Elucidation of Molecular Mechanism of a Selective PPARα Modulator, Pemafibrate, through Combinational Approaches of X-ray Crystallography, Thermodynamic Analysis, and First-Principle Calculations

 , , and
, , and
Abstract
:1. Introduction
2. Results
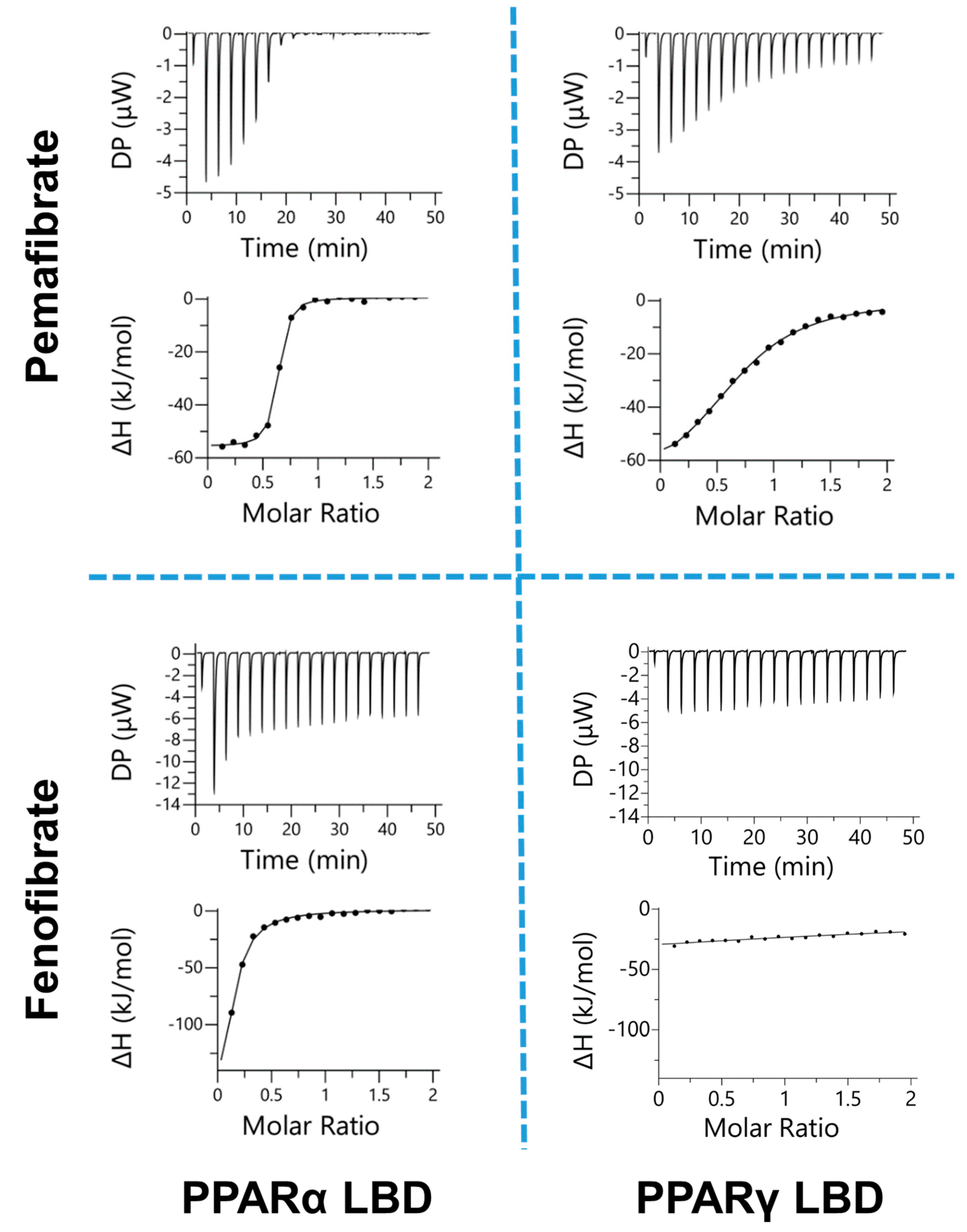
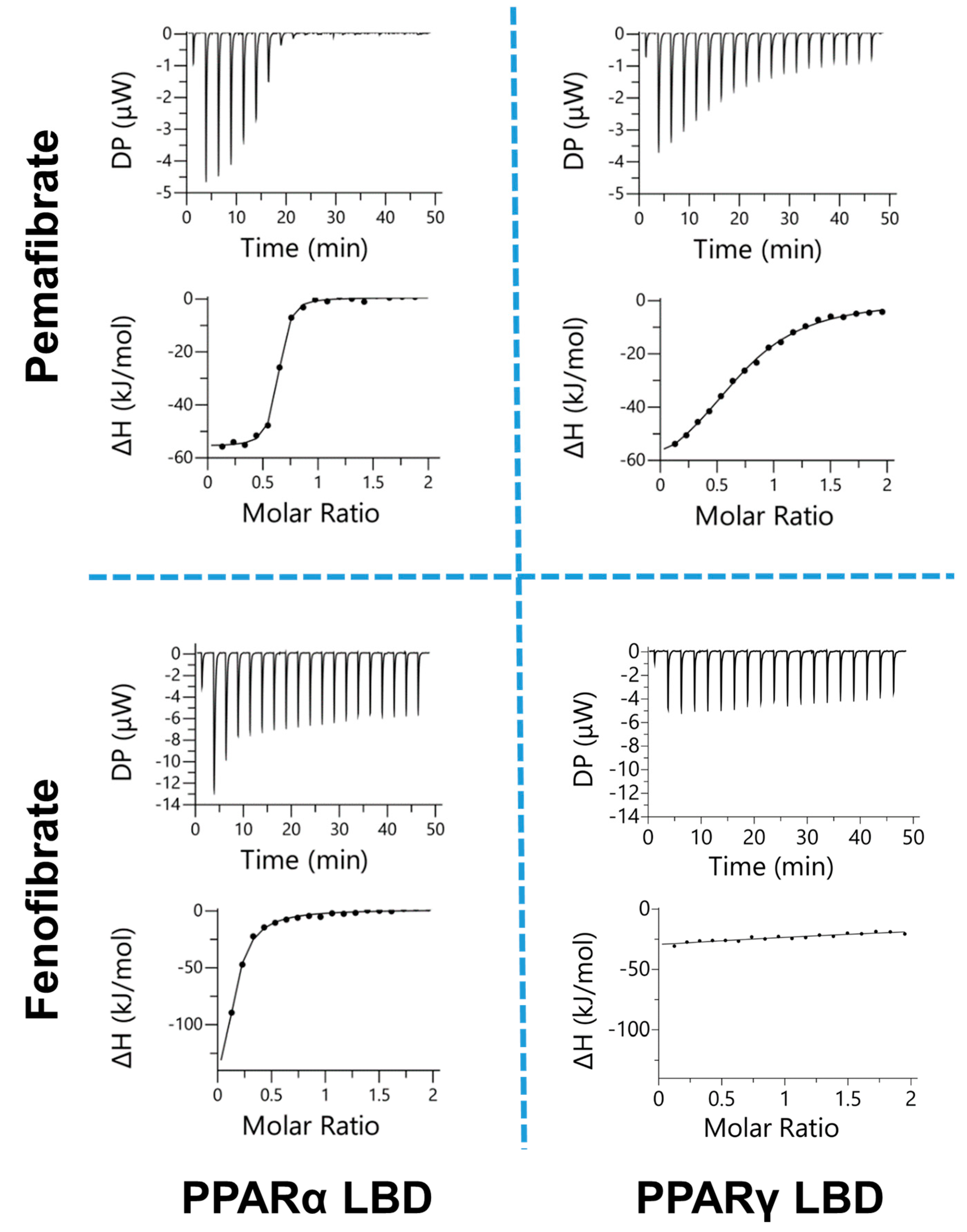
2.1. Estimation of Binding Affinity between PPARs-LBD and Two Fibrates
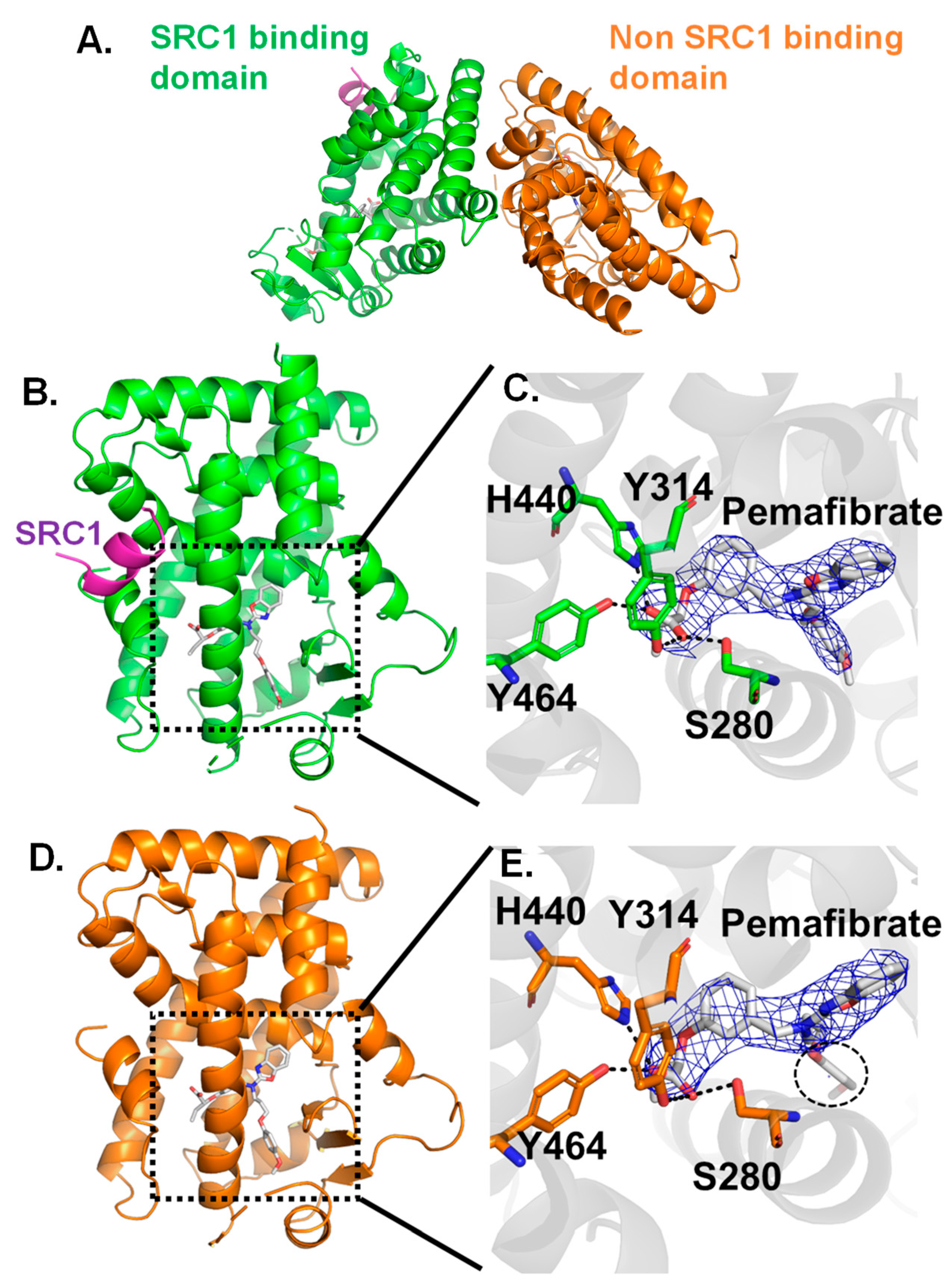
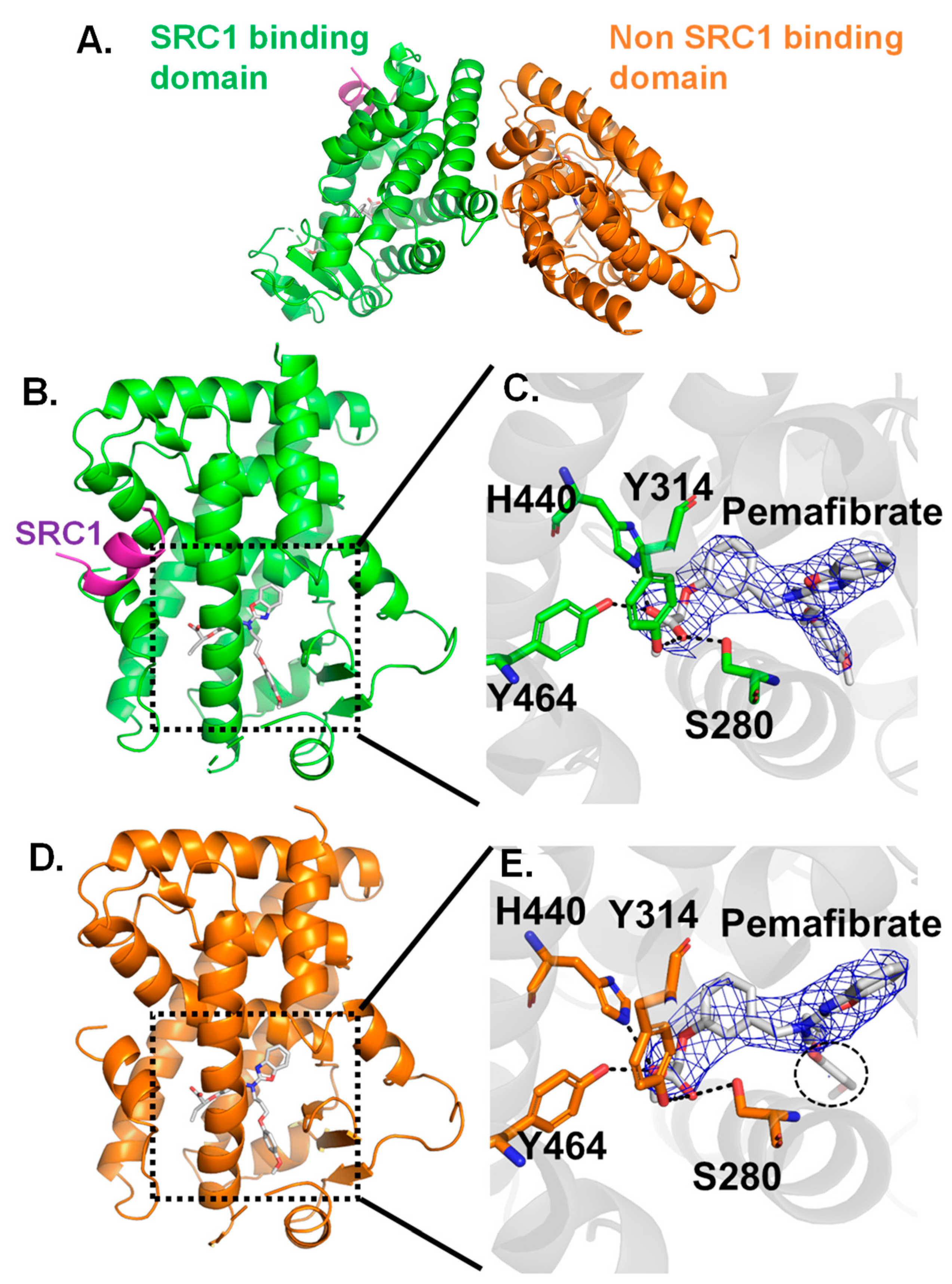
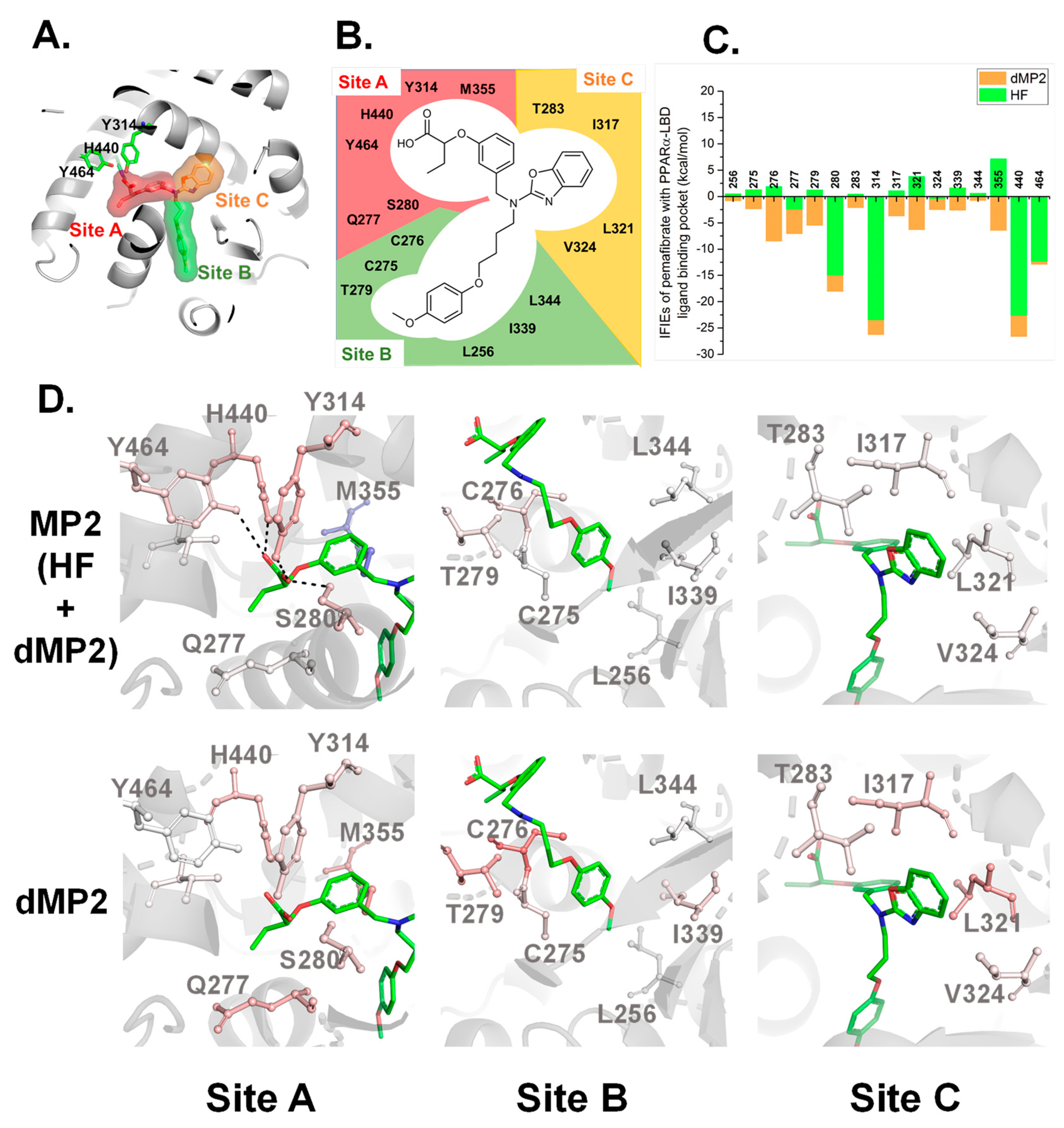
2.2. Crystal Structure Analysis for Pemafibrate and SRC1 Peptide Binding form of PPARα-LBD
2.3. Interaction Energy Analysis between Pemafibrate and PPARα-LBD Based on FMO Method
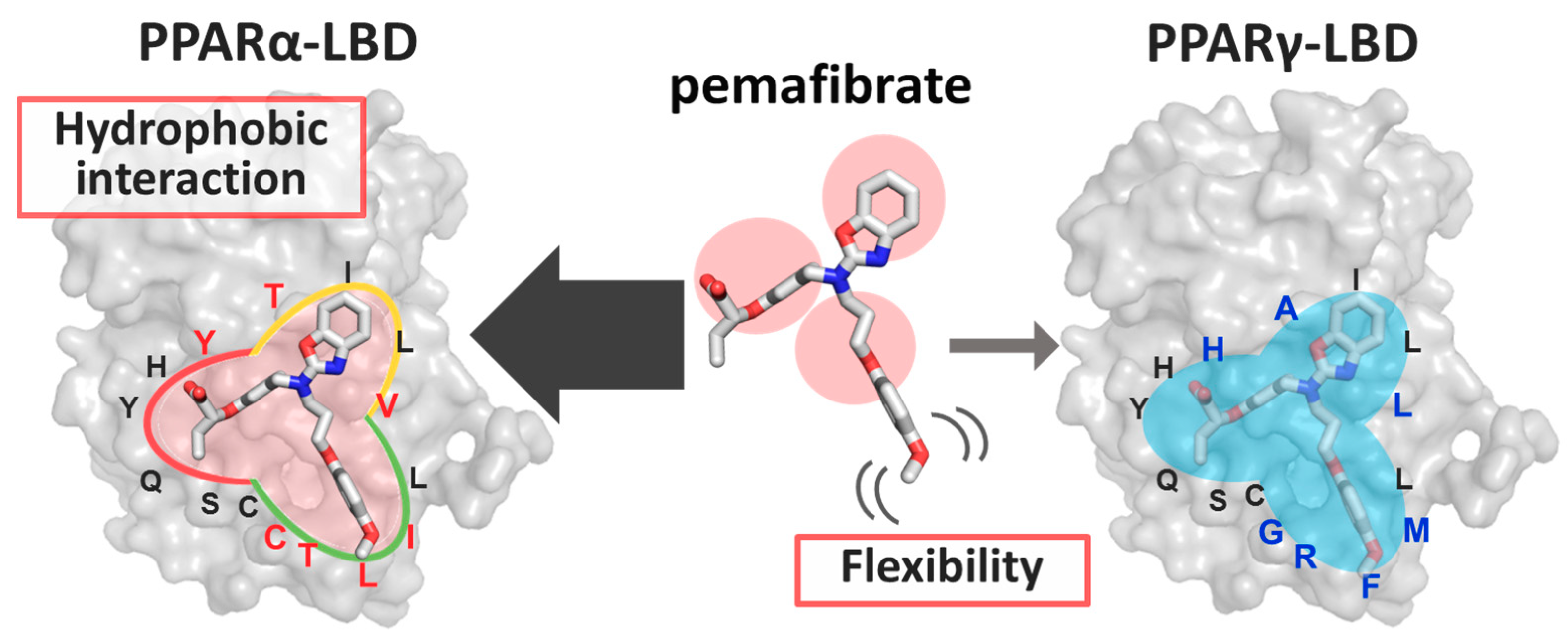
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Preparation of PPARs Ligands and Coactivator Peptides
5.2. Overexpression and Purification of PPARα LBD and PPARγ LBD
5.3. Crystallization and X-ray Data Collection of the PPARα-LBD/Pemafibrate/SRC1 Peptide
5.4. Fragment Molecular Orbital Calculations
5.5. Isothermal Titration Calorimetry
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PPAR | Peroxisome proliferator-activated receptor |
| LBD | Ligand binding domain |
| LBP | Ligand binding pocket |
| SPPARM | Selective PPAR modulator |
| FMO | Fragment molecular orbital |
| ITC | Isothermal titration calorimetry |
| IFIE | Interfragment interaction energy |
References
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From orphan receptors to drug discovery. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xiao, G.; Trujillo, C.; Chang, V.; Blanco, L.; Joseph, S.B.; Bassilian, S.; Saad, M.F.; Tontonoz, P.; Lee, W.N.; et al. Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J. Biol. Chem. 2002, 277, 50237–50244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardes, A.; Souza, P.C.; Muniz, J.R.; Ricci, C.G.; Ayers, S.D.; Parekh, N.M.; Godoy, A.S.; Trivella, D.B.; Reinach, P.; Webb, P.; et al. Molecular mechanism of peroxisome proliferator-activated receptor alpha activation by WY14643: A new mode of ligand recognition and receptor stabilization. J. Mol. Biol. 2013, 425, 2878–2893. [Google Scholar] [CrossRef] [Green Version]
- Cronet, P.; Petersen, J.F.; Folmer, R.; Blomberg, N.; Sjoblom, K.; Karlsson, U.; Lindstedt, E.L.; Bamberg, K. Structure of the PPARalpha and -gamma ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure 2001, 9, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.Y.; Bae, H.; Lee, Y.J.; Choi, Y.I.; Kim, H.J.; Park, S.B.; Suh, S.W.; Kim, S.W.; Han, B.W. Structural Basis for the Enhanced Anti-Diabetic Efficacy of Lobeglitazone on PPARgamma. Sci. Rep. 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosure, S.A.; Shang, J.; Eberhardt, J.; Brust, R.; Zheng, J.; Griffin, P.R.; Forli, S.; Kojetin, D.J. Structural Basis of Altered Potency and Efficacy Displayed by a Major in Vivo Metabolite of the Antidiabetic PPARgamma Drug Pioglitazone. J. Med. Chem. 2019, 62, 2008–2023. [Google Scholar] [CrossRef]
- Ebdrup, S.; Pettersson, I.; Rasmussen, H.B.; Deussen, H.J.; Frost Jensen, A.; Mortensen, S.B.; Fleckner, J.; Pridal, L.; Nygaard, L.; Sauerberg, P. Synthesis and biological and structural characterization of the dual-acting peroxisome proliferator-activated receptor alpha/gamma agonist ragaglitazar. J. Med. Chem. 2003, 46, 1306–1317. [Google Scholar] [CrossRef]
- Oyama, T.; Toyota, K.; Waku, T.; Hirakawa, Y.; Nagasawa, N.; Kasuga, J.I.; Hashimoto, Y.; Miyachi, H.; Morikawa, K. Adaptability and selectivity of human peroxisome proliferator-activated receptor (PPAR) pan agonists revealed from crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 786–795. [Google Scholar] [CrossRef]
