010, 44, 141–157. [Google Scholar]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Duygu, B.; de Windt, L.J.; da Costa Martins, P.A. Targeting microRNAs in heart failure. Trends Cardiovasc. Med. 2016, 26, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Colpaert, R.M.W.; Calore, M. MicroRNAs in Cardiac Diseases. Cells 2019, 8, 737. [Google Scholar] [CrossRef] [Green Version]
- Valášková, Z.; Mladosievičová, B.; Hulín, I.; Maruščáková, L. MicroRNA—information about myocardial damage or biomarker of heart failure? Cardiol. Lett. 2017, 26, 299–302. [Google Scholar]
- Kreutzer, F.P.; Fiedler, J.; Thum, T. Non-coding RNAs: Key players in cardiac disease. J. Physiol. 2019. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef]
- Deng, J.; Zhong, Q. Advanced research on the microRNA mechanism in heart failure. Int. J. Cardiol. 2016, 220, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.Y.; Luo, J.Y.; Wang, L.; Huang, Y. MicroRNAs Regulating Reactive Oxygen Species in Cardiovascular Diseases. Antioxid. Redox Signal. 2018, 29, 1092–1107. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature Part 2 of a 3-Part Series HHS Public Access PATHOPHYSIOLOGICAL ROLE OF OXIDATIVE STRESS IN HEART FAILURE. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.J.; Ahamed, M.; Alhadlaq, H.A.; Alshamsan, A. Mechanism of ROS scavenging and antioxidant signalling by redox metallic and fullerene nanomaterials: Potential implications in ROS associated degenerative disorders. Biochim. Biophys. Acta 2017, 1861, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Palade, F.; Alexa, I.D.; Azoicai, D.; Panaghiu, L.; Ungureanu, G. Oxidative stress in atherosclerosis. Rev. Med. 2003, 107, 502–511. [Google Scholar]
- Halliwell, B. Antioxidants in Human Health and Disease. Annu. Rev. Nutr. 1996, 16, 33–50. [Google Scholar] [CrossRef]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxid. Med. Cell. Longev. 2016, 2016, 1656450. [Google Scholar] [CrossRef] [Green Version]
- Syu, J.-P.; Chi, J.-T.; Kung, H.-N. Nrf2 Contributes to the Poor Prognosis and Chemoresistance. A Master Regul. Oxidative Stress 2016. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Mccord, J.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol. Sci. 2013, 34, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Sanyal, A.J. MicroRNAs. Signal. Pathw. Liver Dis. 2010, 107, 493–499. [Google Scholar]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef] [Green Version]
- Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef]
- Maack, C.; Kartes, T.; Kilter, H.; Schäfers, H.J.; Nickenig, G.; Böhm, M.; Laufs, U. Oxygen free radical, release in human failing myocardium is associated with increased activity of Rac1-GTPase and represents a target for statin treatment. Circulation 2003, 108, 1567–1574. [Google Scholar] [CrossRef] [Green Version]
- Mollnau, H.; Oelze, M.; August, M.; Wendt, M.; Daiber, A.; Schulz, E.; Baldus, S.; Kleschyov, A.L.; Materne, A.; Wenzel, P.; et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2554–2559. [Google Scholar] [CrossRef] [Green Version]
- Bendall, J.K.; Cave, A.C.; Heymes, C.; Gall, N.; Shah, A.M. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 2002, 105, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Nakagami, H.; Takemoto, M.; Liao, J.K. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2003, 35, 851–859. [Google Scholar] [CrossRef]
- Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.; Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin ii-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadicherla, A.K.; Stowe, D.F.; Antholine, W.E.; Yang, M.; Camara, A.K.S. Damage to mitochondrial complex i during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim. Biophys. Acta 2012, 1817, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 2002, 286, 135–141. [Google Scholar] [CrossRef]
- Baldus, S.; Müllerleile, K.; Chumley, P.; Steven, D.; Rudolph, V.; Lund, G.K.; Staude, H.J.; Stork, A.; Köster, R.; Kähler, J.; et al. Inhibition of xanthine oxidase improves myocardial contractility in patients with ischemic cardiomyopathy. Free Radic. Biol. Med. 2006, 41, 1282–1288. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Bitar, M.S.; Wahid, S.; Mustafa, S.; Al-Saleh, E.; Dhaunsi, G.S.; Al-Mulla, F. Nitric oxide dynamics and endothelial dysfunction in type II model of genetic diabetes. Eur. J. Pharmacol. 2005, 511, 53–64. [Google Scholar] [CrossRef]
- Pedro-Botet, J.; Covas, M.I.; Martín, S.; Rubiés-Prat, J. Decreased endogenous antioxidant enzymatic status in essential hypertension. J. Hum. Hypertens. 2000, 14, 343–345. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2749. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Xue, F. Current antioxidant treatments in organ transplantation. Oxid. Med. Cell. Longev. 2016, 2016, 8678510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela, C.; Thomsen, M.; Delbosc, S.; Calise, D.; Cristol, J.P.; Mourad, G. Lipid and Oxidative Stress Disorders in a Rat Model of Chronic Rejection. Transplant. Proc. 2007, 39, 2617–2619. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.; Webb, G.; Kennington, S.; Kelleher, N.; Sheppard, J.; Kuo, J.; Unsworth-White, J. Mannitol in cardioplegia as an oxygen free radical scavenger measured by malondialdehyde. Perfusion 2002, 17, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, E.R. Substrate enhancement of cardioplegic solution: Experimental studies and clinical evaluation. Ann. Thorac. Surg. 1995, 60, 797–800. [Google Scholar] [CrossRef]
