Proteomic Responses to Drought Vary Widely Among Eight Diverse Genotypes of Rice (Oryza sativa)


Abstract
:1. Introduction
2. Results
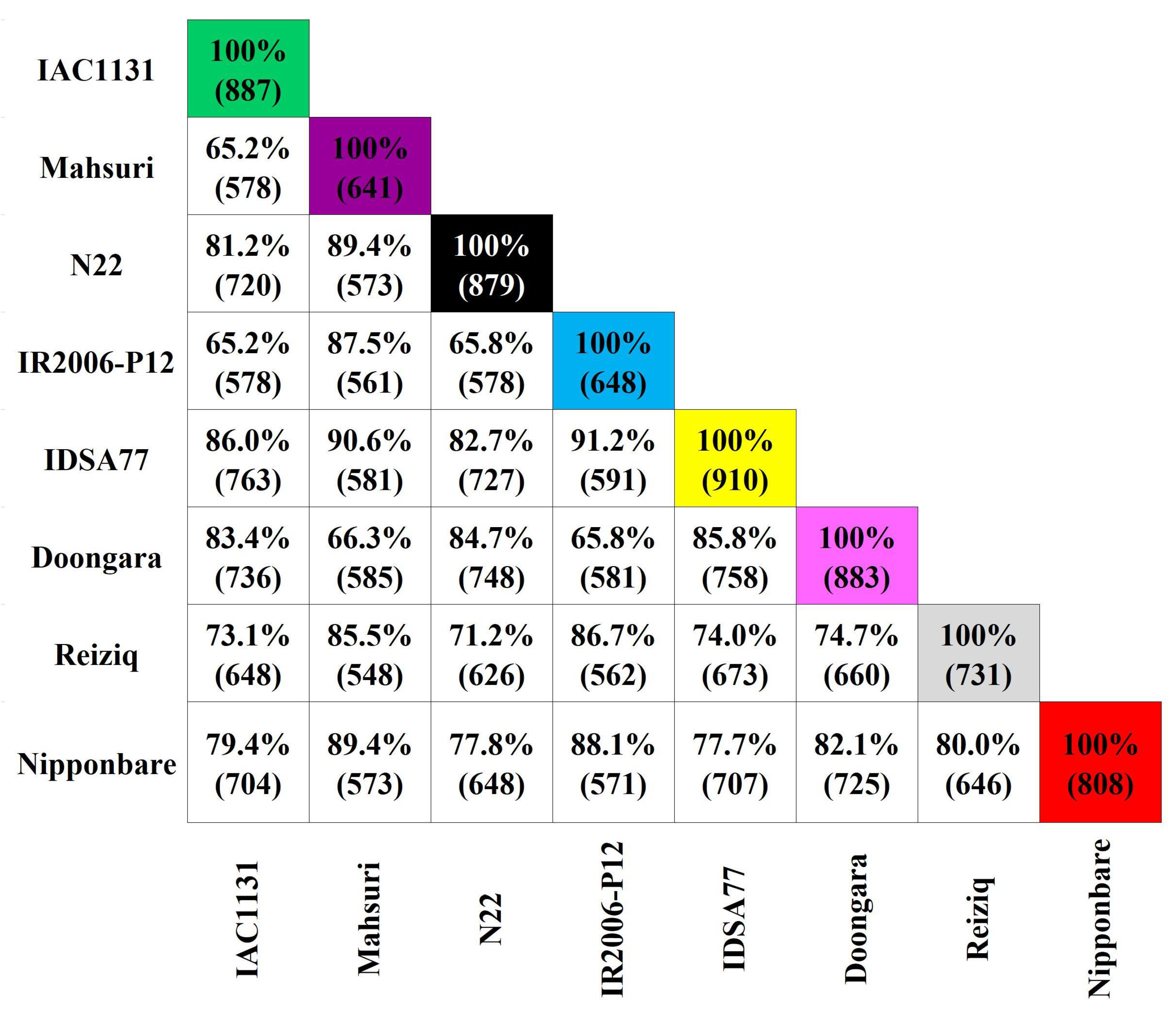
2.1. Label-Free Shotgun Proteomic Data Analysis
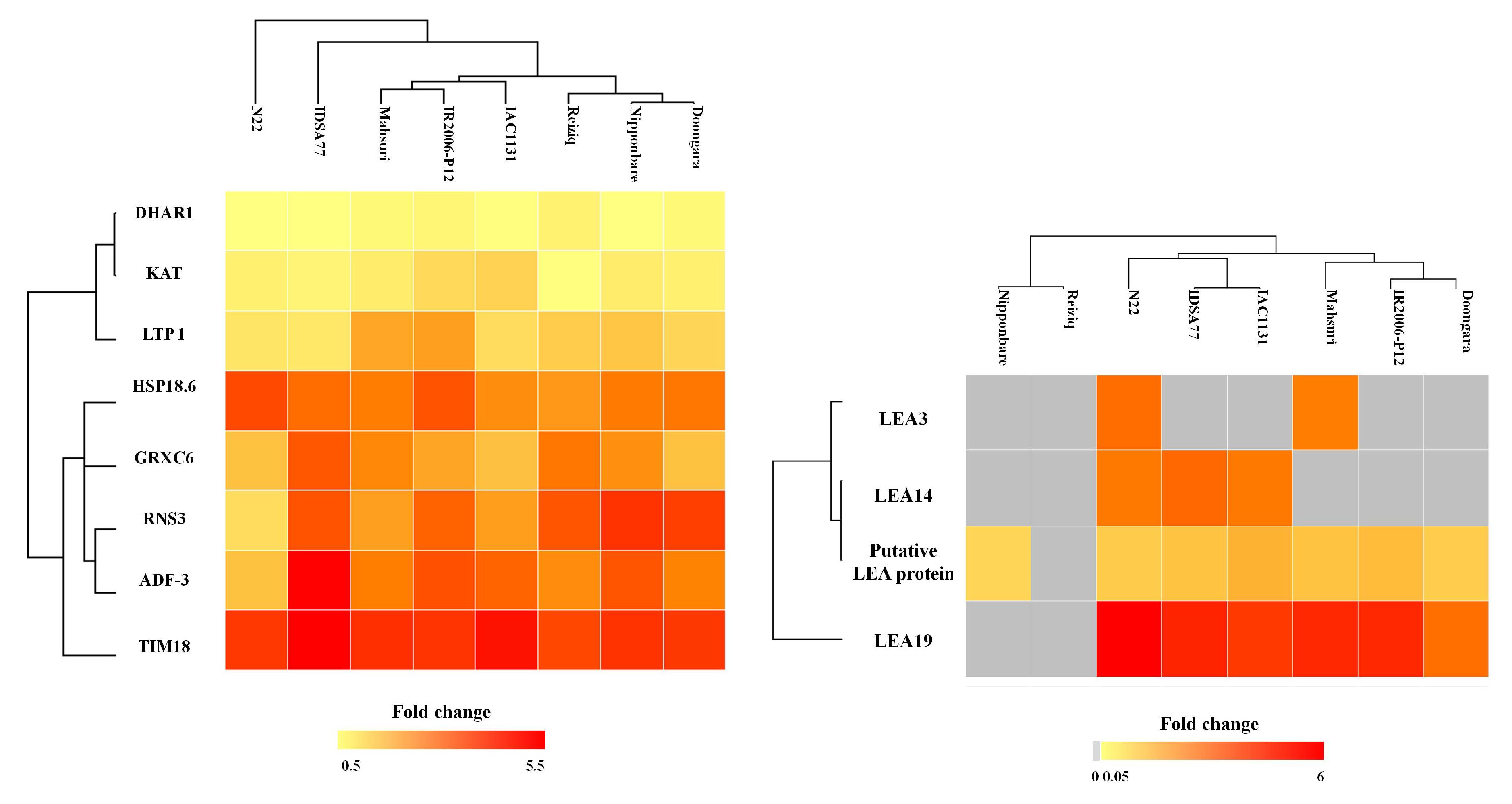
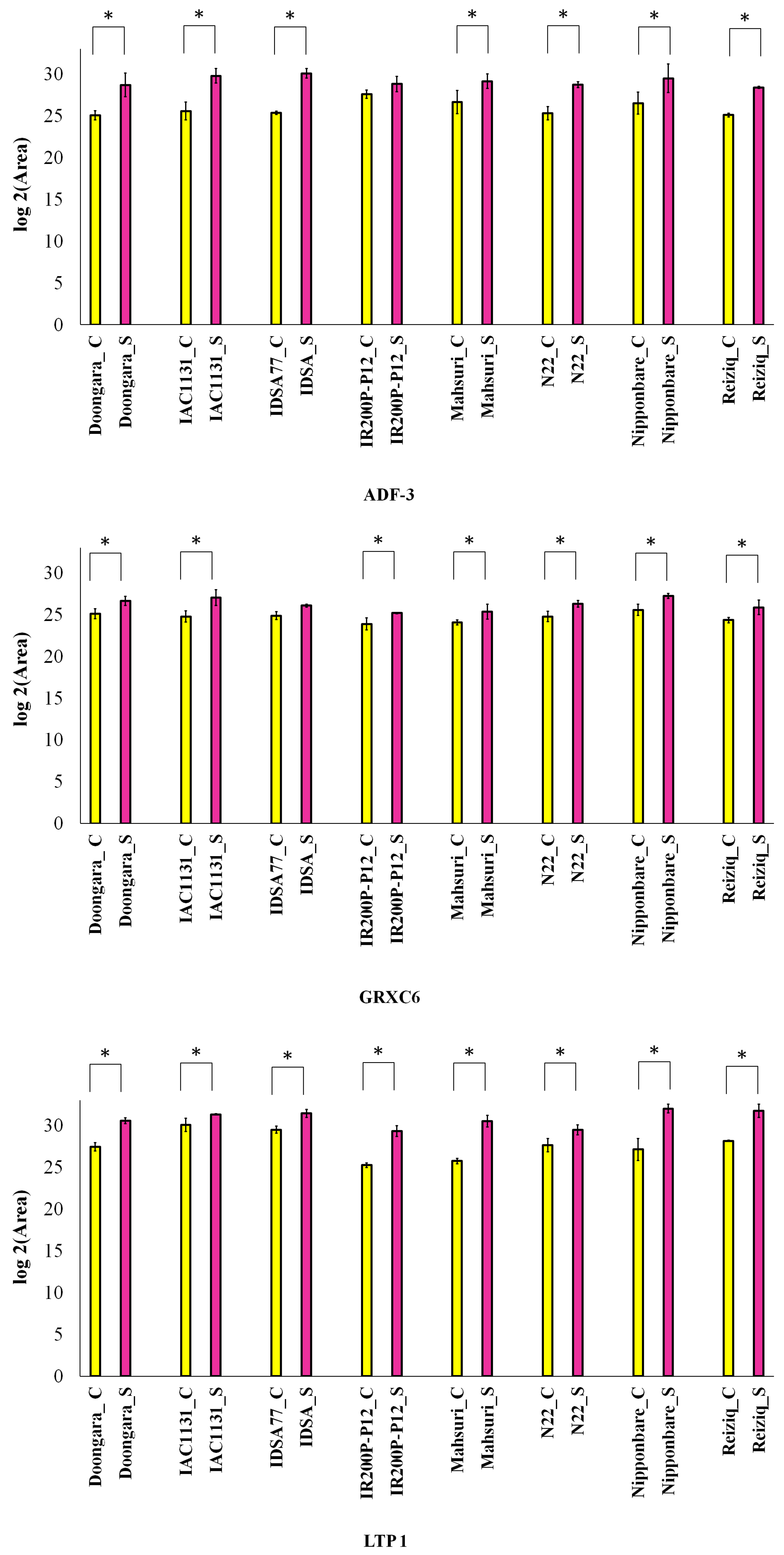