- Kuwabara, N.; Oyama, T.; Tomioka, D.; Ohashi, M.; Yanagisawa, J.; Shimizu, T.; Miyachi, H. Peroxisome proliferator-activated receptors (PPARs) have multiple binding points that accommodate ligands in various conformations: Phenylpropanoic acid-type PPAR ligands bind to PPAR in different conformations. depending on the subtype. J. Med. Chem. 2012, 55, 893–902. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T., Jr.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef] [Green Version]
- Fruchart, J.C. Selective peroxisome proliferator-activated receptor alpha modulators (SPPARMalpha): The next generation of peroxisome proliferator-activated receptor alpha-agonists. Cardiovasc. Diabetol. 2013, 12, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yew, T.; Toh, S.A.; Millar, J.S. Selective peroxisome proliferator-activated receptor-gamma modulation to reduce cardiovascular risk in patients with insulin resistance. Recent Pat. Cardiovasc. Drug Discov. 2012, 7, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Einstein, M.; Akiyama, T.E.; Castriota, G.A.; Wang, C.F.; McKeever, B.; Mosley, R.T.; Becker, J.W.; Moller, D.E.; Meinke, P.T.; Wood, H.B.; et al. The differential interactions of peroxisome proliferator-activated receptor gamma ligands with Tyr473 is a physical basis for their unique biological activities. Mol. Pharmacol. 2008, 73, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, R.; Hoener, P.A.; Jow, L.; Bilakovics, J.; Klausing, K.; Mais, D.E.; Faulkner, A.; Croston, G.E.; Paterniti, J.R., Jr. A selective peroxisome proliferator-activated receptor-gamma (PPARgamma) modulator blocks adipocyte differentiation but stimulates glucose uptake in 3T3-L1 adipocytes. Mol. Endocrinol. 2000, 14, 1425–1433. [Google Scholar]