- Fudulu, D.; Angelini, G. Oxidative Stress after Surgery on the Immature Heart. Oxid. Med. Cell. Longev. 2016, 2016, 1971452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.B.; Hicks, M.; Cropper, J.R.; Nicholson, A.; Kesteven, S.H.; Wilson, M.K.; Feneley, M.P.; Macdonald, P.S. Lazaroid (U74389G)-supplemented cardioplegia: Results of a double-blind, randomized, controlled trial in a porcine model of orthotopic heart transplantation. J. Hear. Lung Transplant. 2003, 22, 347–356. [Google Scholar] [CrossRef]
- Watson, A.J.; Gao, L.; Sun, L.; Tsun, J.; Jabbour, A.; Ru Qiu, M.; Jansz, P.C.; Hicks, M.; MacDonald, P.S. Enhanced preservation of the rat heart after prolonged hypothermic ischemia with erythropoietin-supplemented Celsior solution. J. Hear. Lung Transplant. 2013, 32, 633–640. [Google Scholar] [CrossRef]
- Villanueva, J.E.; Gao, L.; Chew, H.C.; Hicks, M.; Doyle, A.; Qui, M.R.; Dhital, K.K.; MacDonald, P.S.; Jabbour, A. Functional recovery after dantrolenesupplementation of cold stored hearts using an ex vivo isolated working rat heart model. PLoS ONE 2018, 13, e0205850. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Engedal, N.; Žerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurrò, V.; Betti, M.; Albertini, M.C. From oxidative stress damage to pathways, networks, and autophagy via microRNAs. Oxid. Med. Cell. Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef]
- Wan, Y.; Cui, R.; Gu, J.; Zhang, X.; Xiang, X.; Liu, C.; Qu, K.; Lin, T. Identification of Four Oxidative Stress-Responsive MicroRNAs, miR-34a-5p, miR-1915-3p, miR-638, and miR-150-3p, in Hepatocellular Carcinoma. Oxid. Med. Cell. Longev. 2017, 2017, 5189138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, J.; Khanna, S.; Bhattacharya, A. MicroRNA Regulation of Oxidative Stress. Oxid. Med. Cell. Longev. 2017, 2017, 2872156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Y.; Zhu, G.Y.; Su, X.H.; Wang, R.; Liu, J.; Liao, K.; Ren, R.; Li, T.; Liu, L. 7-deacetylgedunin suppresses inflammatory responses through activation of Keap1/Nrf2/HO-1 signaling. Oncotarget 2017, 8, 55051–55063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Wang, S.; Qi, W.; Xu, X.; Liang, Y. Overexpression of miR-153 promotes oxidative stress in MPP+-induced PD model by negatively regulating the Nrf2/HO-1 signaling pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 4179–4187. [Google Scholar]
- Sangokoya, C.; Telen, M.J.; Chi, J.T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010, 116, 4338–4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Zhao, X.; Yang, Z.; Min, X. Downregulation of miR-93 elevates Nrf2 expression and alleviates reactive oxygen species and cell apoptosis in diabetic retinopathy. Int. J. Clin. Exp. Med. 2019, 12, 10235–10243. [Google Scholar]
- Cheng, L.B.; Li, K.; Yi, N.; Li, X.M.; Wang, F.; Xue, B.; Pan, Y.; Yao, J.; Jiang, Q.; Wu, Z.F. miRNA-141 attenuates UV-induced oxidative stress via activating Keap1-Nrf2 signaling in human retinal pigment epithelium cells and retinal ganglion cells. Oncotarget 2017, 8, 13186–13194. [Google Scholar] [CrossRef]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Junn, E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic. Biol. Med. 2015, 89, 548–556. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wu, W.; Jiao, G.; Li, C.; Liu, H. MiR-455-3p activates Nrf2/ARE signaling via HDAC2 and protects osteoblasts from oxidative stress. Int. J. Biol. Macromol. 2018, 107, 2094–2101. [Google Scholar] [CrossRef]
- Yang, J.J.; Tao, H.; Hu, W.; Liu, L.P.; Shi, K.H.; Deng, Z.Y.; Li, J. MicroRNA-200a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis. Cell. Signal. 2014, 26, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.; Kim, Y.N.; Lee, I.H. Deacetylation of CHK2 by SIRT1 protects cells from oxidative stress-dependent DNA damage response. Exp. Mol. Med. 2019, 51, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between oxidative stress and SIRT1: Impact on the aging process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, A.; Molkentin, J.D.; Paik, J.H.; DePinho, R.A.; Yutzey, K.E. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 2011, 286, 7468–7478. [Google Scholar] [CrossRef] [Green Version]
- Badi, I.; Burba, I.; Ruggeri, C.; Zeni, F.; Bertolotti, M.; Scopece, A.; Pompilio, G.; Raucci, A. MicroRNA-34a Induces Vascular Smooth Muscle Cells Senescence by SIRT1 Downregulation and Promotes the Expression of Age-Associated Pro-inflammatory Secretory Factors. J. Gerontol. 2015, 70, 1304–1311. [Google Scholar] [CrossRef]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Jin, Y.; Li, L.; Xu, L.; Tang, Z.; Qi, Y.; Yin, L.; Peng, J. MicroRNA-128-3p aggravates doxorubicin-induced liver injury by promoting oxidative stress via targeting Sirtuin-1. Pharmacol. Res. 2019, 146, 104276. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, Y.; Wang, Y.; Li, J.; Schiller, P.W.; Peng, T. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc. Res. 2011, 92, 75–84. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, S.; Cetrullo, S.; Guidotti, S.; Borzì, R.M.; Flamigni, F. Hydroxytyrosol modulates the levels of microRNA-9 and its target sirtuin-1 thereby counteracting oxidative stress-induced chondrocyte death. Osteoarthr. Cartil. 2017, 25, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Gaspar-Pereira, S.; Fullard, N.; Townsend, P.A.; Banks, P.S.; Ellis, E.L.; Fox, C.; Maxwell, A.G.; Murphy, L.B.; Kirk, A.; Bauer, R.; et al. The NF-κB subunit c-Rel stimulates cardiac hypertrophy and fibrosis. Am. J. Pathol. 2012, 180, 929–939. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-κB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Tong, L.; Wu, S. MicroRNA and NF-kappa B. Adv. Exp. Med. Biol. 2015, 887, 157–170. [Google Scholar] [PubMed]