2.2. Proteins Induced in Response to Drought Stress in All Eight Genotypes
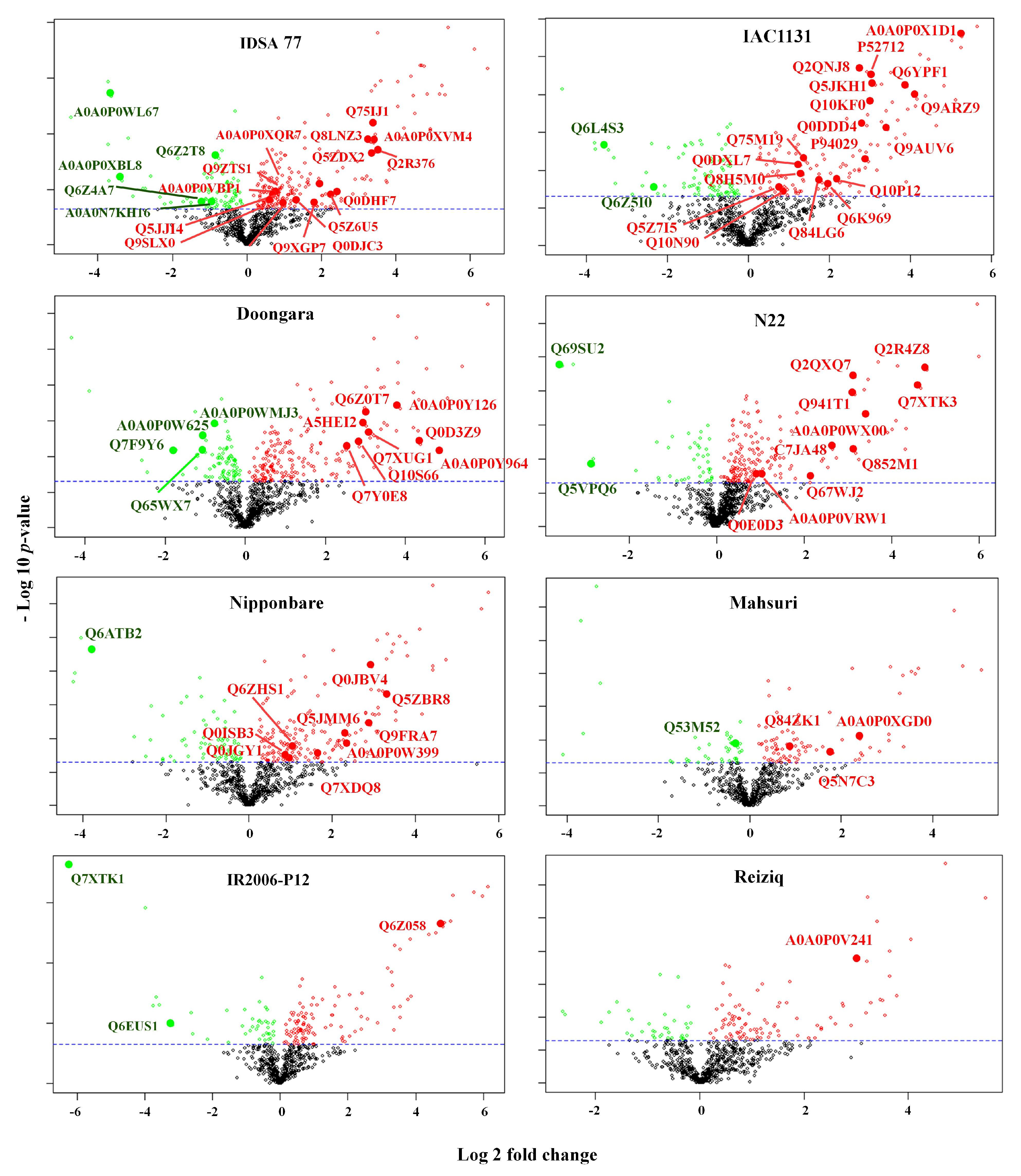
2.3. Proteins Induced in Response to Drought Stress Uniquely in a Single Rice Genotype
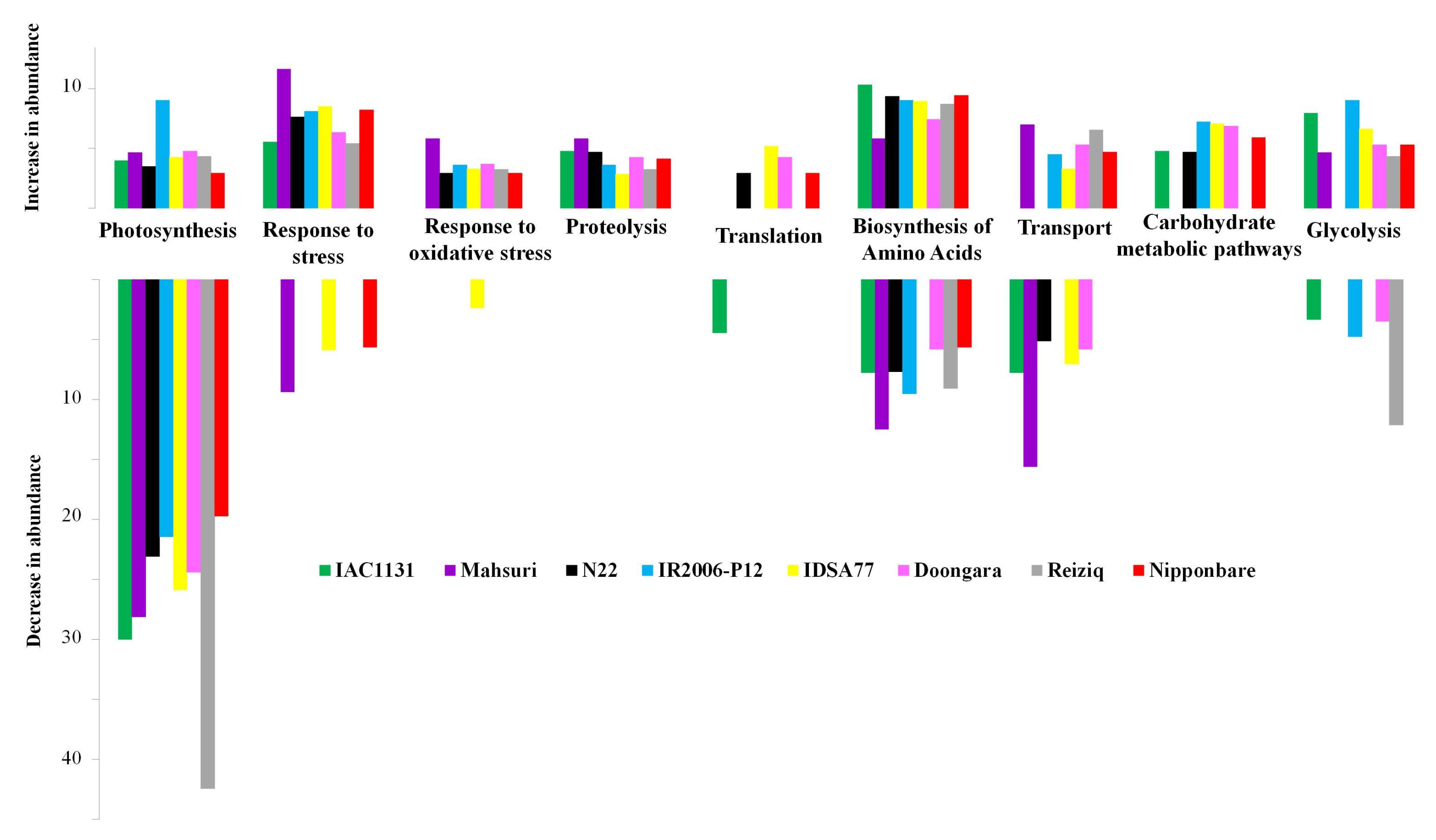
2.4. GO Functional Classification of Significantly Altered Protein in Response to Drought
2.5. Parallel Reaction Monitoring (PRM) Validation
3. Discussion
4. Materials and Methods
4.1. Plant Material and Sample Preparation
4.2. Protein Extraction and Protein Assay
4.3. In-Solution Digestion and Peptide Extraction
4.4. Nanoflow Liquid Chromatography–Tandem Mass Spectrometry
4.5. Parallel Reaction Monitoring (PRM) Analysis
4.6. Protein Identification
4.7. Data Processing and Quantitation
4.8. Functional Protein Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| FDR | False discovery rate |
| GO | Gene ontology |
| LEA | Late embryogenesis abundant |
| nLC–MS/M | nanoflow liquid chromatography–tandem mass spectrometry |
| NSAF | Normalized spectral abundance factor |
| PRM | Parallel reaction monitoring |
| ROS | Reactive oxygen species |
References
- FAO Staple Foods. What do People Eat? Available online: http://www.fao.org/3/u8480e/U8480E07.htm (accessed on 2 December 2019).