- Higgins, L.S.; Depaoli, A.M. Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am. J. Clin. Nutr. 2010, 91, 267S–272S. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, Y.; Shimura, Y.; Ikeda, H. Effects of pioglitazone on hepatic and peripheral insulin resistance in Wistar fatty rats. Arzneimittelforschung 1990, 40, 436–440. [Google Scholar]
- Young, P.W.; Buckle, D.R.; Cantello, B.C.; Chapman, H.; Clapham, J.C.; Coyle, P.J.; Haigh, D.; Hindley, R.M.; Holder, J.C.; Kallender, H.; et al. Identification of high-affinity binding sites for the insulin sensitizer rosiglitazone (BRL-49653) in rodent and human adipocytes using a radioiodinated ligand for peroxisomal proliferator-activated receptor gamma. J. Pharmacol. Exp. Ther. 1998, 284, 751–759. [Google Scholar]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Alagona, P., Jr. Fenofibric acid: A new fibrate approved for use in combination with statin for the treatment of mixed dyslipidemia. Vasc. Health Risk Manag. 2010, 6, 351–362. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef] [Green Version]
- Fazio, S.; Linton, M.F. The role of fibrates in managing hyperlipidemia: Mechanisms of action and clinical efficacy. Curr. Atheroscler. Rep. 2004, 6, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Remick, J.; Weintraub, H.; Setton, R.; Offenbacher, J.; Fisher, E.; Schwartzbard, A. Fibrate therapy: An update. Cardiol. Rev. 2008, 16, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Boissonnat, P.; Salen, P.; Guidollet, J.; Ferrera, R.; Dureau, G.; Ninet, J.; Renaud, S.; de Lorgeril, M. The long-term effects of the lipid-lowering agent fenofibrate in hyperlipidemic heart transplant