- Gu, X.L. MicroRNA-124 Prevents H 2 O 2 -Induced Apoptosis and Oxidative Stress in Human Lens Epithelial Cells via Inhibition of the NF-κB Signaling Pathway. Pharmacology 2018, 102, 213–222. [Google Scholar] [CrossRef]
- Wei, C.; Li, L.; Kim, I.K.; Sun, P.; Gupta, S. NF-κB mediated miR-21 regulation in cardiomyocytes apoptosis under oxidative stress. Free Radic. Res. 2014, 48, 282–291. [Google Scholar] [CrossRef]
- Xie, Y.; Chu, A.; Feng, Y.; Chen, L.; Shao, Y.; Luo, Q.; Deng, X.; Wu, M.; Shi, X.; Chen, Y. MicroRNA-146a: A comprehensive indicator of inflammation and oxidative stress status induced in the brain of chronic T2DM rats. Front. Pharmacol. 2018, 9, 478. [Google Scholar] [CrossRef]
- Farías, J.G.; Molina, V.M.; Carrasco, R.A.; Zepeda, A.B.; Figueroa, E.; Letelier, P.; Castillo, R.L. Antioxidant therapeutic strategies for cardiovascular conditions associated with oxidative stress. Nutrients 2017, 9, 966. [Google Scholar] [CrossRef]
- Sekhon, M.S.; Ainslie, P.N.; Griesdale, D.E. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: A “two-hit” model. Crit. Care 2017, 21, 90. [Google Scholar] [CrossRef] [Green Version]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009, 21, 452–460. [Google Scholar] [CrossRef]
- Dong, Y.; Xu, W.; Liu, C.; Liu, P.; Li, P.; Wang, K. Reactive oxygen species related noncoding RNAs as regulators of cardiovascular diseases. Int. J. Biol. Sci. 2019, 15, 680–687. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.-H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 Negatively Regulates Expression of the Hypertrophy-Associated Calmodulin and Mef2a Genes. Mol. Cell. Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef] [Green Version]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kamińska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [Green Version]
- Rawal, S.; Nagesh, P.T.; Coffey, S.; Van Hout, I.; Galvin, I.F.; Bunton, R.W.; Davis, P.; Williams, M.J.A.; Katare, R. Early dysregulation of cardiac-specific microRNA-208a is linked to maladaptive cardiac remodelling in diabetic myocardium. Cardiovasc. Diabetol. 2019, 18, 13. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Chen, Y.; Li, F. Attenuation of MicroRNA-495 Derepressed PTEN to Effectively Protect Rat Cardiomyocytes from Hypertrophy. Cardiology 2018, 139, 245–254. [Google Scholar] [CrossRef]
- Da Costa Martins, P.A.; Salic, K.; Gladka, M.M.; Armand, A.S.; Leptidis, S.; El Azzouzi, H.; Hansen, A.; Coenen-De Roo, C.J.; Bierhuizen, M.F.; Van Der Nagel, R.; et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat. Cell Biol. 2010, 12, 1220–1227. [Google Scholar] [CrossRef]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. MiR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017, 24, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Kura, B.; Parikh, M.; Slezak, J.; Pierce, G.N. The influence of diet on microRNAs that impact cardiovascular disease. Molecules 2019, 24, 1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Yang, B. Role of microRNAs in cardiac hypertrophy, myocardial fibrosis and heart failure. Acta Pharm. Sin. B 2011, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cai, J. The role of microRNAs in heart failure. Biochim. Biophys. Acta 2017, 1863, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Van Empel, V.P.M.; De Windt, L.J.; Da Costa Martins, P.A. Circulating miRNAs: Reflecting or affecting cardiovascular disease. Curr. Hypertens. Rep. 2012, 14, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Sassi, Y.; Avramopoulos, P.; Ramanujam, D.; Grüter, L.; Werfel, S.; Giosele, S.; Brunner, A.D.; Esfandyari, D.; Papadopoulou, A.S.; De Strooper, B.; et al. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat. Commun. 2017, 8, 1614. [Google Scholar] [CrossRef]
- Piper, H.M.; Meuter, K.; Schäfer, C. Cellular mechanisms of ischemia-reperfusion injury. Ann. Thorac. Surg. 2003, 75, S644–S648. [Google Scholar] [CrossRef]
- Xiao, X.; Lu, Z.; Lin, V.; May, A.; Shaw, D.H.; Wang, Z.; Che, B.; Tran, K.; Du, H.; Shaw, P.X. MicroRNA miR-24-3p reduces apoptosis and regulates Keap1-Nrf2 pathway in mouse cardiomyocytes responding to ischemia/reperfusion injury. Oxid. Med. Cell. Longev. 2018, 2018, 7042105. [Google Scholar] [CrossRef]
- Jiang, H.; Lu, Z. MicroRNA-144 attenuates cardiac ischemia/reperfusion injury by targeting FOXO1. Exp. Ther. Med. 2019, 17, 2152–2160. [Google Scholar]
- Puthanveetil, P.; Zhang, D.; Wang, Y.; Wang, F.; Wan, A.; Abrahani, A.; Rodrigues, B. Diabetes triggers a PARP1 mediated death pathway in the heart through participation of FoxO1. J. Mol. Cell. Cardiol. 2012, 53, 677–686. [Google Scholar] [CrossRef]