- Leng, G.; Hall, J. Crop yield sensitivity of global major agricultural countries to droughts and the projected changes in the future. Sci. Total Environ. 2019, 654, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Li, X.; Xiong, L. Insight into differential responses of upland and paddy rice to drought stress by comparative expression profiling analysis. Int. J. Mol. Sci. 2013, 14, 5214–5238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, D.Q.; Weisskopf, A.R.; Castillo, C.C. Pathways of Rice Diversification across Asia. Archaeol. Int. 2016, 19, 84–96. [Google Scholar] [CrossRef]
- Demirevska, K.; Zasheva, D.; Dimitrov, R.; Simova-Stoilova, L.; Stamenova, M.; Feller, U. Drought stress effects on Rubisco in wheat: Changes in the rubisco large subunit. Acta Physiol. Plant. 2009, 31, 1129–1138. [Google Scholar] [CrossRef]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome ( Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Rahiminejad, M.; Ledari, M.T.; Mirzaei, M.; Ghorbanzadeh, Z.; Kavousi, K.; Ghaffari, M.R.; Haynes, P.A.; Komatsu, S.; Salekdeh, G.H. The Quest for Missing Proteins in Rice. Mol. Plant 2019, 12, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Bernier, J.; Atlin, G.N.; Serraj, R.; Kumar, A.; Spaner, D. Breeding upland rice for drought resistance. J. Sci. Food Agric. 2008, 88, 927–939. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, X.; Jiao, Y.; Qin, Y.; Liu, X.; He, K.; Chen, C.; Ma, L.; Wang, J.; Xiong, L.; et al. Global genome expression analysis of rice in response to drought and high-salinity stresses in shoot, flag leaf, and panicle. Plant Mol. Biol. 2007, 63, 591–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenkolbe, T.; Do, P.T.; Zuther, E.; Repsilber, D.; Walther, D.; Hincha, D.K.; Köhl, K.I. Expression profiling of rice cultivars differing in their tolerance to long-term drought stress. Plant Mol. Biol. 2009, 69, 133–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Pan, Y.; Zhao, X.; Zhu, L.; Fu, B.; Li, Z. Genome-wide temporal-spatial gene expression profiling of drought responsiveness in rice. BMC Genom. 2011, 12, 149. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhang, F.; Zhang, F.; Wang, W.; Zhou, Y.; Fu, B.; Li, Z. Comparative transcriptome sequencing of tolerant rice introgression line and its parents in response to drought stress. BMC Genom. 2014, 15, 1026. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, X.; Lu, C.; Zeng, X.; Li, Y.; Fu, D.; Wu, G. Non-specific lipid transfer proteins in plants: Presenting new advances and an integrated functional analysis. Exp. Bot. 2015, 66, 5663–5681. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.O.; Lee, J.R.; Yoo, J.Y.; Jang, H.H.; Moon, J.C.; Jung, B.G.; Chi, Y.H.; Park, S.K.; Lee, S.S.; Lim, C.O.; et al. GSH-dependent peroxidase activity of the rice (Oryza sativa) glutaredoxin, a thioltransferase. Biochem. Biophys. Res. Commun. 2002, 296, 1152–1156. [Google Scholar] [CrossRef]
- Qian, D.; Zhang, Z.; He, J.; Zhang, P.; Ou, X.; Li, T.; Niu, L.; Nan, Q.; Niu, Y. Arabidopsis ADF5 promotes stomatal closure by regulating actin cytoskeleton remodeling in response to ABA and drought stress. Exp. Bot. 2019, 70, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Zhang, X.-F.; Wang, X.-F.; Zhang, D.-P. Arabidopsis 3-Ketoacyl-CoA Thiolase-2 (KAT2), an enzyme of fatty acid β-Oxidation, is involved in ABA signal transduction. Plant Cell Physiol. 2011, 52, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Do, H.; Kim, I.-S.; Jeon, B.W.; Lee, C.W.; Park, A.K.; Wi, A.R.; Shin, S.C.; Park, H.; Kim, Y.-S.; Yoon, H.-S.; et al. Structural understanding of the recycling of oxidized ascorbate by dehydroascorbate reductase (OsDHAR) from Oryza sativa L. japonica. Sci. Rep. 2016, 6, 19498. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, C.; Krüger, M.M.; dos Santos, R.S.; Mertz, L.M.; da Maia, L.C.; Rombaldi, C.V.; de Oliveira, A.C. Transcription of mitochondrial and chloroplast related genes in rice plants under anoxic stress. Plant Omics 2012, 5, 547–552. [Google Scholar]