recipients. Transplantation 1994, 58, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Duez, H.; Lefebvre, B.; Poulain, P.; Torra, I.P.; Percevault, F.; Luc, G.; Peters, J.M.; Gonzalez, F.J.; Gineste, R.; Helleboid, S.; et al. Regulation of human apoA-I by gemfibrozil and fenofibrate through selective peroxisome proliferator-activated receptor alpha modulation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 585–591. [Google Scholar] [CrossRef]
- Robins, S.J. PPARalpha ligands and clinical trials: Cardiovascular risk reduction with fibrates. J. Cardiovasc. Risk 2001, 8, 195–201. [Google Scholar] [CrossRef]
- Mychaleckyj, J.C.; Craven, T.; Nayak, U.; Buse, J.; Crouse, J.R.; Elam, M.; Kirchner, K.; Lorber, D.; Marcovina, S.; Sivitz, W.; et al. Reversibility of fenofibrate therapy-induced renal function impairment in ACCORD type 2 diabetic participants. Diabetes Care 2012, 35, 1008–1014. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Abe, K.; Toma, T.; Nishikawa, M.; Ozawa, H.; Okuda, A.; Araki, T.; Oda, S.; Inoue, K.; Shibuya, K.; et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor alpha agonists. Bioorg. Med. Chem. Lett. 2007, 17, 4689–4693. [Google Scholar] [CrossRef]
- Fruchart, J.C. Pemafibrate (K-877). a novel selective peroxisome proliferator-activated receptor alpha modulator for management of atherogenic dyslipidaemia. Cardiovasc. Diabetol. 2017, 16, 124. [Google Scholar] [CrossRef]
- Raza-Iqbal, S.; Tanaka, T.; Anai, M.; Inagaki, T.; Matsumura, Y.; Ikeda, K.; Taguchi, A.; Gonzalez, F.J.; Sakai, J.; Kodama, T. Transcriptome Analysis of K-877 (a Novel Selective PPARalpha Modulator (SPPARMalpha))-Regulated Genes in Primary Human Hepatocytes and the Mouse Liver. J. Atheroscler. Thromb. 2015, 22, 754–772. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, S.; Yamashita, S.; Arai, H.; Araki, E.; Yokote, K.; Suganami, H.; Fruchart, J.C.; Kodama, T. Effects of K-877. a novel selective PPARalpha modulator (SPPARMalpha), in dyslipidaemic patients: A randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 2016, 249, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Yokote, K.; Yamashita, S.; Arai, H.; Araki, E.; Suganami, H.; Ishibashi, S. A Pooled Analysis of Pemafibrate Phase II/III Clinical Trials Indicated Significant Improvement in Glycemic and Liver Function-related Parameters. Atheroscler. Suppl. 2018, 32, 154–155. [Google Scholar] [CrossRef]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S. Efficacy and safety of K-877. a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), in combination with statin treatment: Two randomised, double-blind, placebo-controlled clinical trials in patients with dyslipidaemia. Atherosclerosis 2017, 261, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Takei, K.; Arulmozhiraja, S.; Sladek, V.; Matsuo, N.; Han, S.I.; Matsuzaka, T.; Sekiya, M.; Tokiwa, T.; Shoji, M.; et al. Molecular association model of PPARalpha and its new specific and efficient ligand. pemafibrate: Structural basis for SPPARMalpha. Biochem. Biophys. Res. Commun. 2018, 499, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Hennuyer, N.; Duplan, I.; Paquet, C.; Vanhoutte, J.; Woitrain, E.; Touche, V.; Colin, S.; Vallez, E.; Lestavel, S.; Lefebvre, P.; et al. The novel selective PPARalpha modulator (SPPARMalpha) pemafibrate improves dyslipidemia. enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis 2016, 249, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today 2008, 13, 869–874. [Google Scholar] [CrossRef]