- Chen, C.J.; Yu, W.; Fu, Y.C.; Wang, X.; Li, J.L.; Wang, W. Resveratrol protects cardiomyocytes from hypoxia-induced apoptosis through the SIRT1-FoxO1 pathway. Biochem. Biophys. Res. Commun. 2009, 378, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.C.; Yeh, C.H. Inhibition of MIR-302 Suppresses Hypoxia-Reoxygenation-Induced H9c2 Cardiomyocyte Death by Regulating Mcl-1 Expression. Oxid. Med. Cell. Longev. 2017, 2017, 7968905. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.C.; Wongsurawat, T.; Lin, X.Y.; Sorokin, V. The miRNA 30B-5P targeting mRNA MBNL1 leads to pro-myogenic VSMC phenotype modulation in myocardial infarction patients. Atherosclerosis 2018, 275, e46. [Google Scholar] [CrossRef]
- Woo, C.C.; Wongsurawat, T.; Soong, R.; Lee, C.N.; Richards, M.; Kuznetsov, V.; Sorokin, V. Distinctive pattern of LET-7B and MIR-30B in human aortic smooth muscle cells of myocardial infarction patients. Atherosclerosis 2017, 263, e63. [Google Scholar] [CrossRef]
- Sorokin, V.; Woo, C.C.; Lin, X.Y.; Kofidis, T.; Lee, C.N. Role of Micro RNA in Arterial Wall Remodeling in Patients with Advanced Coronary Artery Disease Undergoing Bypass. In Proceedings of the 22nd Annual Meeting of the Asian Society for Cardiovascular and Thoracic Surgery (ASCVTS’14), Istanbul, Turkey, 18–22 August 2014. [Google Scholar]
- Long, B.; Gan, T.Y.; Zhang, R.C.; Zhang, Y.H. miR-23a regulates cardiomyocyte apoptosis by targeting manganese superoxide dismutase. Mol. Cells 2017, 40, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yuan, Y.; Li, J.; Ren, H.; Cai, Q.; Chen, X.; Liang, H.; Shan, H.; Fu, Z.D.; Gao, X.; et al. MicroRNA-1 aggravates cardiac oxidative stress by post-transcriptional modification of the antioxidant network. Cell Stress Chaperones 2015, 20, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Du, Y.; Cao, J.; Gao, Q.; Li, H.; Chen, Y.; Lu, N. MiR-130a inhibition protects rat cardiac myocytes from hypoxia-triggered apoptosis by targeting Smad4. Kardiol. Pol. 2018, 76, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Liu, H.; Guo, J.; Yu, Y.; Yang, D.; He, F.; Du, Z. MicroRNA-98 negatively regulates myocardial infarction-induced apoptosis by down-regulating Fas and caspase-3. Sci. Rep. 2017, 7, 7460. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Sun, Y.; Yu, B. Microrna-208a correlates apoptosis and oxidative stress induced by h2o2 through protein tyrosine kinase/phosphatase balance in cardiomyocytes. Int. Heart J. 2018, 59, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zang, G.; Zhang, G.; Wang, H.; Zhang, X.; Johnston, N.; Min, W.; Luke, P.; Jevnikar, A.; Haig, A.; et al. MicroRNA and mRNA signatures in ischemia reperfusion injury in heart transplantation. PLoS ONE 2013, 8, e79805. [Google Scholar] [CrossRef]
- Shah, P.; Bristow, M.R.; Port, J.D. MicroRNAs in Heart Failure, Cardiac Transplantation, and Myocardial Recovery: Biomarkers with Therapeutic Potential. Curr. Heart Fail. Rep. 2017, 14, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Hamdorf, M.; Kawakita, S.; Everly, M. The Potential of MicroRNAs as Novel Biomarkers for Transplant Rejection. J. Immunol. Res. 2017, 2017, 4072364. [Google Scholar] [CrossRef] [PubMed]
- Van Huyen, J.P.D.; Tible, M.; Gay, A.; Guillemain, R.; Aubert, O.; Varnous, S.; Iserin, F.; Rouvier, P.; François, A.; Vernerey, D.; et al. MicroRNAs as non-invasive biomarkers of heart transplant rejection. Eur. Heart J. 2014, 35, 3194–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Shi, C.; Manduchi, E.; Civelek, M.; Davies, P.F. MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 13450–13455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez, Y.; Wang, C.; Manes, T.D.; Pober, J.S. Cutting Edge: TNF-Induced MicroRNAs Regulate TNF-Induced Expression of E-Selectin and Intercellular Adhesion Molecule-1 on Human Endothelial Cells: Feedback Control of Inflammation. J. Immunol. 2010, 184, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhou, M.; Wang, Y.; Huang, W.; Qin, G.; Weintraub, N.L.; Tang, Y. MiR-92a inhibits vascular smooth muscle cell apoptosis: Role of the MKK4-JNK pathway. Apoptosis 2014, 19, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Wang, M.; Qu, X.; Mah, A.; Xiong, X.; Harris, A.G.C.; Phillips, L.K.; Martinez, O.M.; Krams, S.M. Differential expression of microRNAs during allograft rejection. Am. J. Transplant. 2012, 12, 1113–1123. [Google Scholar] [CrossRef]
- Malakar, A.K.; Choudhury, D.; Halder, B.; Paul, P.; Uddin, A.; Chakraborty, S. A review on coronary artery disease, its risk factors, and therapeutics. J. Cell. Physiol. 2019, 234, 16812–16823. [Google Scholar] [CrossRef]
- Li, T.; Song, X.; Zhang, J.; Zhao, L.; Shi, Y.; Li, Z.; Liu, J.; Liu, N.; Yan, Y.; Xiao, Y.; et al. Protection of Human Umbilical Vein Endothelial Cells against Oxidative Stress by MicroRNA-210. Oxid. Med. Cell. Longev. 2017, 2017, 3565613. [Google Scholar] [CrossRef]
- Fu, X.M.; Zhou, Y.Z.; Cheng, Z.; Liao, X.B.; Zhou, X.M. MicroRNAs: Novel players in aortic aneurysm. Biomed. Res. Int. 2015, 2015, 831641. [Google Scholar] [CrossRef] [Green Version]