- Meyer, Y.; Buchanan, B.B.; Vignols, F.; Reichheld, J.-P. Thioredoxins and Glutaredoxins: Unifying Elements in Redox Biology. Annu. Rev. Genet. 2009, 43, 335–367. [Google Scholar] [CrossRef]
- MacIntosh, G.C.; Hillwig, M.S.; Meyer, A.; Flagel, L. RNase T2 genes from rice and the evolution of secretory ribonucleases in plants. Mol. Genet. Genom. 2010, 283, 381–396. [Google Scholar] [CrossRef]
- Sarkar, N.K.; Kim, Y.-K.; Grover, A. Rice sHsp genes: Genomic organization and expression profiling under stress and development. BMC Genom. 2009, 10, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Lan, T. Functional characterization of the late embryogenesis abundant (LEA) protein gene family from Pinus tabuliformis (Pinaceae) in Escherichia coli. Sci. Rep. 2016, 6, 19467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Chakraborty, A.; Bhunia, R.K.; Sen, S.K.; Ghosh, A.K. Tolerance to soil water stress by Oryza sativa cv. IR20 was improved by expression of Wsi18 gene locus from Oryza nivara. Biol. Plant. 2014, 62, 129–139. [Google Scholar] [CrossRef]
- Hundertmark, M.; Hincha, D.K. LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genom. 2008, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomé, F.; Nägele, T.; Adamo, M.; Garg, A.; Marco-llorca, C.; Nukarinen, E.; Pedrotti, L.; Peviani, A.; Simeunovic, A.; Tatkiewicz, A.; et al. The low energy signaling network. Front. Plant Sci. 2014, 5, 353. [Google Scholar] [PubMed] [Green Version]
- Głowacka, K.; Kromdijk, J.; Kucera, K.; Xie, J.; Cavanagh, A.P.; Leonelli, L.; Leakey, A.D.B.; Ort, D.R.; Niyogi, K.K.; Long, S.P. Photosystem II Subunit S overexpression increases the efficiency of water use in a field-grown crop. Nat. Commun. 2018, 9, 868. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Breusegem, F. Van Reactive oxygen gene network of plants. Trends Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, C.; Planchon, S.; Pinheiro, C.; Renaut, J.; Ricardo, C.P.; Oliveira, M.M.; Batista, R. Maize IgE binding proteins: Each plant a different profile? Proteome Sci. 2014, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Kurepa, J.; Wang, S.; Li, Y.; Smalle, J. Proteasome regulation, plant growth and stress tolerance. Plant Signal. Behav. 2009, 4, 924–927. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, A.K.; Mishra, A.; Tiwari, V.; Jha, B. Cloning and transcript analysis of type 2 metallothionein gene (SbMT-2) from extreme halophyte Salicornia brachiata and its heterologous expression in E. coli. Gene 2012, 499, 280–287. [Google Scholar] [CrossRef]
- Liu, B.; Qian, S.-B. Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 2014, 5, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cai, X.; Xu, C.; Wang, Q.; Dai, S. Drought-Responsive Mechanisms in Plant Leaves Revealed by Proteomics. Int. J. Mol. Sci 2016, 17, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascovici, D.; Handler, D.C.L.; Wu, J.X.; Haynes, P.A. Multiple testing corrections in quantitative proteomics: A useful but blunt tool. Proteomics 2016, 16, 2448–2453. [Google Scholar] [CrossRef]