- Chaires, J.B. Calorimetry and thermodynamics in drug design. Annu. Rev. Biophys. 2008, 37, 135–151. [Google Scholar] [CrossRef] [Green Version]
- Egawa, D.; Itoh, T.; Akiyama, Y.; Saito, T.; Yamamoto, K. 17-OxoDHA Is a PPARalpha/gamma Dual Covalent Modifier and Agonist. ACS Chem. Biol. 2016, 11, 2447–2455. [Google Scholar] [CrossRef]
- Jin, L.; Lin, S.; Rong, H.; Zheng, S.; Jin, S.; Wang, R.; Li, Y. Structural basis for iloprost as a dual peroxisome proliferator-activated receptor alpha/delta agonist. J. Biol. Chem. 2011, 286, 31473–31479. [Google Scholar] [CrossRef] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. Part A 1997, 276, 307–326. [Google Scholar]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delano, W.L. The PyMOL Molecular Graphics System 2002. Available online: http://pymol.sourceforge.net/overview/sld001.htm (accessed on 2 December 2019).
- Nakano, S.; Yasukawa, K.; Tokiwa, T.; Ishikawa, T.; Ishitsubo, E.; Matsuo, N.; Ito, S.; Tokiwa, H.; Asano, Y. Origin of Stereoselectivity and Substrate/Ligand Recognition in an FAD-Dependent R-Selective Amine Oxidase. J. Phys. Chem. B 2016, 120, 10736–10743. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, T.; Nakano, S.; Yamamoto, Y.; Tokiwa, H.; Asano, Y.; Ito, S. Product Release Mechanism Associated with Structural Changes in Monomeric l-Threonine 3-Dehydrogenase. Biochemistry 2017, 56, 5758–5770. [Google Scholar] [CrossRef] [PubMed]
- Arulmozhiraja, S.; Matsuo, N.; Ishitsubo, E.; Okazaki, S.; Shimano, H.; Tokiwa, H. Comparative Binding Analysis of Dipeptidyl Peptidase IV (DPP-4) with Antidiabetic Drugs—An Ab Initio Fragment Molecular Orbital Study. PLoS ONE 2016, 11, e0166275. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment 2013 (MOE, Montreal, QC, Canada). Available online: https://www.chemcomp.com/Products.htm (accessed on 20 December 2019).