- Marcil, V.; Delvin, E.; Sané, A.T.; Tremblay, A.; Levy, E. Oxidative stress influences cholesterol efflux in THP-1 macrophages: Role of ATP-binding cassette A1 and nuclear factors. Cardiovasc. Res. 2006, 72, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, M.; Wang, D.-Z. Non-Coding RNAs Including miRNAs and lncRNAs in Cardiovascular Biology and Disease. Cells 2014, 3, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Du, Y.; Shu, Y.; Gao, M.; Sun, F.; Luo, S.; Yang, T.; Zhan, L.; Yuan, Y.; Chu, W.; et al. MIAT is a Pro-fibrotic Long Non-coding RNA Governing Cardiac Fibrosis in Post-infarct Myocardium. Sci. Rep. 2017, 7, 42657. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.; Liu, G.; Qi, X.; Cao, X. MicroRNA-24 inhibits the proliferation and migration of endothelial cells in patients with atherosclerosis by targeting importin-α3 and regulating inflammatory responses. Exp. Ther. Med. 2018, 15, 338–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cai, W.; Fan, Z.; Yang, C.; Wang, W.; Xiong, M.; Ma, C.; Yang, J. MicroRNA-24 inhibits the oxidative stress induced by vascular injury by activating the Nrf2/Ho-1 signaling pathway. Atherosclerosis 2019, 290, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Gou, L.; Zhao, L.; Song, W.; Wang, L.; Liu, J.; Zhang, H.; Lau, C.W.; Yao, X.; Tian, X.Y.; Wong, W.T.; et al. Inhibition of miR-92a Suppresses Oxidative Stress and Improves Endothelial Function by Upregulating Heme Oxygenase-1 in db/db Mice. Antioxid. Redox Signal. 2018, 28, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamac, A.H.; Huyut, M.A.; Yilmaz, E.; Celikkale, I.; Bacaksiz, A.; Demir, Y.; Demir, A.R.; Erturk, M.; Bakhshaliyev, N.; Ozdemir, R.; et al. MicroRNA 199a is downregulated in patients after coronary artery bypass graft surgery and is associated with increased levels of sirtuin 1 (SIRT 1) protein and major adverse cardiovascular events at 3-year follow-up. Med. Sci. Monit. 2018, 24, 6245–6254. [Google Scholar] [CrossRef]
- Guo, M.; Mao, X.; Ji, Q.; Lang, M.; Li, S.; Peng, Y.; Zhou, W.; Xiong, B.; Zeng, Q. MiR-146a in PBMCs modulates Th1 function in patients with acute coronary syndrome. Immunol. Cell Biol. 2010, 88, 555–564. [Google Scholar] [CrossRef]
- O′Sullivan, J.F.; Neylon, A.; McGorrian, C.; Blake, G.J. miRNA-93-5p and other miRNAs as predictors of coronary artery disease and STEMI. Int. J. Cardiol. 2016, 224, 310–316. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart. Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Dickstein, K.; Cohen-Solal, A.; Filippatos, G.; McMurray, J.J.V.; Ponikowski, P.; Poole-Wilson, P.A.; Strömberg, A.; van Veldhuisen, D.J.; Atar, D.; Hoes, A.W.; et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008. The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart. Eur. J. Heart Fail. 2008, 10, 933–989. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.A.; Abraham, W.T.; Chin, M.H.; Feldman, A.M.; Francis, G.S.; Ganiats, T.G.; Jessup, M.; Konstam, M.A.; Mancini, D.M.; Michl, K.; et al. 2009 Focused Update Incorporated Into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed. J. Am. Coll. Cardiol. 2009, 53, e1–e90. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, S.; Kinugawa, S.; Tsuchihashi-Makaya, M.; Goto, K.; Goto, D.; Yokota, T.; Yamada, S.; Yokoshiki, H.; Takeshita, A.; Tsutsui, H. Spironolactone use at discharge was associated with improved survival in hospitalized patients with systolic heart failure. Am. Heart J. 2010, 160, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, S.; Tsuchihashi-Makaya, M.; Kinugawa, S.; Yokota, T.; Ide, T.; Takeshita, A.; Tsutsui, H. Chronic kidney disease as an independent risk for long-term adverse outcomes in patients hospitalized with heart failure in Japan—Report from the Japanese Cardiac Registry of Heart Failure in Cardiology (JCARE-CARD). Circ. J. 2009, 73, 1442–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, H.; Tsuchihashi-Makaya, M.; Kinugawa, S.; Goto, D.; Takeshita, A. Clinical characteristics and outcome of hospitalized patients with heart failure in Japan—Rationale and design of Japanese Cardiac Registry Of Heart Failure In Cardiology (JCARE-CARD). Circ. J. 2006, 70, 1617–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchihashi-Makaya, M.; Hamaguchi, S.; Kinugawa, S.; Yokota, T.; Goto, D.; Yokoshiki, H.; Kato, N.; Takeshita, A.; Tsutsui, H. Characteristics and outcomes of hospitalized patients with heart failure and reduced vs preserved ejection fraction—A report from the Japanese Cardiac Registry of Heart Failure in Cardiology (JCARE-CARD). Circ. J. 2009, 73, 1893–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieve, D.J.; Shah, A.M. Oxidative stress in heart failure. More than just damage. Eur. Heart J. 2003, 24, 2161–2163. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, D.B. Oxidative stress in heart failure: What are we missing? Am. J. Med. Sci. 2011, 342, 120–124. [Google Scholar] [CrossRef] [Green Version]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Shan, H.; Zhang, Y.; Lu, Y.; Zhang, Y.; Pan, Z.; Cai, B.; Wang, N.; Li, X.; Feng, T.; Hong, Y.; et al. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc. Res. 2009, 83, 465–472. [Google Scholar] [CrossRef]