- Neilson, K.A.; Keighley, T.; Pascovici, D.; Cooke, B.; Haynes, P.A. Label-Free Quantitative Shotgun Proteomics Using Normalized Spectral Abundance Factors. In Proteomics for Biomarker Discovery; Humana Press: Totowa, NJ, USA, 2013; pp. 205–222. ISBN 978-1-62703-360-2. [Google Scholar]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Hussain, S.; Fujii, T.; McGoey, S.; Yamada, M.; Ramzan, M.; Akmal, M. Evaluation of different rice varieties for growth and yield characteristics. J. Anim. Plant Sci. 2014, 24, 1504–1510. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Row | Rice Genotype | Total Proteins | Upregulated Proteins | Downregulated Proteins | Protein FDR (%) | Peptide FDR (%) |
|---|---|---|---|---|---|---|
| 1 | Doongara | 883 | 192 | 87 | 1.12 | 0.02 |
| 2 | IAC1131 | 887 | 129 | 91 | 1.11 | 0.32 |
| 3 | IDSA77 | 910 | 214 | 85 | 0.07 | 0.02 |
| 4 | IR2006-P12 | 648 | 112 | 43 | 1.36 | 0.05 |
| 5 | Mahsuri | 641 | 86 | 39 | 1.53 | 0.06 |
| 6 | N22 | 879 | 171 | 42 | 1.12 | 0.03 |
| 7 | Nipponbare | 808 | 105 | 70 | 0.98 | 0.37 |
| 8 | Reiziq | 731 | 93 | 35 | 1.08 | 0.23 |
| Row | Rice Genotype | Uniprot ID | Protein Name | NSAF Fold Change | Homologous Protein | Identity (Percent) |
|---|---|---|---|---|---|---|
| 1 | IDSA77 | Q2R376 | Expressed protein (Os11g0533400 protein) | 12.03 | - | - |
| 2 | IDSA77 | Q0DHF7 | Os05g0468800 protein | 5.19 | putative cold regulated protein [Oryza sativa japonica Group] | 89% |
| 3 | IDSA77 | A0A0P0XQR7 | Os09g0535900 protein | 2.34 | DNA-(apurinic or apyrimidinic site) lyase 2 isoform X2 [Panicum miliaceum] | 88% |
| 4 | IDSA77 | Q0DJC3 | Os05g0301700 protein | 2.3 | cytochrome c1-2, heme protein, mitochondrial [Oryza sativa japonica Group] | 100% |
| 5 | IDSA77 | A0A0P0VBP1 | Os01g0895600 protein | 1.8 | calreticulin-3 [Oryza sativa Japonica Group] | 100% |
| 6 | IAC1131 | Q0DDD4 | Os06g0232100 protein | 7.62 | probable serine/threonine-protein kinase SIS8 | 100% |
| 7 | IAC1131 | Q8H5M0 | Os07g0585000 protein | 2.55 | calcium-dependent lipid-binding (CaLB domain) family protein [Zea mays] | 75% |
| 8 | Mahsuri | A0A0P0XGD0 | Os08g0425800 protein | 5.39 | - | - |
| 9 | Reiziq | A0A0P0V241 | Os01g0332900 protein | 8.19 | Predicted acidic leucine-rich nuclear phosphoprotein 32-related protein [Oryza brachyantha] | 84% |
| 10 | N22 | Q2QXQ7 | Expressed protein (Os12g0147200 protein) | 2.09 | - | - |
| Row | Rice Genotype | Ecosystem | Description |
|---|---|---|---|
| 1 | Doongara | Lowland | Australian japonica |
| 2 | IAC1131 | Upland | japonica, drought tolerant |
| 3 | IDSA77 | Upland | Tolerant to high temperature |
| 4 | IR2006-P12 | Upland | indica, tolerant to high temperature |
| 5 | Mahsuri | Lowland | Indian traditional rice genotype, drought sensitive |
| 6 | N22 | Upland | indica, tolerant to high temperature and drought |
| 7 | Nipponbare | Lowland | japonica, drought sensitive |
| 8 | Reiziq | Lowland | Australian japonica |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamzelou, S.; Pascovici, D.; Kamath, K.S.; Amirkhani, A.; McKay, M.; Mirzaei, M.; Atwell, B.J.; Haynes, P.A. Proteomic Responses to Drought Vary Widely Among Eight Diverse Genotypes of Rice (Oryza sativa). Int. J. Mol. Sci. 2020, 21, 363. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010363
Hamzelou S, Pascovici D, Kamath KS, Amirkhani A, McKay M, Mirzaei M, Atwell BJ, Haynes PA. Proteomic Responses to Drought Vary Widely Among Eight Diverse Genotypes of Rice (Oryza sativa). International Journal of Molecular Sciences. 2020; 21(1):363. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010363
Chicago/Turabian StyleHamzelou, Sara, Dana Pascovici, Karthik Shantharam Kamath, Ardeshir Amirkhani, Matthew McKay, Mehdi Mirzaei, Brian J. Atwell, and Paul A. Haynes. 2020. "Proteomic Responses to Drought Vary Widely Among Eight Diverse Genotypes of Rice (Oryza sativa)" International Journal of Molecular Sciences 21, no. 1: 363. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010363