- Ohtake, K.; Yamaguchi, A.; Mukai, T.; Kashimura, H.; Hirano, N.; Haruki, M.; Kohashi, S.; Yamagishi, K.; Murayama, K.; Tomabechi, Y.; et al. Protein stabilization utilizing a redefined codon. Sci. Rep. 2015, 5, 9762. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Kuwata, K. Fragment molecular orbital calculation using the RI-MP2 method. Chem. Phys. Lett. 2009, 474, 195–198. [Google Scholar] [CrossRef]
- Valiron, P.; Mayer, I. Hierarchy of counterpoise corrections for N-body clusters: Generalization of the Boys-Bernardi scheme. Chem. Phys. Lett. 1997, 275, 46–55. [Google Scholar] [CrossRef]
- Kamiya, M.; Hirata, S.; Valiev, M. Fast electron correlation methods for molecular clusters without basis set superposition errors. J. Chem. Phys. 2008, 128, 074103. [Google Scholar] [CrossRef]
- Tokiwa, T.; Nakano, S.; Yamamoto, Y.; Ishikawa, T.; Ito, S.; Sladek, V.; Fukuzawa, K.; Mochizuki, Y.; Tokiwa, H.; Misaizu, F.; et al. Development of an Analysis Toolkit. AnalysisFMO, to Visualize Interaction Energies Generated by Fragment Molecular Orbital Calculations. J. Chem. Inf. Model. 2019, 59, 25–30. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Kd | ΔG | ΔH | −TΔS | |
|---|---|---|---|---|---|
| μM | kcal/mol | ||||
| Pemafibrate | |||||
| PPARα-LBD | 0.61 ± 0.03 | 0.13 ± 0.04 | −9.37 ± 0.02 | −12.3 ± 0.6 | 3.13 ± 0.69 |
| PPARγ-LBD | 0.65 ± 0.05 | 9.58 ± 1.85 | −6.83 ± 0.41 | −17.1 ± 1.0 | 10.3 ± 1.2 |
| Fenofibric Acid | |||||
| PPARα-LBD | 0.27 ± 0.04 | 7.37 ± 2.68 | −7.02 ± 0.25 | −23.2 ± 2.3 | 16.1 ± 2.5 |
| PPARγ-LBD | n.d. b | n.d. | n.d. | n.d. | n.d. |
| PPARα-LBD/Pemafibrate/SRC1 | |
|---|---|
| Space group | P3121 |
| Unit cell parameters | |
| a (Å) | 82.74 |
| b (Å) | 82.74 |
| c (Å) | 177.5 |
| α (degree) | 90.0 |
| β (degree) | 90.0 |
| γ (degree) | 120.0 |
| X-ray source | PF BL5A |
| Wavelength (Å) | 1.00 |
| Resolution (Å) | 45.7–3.2 (3.26–3.2) |
| No. of reflections a | 131,418 |
| No. of unique reflections | 224,57 |
| Completeness (%) | 100 (100) |
| I/sig(I) | 20.8 (1.5) |
| Rmergeb | 0.080 (0.678) |
| CC1/2 | 0.996 (0.800) |
| Rc | 0.190 |
| Rfreed | 0.253 |
| RMSD of geometry | |
| Bond length (Å) | 0.013 |
| Bond angle (degree) | 1.656 |
| Geometry | |
| Ramachandran outlier (%) | 0.4 |
| Ramachandran favored (%) | 99.6 |
| PDB code | 6L96 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawasaki, M.; Kambe, A.; Yamamoto, Y.; Arulmozhiraja, S.; Ito, S.; Nakagawa, Y.; Tokiwa, H.; Nakano, S.; Shimano, H. Elucidation of Molecular Mechanism of a Selective PPARα Modulator, Pemafibrate, through Combinational Approaches of X-ray Crystallography, Thermodynamic Analysis, and First-Principle Calculations. Int. J. Mol. Sci. 2020, 21, 361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010361
Kawasaki M, Kambe A, Yamamoto Y, Arulmozhiraja S, Ito S, Nakagawa Y, Tokiwa H, Nakano S, Shimano H. Elucidation of Molecular Mechanism of a Selective PPARα Modulator, Pemafibrate, through Combinational Approaches of X-ray Crystallography, Thermodynamic Analysis, and First-Principle Calculations. International Journal of Molecular Sciences. 2020; 21(1):361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010361
Chicago/Turabian StyleKawasaki, Mayu, Akira Kambe, Yuta Yamamoto, Sundaram Arulmozhiraja, Sohei Ito, Yoshimi Nakagawa, Hiroaki Tokiwa, Shogo Nakano, and Hitoshi Shimano. 2020. "Elucidation of Molecular Mechanism of a Selective PPARα Modulator, Pemafibrate, through Combinational Approaches of X-ray Crystallography, Thermodynamic Analysis, and First-Principle Calculations" International Journal of Molecular Sciences 21, no. 1: 361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010361