- Ye, Y.; Perez-polo, J.R.; Qian, J.; Birnbaum, Y.; Ye, Y.; Perez-polo, J.R.; Qian, J.; Birnbaum, Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiol. Genom. 2010, 43, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Price, N.L.; Fernández-Hernando, C. Non-coding RNAs in lipid metabolism. Vascul. Pharmacol. 2019, 114, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Schulte, C.; Karakas, M.; Zeller, T. MicroRNAs in cardiovascular disease—Clinical application. Clin. Chem. Lab. Med. 2017, 55, 687–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.S.; Kala, C.; Abid, M.; Ahmad, N.; Sharma, U.S.; Khan, N.A. Pathological microRNAs in acute cardiovascular diseases and microRNA therapeutics. J. Acute Dis. 2016, 5, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Sue, Y.M.; Chou, Y.; Cheng, C.F.; Chang, C.C.; Li, H.F.; Chen, C.C.; Juan, S.H. Activation of a nuclear factor of activated T-lymphocyte-3 (NFAT3) by oxidative stress in carboplatin-mediated renal apoptosis. Br. J. Pharmacol. 2010, 161, 1661–1676. [Google Scholar] [CrossRef] [Green Version]
- Kakita, T.; Hasegawa, K.; Iwai-Kanai, E.; Adachi, S.; Morimoto, T.; Wada, H.; Kawamura, T.; Yanazume, T.; Sasayama, S. Calcineurin pathway is required for endothelin-1-mediated protection against oxidant stress-induced apoptosis in cardiac myocytes. Circ. Res. 2001, 88, 1239–1246. [Google Scholar] [CrossRef]
- Wang, X.; Lian, Y.; Wen, X.; Guo, J.; Wang, Z.; Jiang, S.; Hu, Y. Expression of miR-126 and its potential function in coronary artery disease. Afr. Health Sci. 2017, 17, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Reddy, L.L.; Shah, S.A.V.; Ponde, C.K.; Rajani, R.M.; Ashavaid, T.F. Circulating miRNA-33: A potential biomarker in patients with coronary artery disease. Biomarkers 2019, 24, 36–42. [Google Scholar] [CrossRef]
- Schulte, C.; Zeller, T. microRNA-based diagnostics and therapy in cardiovascular disease-Summing up the facts. Cardiovasc. Diagn. Ther. 2015, 5, 17–36. [Google Scholar]
- Cakmak, H.A.; Barman, H.A.; Coskunpinar, E.; Oltulu, Y.M.; Ikitimur, B.; Can, G.; Ozcan, S.; Vural, V.A. The Diagnostic Importance of MicroRNAs in Congestive Heart Failure. J. Am. Coll. Cardiol. 2013, 62, C17–C18. [Google Scholar] [CrossRef] [Green Version]
- Murach, K.A.; McCarthy, J.J. MicroRNAs, heart failure, and aging: Potential interactions with skeletal muscle. Heart Fail. Rev. 2017, 22, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, E.F.; Ohashi, P.S. Mir-155, a central modulator of T-cell responses. Eur. J. Immunol. 2014, 44, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Jones Buie, J.N.; Goodwin, A.J.; Cook, J.A.; Halushka, P.V.; Fan, H. The role of miRNAs in cardiovascular disease risk factors. Atherosclerosis 2016, 254, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilad, S.; Meiri, E.; Yogev, Y.; Benjamin, S.; Lebanony, D.; Yerushalmi, N.; Benjamin, H.; Kushnir, M.; Cholakh, H.; Melamed, N.; et al. Serum microRNAs are promising novel biomarkers. PLoS ONE 2008, 3, e3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebolts, U.; Vamholt, H.; Drebber, U.; Dienes, H.P.; Wickenhauser, C.; Odenthal, M. Tissues from routine pathology archives are suitable for microRNA analyses by quantitative PCR. J. Clin. Pathol. 2009, 62, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaarz, A.; Debey-Pascher, S.; Classen, S.; Eggle, D.; Gathof, B.; Chen, J.; Fan, J.B.; Voss, T.; Schultze, J.L.; Staratschek-Jox, A. Bead array-based microRNA expression profiling of peripheral blood and the impact of different RNA isolation approaches. J. Mol. Diagnostics 2010, 12, 335–344. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci. Transl. Med. 2014, 6, 239ps3. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.R.; Wu, M.; Yu, H.; Long, S.; Stevens, A.; Engers, D.W.; Sackin, H.; Daniels, J.S.; Dawson, E.S.; Hopkins, C.R.; et al. Selective inhibition of the K ir2 family of inward rectifier potassium channels by a small molecule probe: The discovery, SAR, and pharmacological characterization of ML133. ACS Chem. Biol. 2011, 6, 845–856. [Google Scholar] [CrossRef] [Green Version]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with antagomirs. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Oliveira-Carvalho, V.; Carvalho, V.O.; Silva, M.M.; Guimarães, G.V.; Bocchi, E.A. MicroRNAs: A new paradigm in the treatment and diagnosis of heart failure? Arq. Bras. Cardiol. 2012, 98, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, J.A.; Zamore, P.D. MicroRNA therapeutics. Gene Ther. 2011, 18, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Caroli, A.; Cardillo, M.T.; Galea, R.; Biasucci, L.M. Potential therapeutic role of microRNAs in ischemic heart disease. J. Cardiol. 2013, 61, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Impact of oxidative stress on the heart. Overproduction of reactive oxygen species (ROS) contributes to different cardiac pathologies, e.g., hypertrophy or fibrosis. These pathological changes in the heart may result in cardiovascular diseases, which may subsequently contribute to the production of ROS.
Figure 1.
Impact of oxidative stress on the heart. Overproduction of reactive oxygen species (ROS) contributes to different cardiac pathologies, e.g., hypertrophy or fibrosis. These pathological changes in the heart may result in cardiovascular diseases, which may subsequently contribute to the production of ROS.
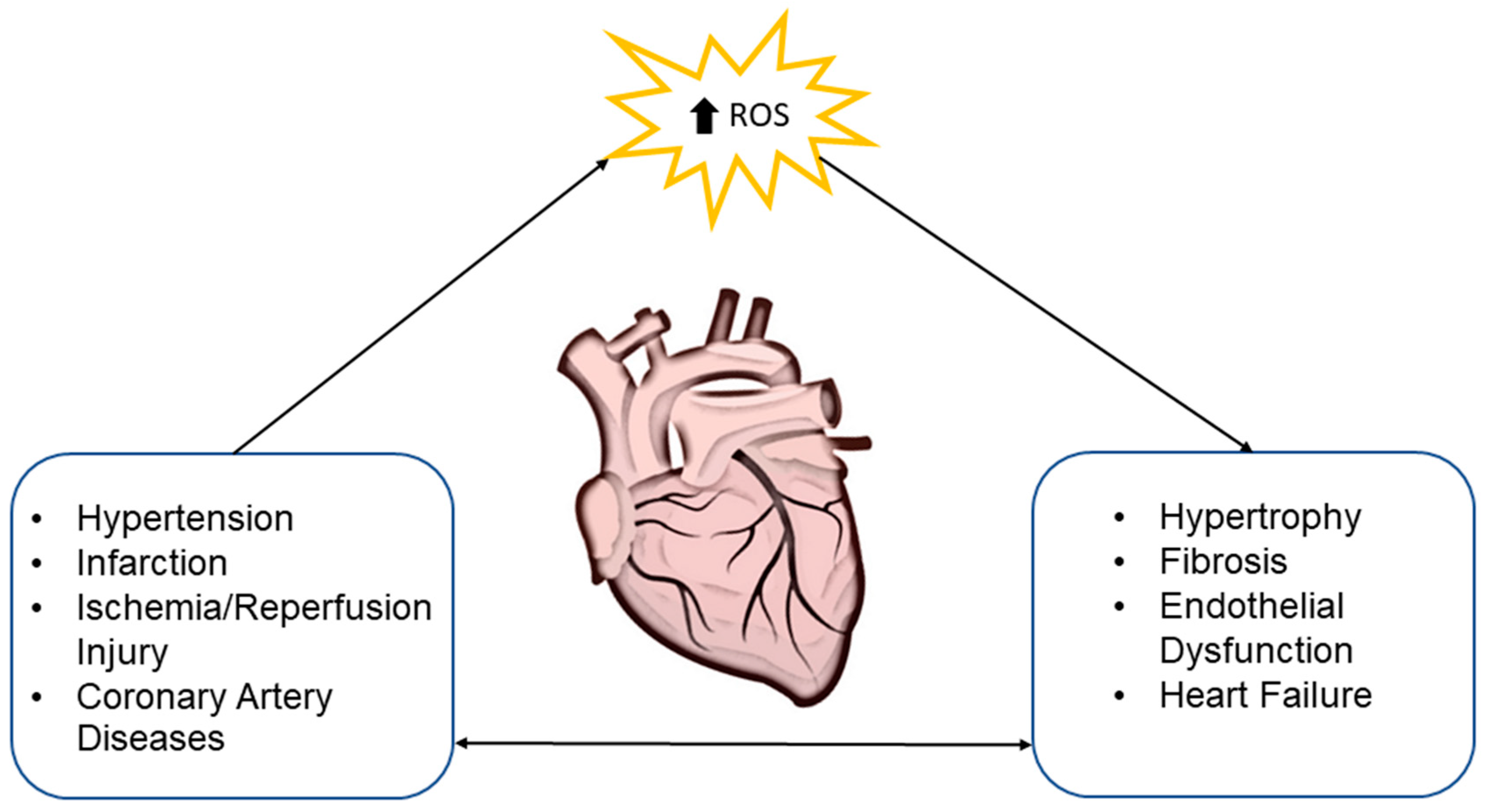
Figure 2.
Selected signaling pathways (Nrf2, SIRT1, and NF-κB) influenced by miRNAs in situations with oxidative stress. ROS either inhibit or induce miRNA expression level. This leads to subsequent regulation of their target genes. Upper arrow represents increase of oxidative stress/activation of signaling pathway.
Figure 2.
Selected signaling pathways (Nrf2, SIRT1, and NF-κB) influenced by miRNAs in situations with oxidative stress. ROS either inhibit or induce miRNA expression level. This leads to subsequent regulation of their target genes. Upper arrow represents increase of oxidative stress/activation of signaling pathway.
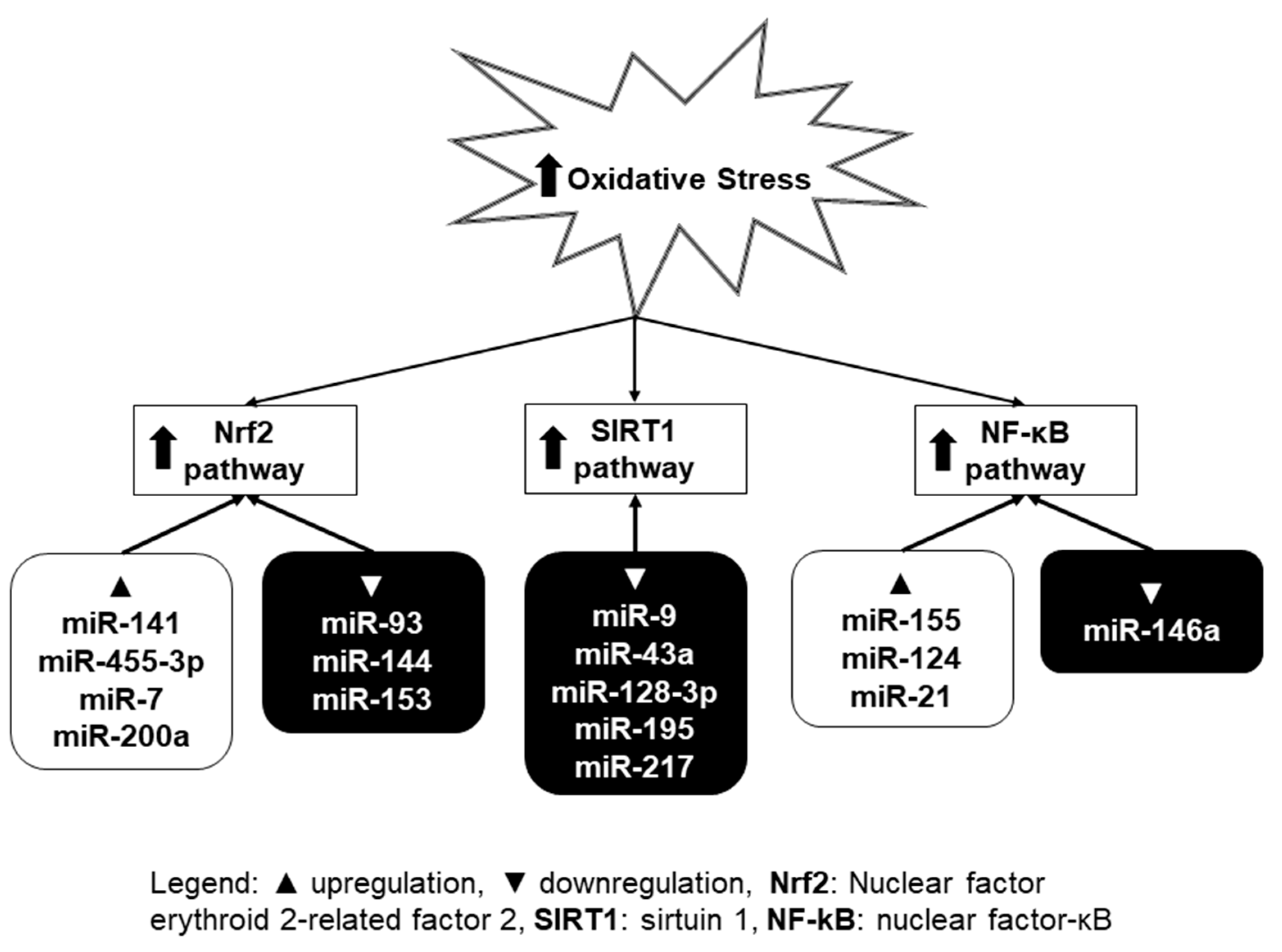

{kind=link}
{kind=link}
Table 1.
List of selected miRNAs with function in oxidative-stress-induced cardiovascular diseases.
| Disease | miRNA | Expression | Target | References |
|---|---|---|---|---|
| Cardiac hypertrophy | miRNA-1 | Downregulated | Mef2a; Gata4 | [93,94,95] |
| miRNA-133 | Downregulated | GDP–GTP exchange protein; Cdc42 | [93,96,97] | |
| miRNA-208a | Downregulated | Myh7 | [18,98,99] | |
| Ischemia/reperfusion injury | miRNA-24-3p | Downregulated | Keap1/Nrf2 | [109] |
| miRNA-144 | Downregulated | FoxO1 | [110,111] | |
| miRNA-302 | Upregulated | Mcl-1 | [113] | |
| miRNA-23a | Upregulated | MnSOD | [117] | |
| Heart transplantation | miRNA-10a | Downregulated | NF-κB | [126] |
| miRNA-31 | Upregulated | TNF-α | [127] | |
| miRNA-92a | Upregulated | Integrin a5, S1P1, MKK4, eNOS | [128] | |
| miRNA-155 | Upregulated | T-cell receptor, IFN receptor | [164] | |
| Coronary artery diseases | miRNA-24 | Downregulated | Ogt, Keap1/Nrf2 | [132,134,135,136,137] |
| miRNA-92a | Upregulated | HO-1 | [138] | |
| miRNA-199a | Downregulated | SIRT1 | [139] | |
| Heart failure | miRNA-199b | Upregulated | calcineurin/NFAT | [101,157,158,159] |
| miRNA-21 | Upregulated | natriuretic peptide B | [156,163] |
Mef2a—Myocyte-specific enhancer factor 2A, Gata4—Transcription factor GATA-4, GDP–GTP exchange protein–guanosine diphosphate–guanosine triphosphate exchange protein, Cdc42—Cell division control protein 42, Myh7—beta myosin heavy chain, Keap1/Nrf2—Kelch-like ECH-associated protein 1/Nuclear factor erythroid 2-related factor 2, FoxO1–Forkhead box protein O1, Mcl-1—Myeloid cell leukemia 1, MnSOD—manganese-dependent superoxide dismutase, NF-κB–nuclear factor kappa B, TNF-α–tumor necrosis factor alpha, S1P1–Sphingosine-1-phosphate receptor 1, MKK4—Mitogen-activated protein kinase kinase 4, eNOS—Endothelial nitric oxide synthase, INF receptor–interferon receptor, Ogt—O-linked β-N-acetylglucosamine transferase, HO-1—Heme oxygenase 1, SIRT1—sirtuin 1, NFAT—Nuclear factor of activated T cells.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kura, B.; Szeiffova Bacova, B.; Kalocayova, B.; Sykora, M.; Slezak, J. Oxidative Stress-Responsive MicroRNAs in Heart Injury. Int. J. Mol. Sci. 2020, 21, 358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010358
AMA Style
Kura B, Szeiffova Bacova B, Kalocayova B, Sykora M, Slezak J. Oxidative Stress-Responsive MicroRNAs in Heart Injury. International Journal of Molecular Sciences. 2020; 21(1):358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010358
Chicago/Turabian StyleKura, Branislav, Barbara Szeiffova Bacova, Barbora Kalocayova, Matus Sykora, and Jan Slezak. 2020. "Oxidative Stress-Responsive MicroRNAs in Heart Injury" International Journal of Molecular Sciences 21, no. 1: 358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010358
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.