iQSP: A Sequence-Based Tool for the Prediction and Analysis of Quorum Sensing Peptides Using Informative Physicochemical Properties

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
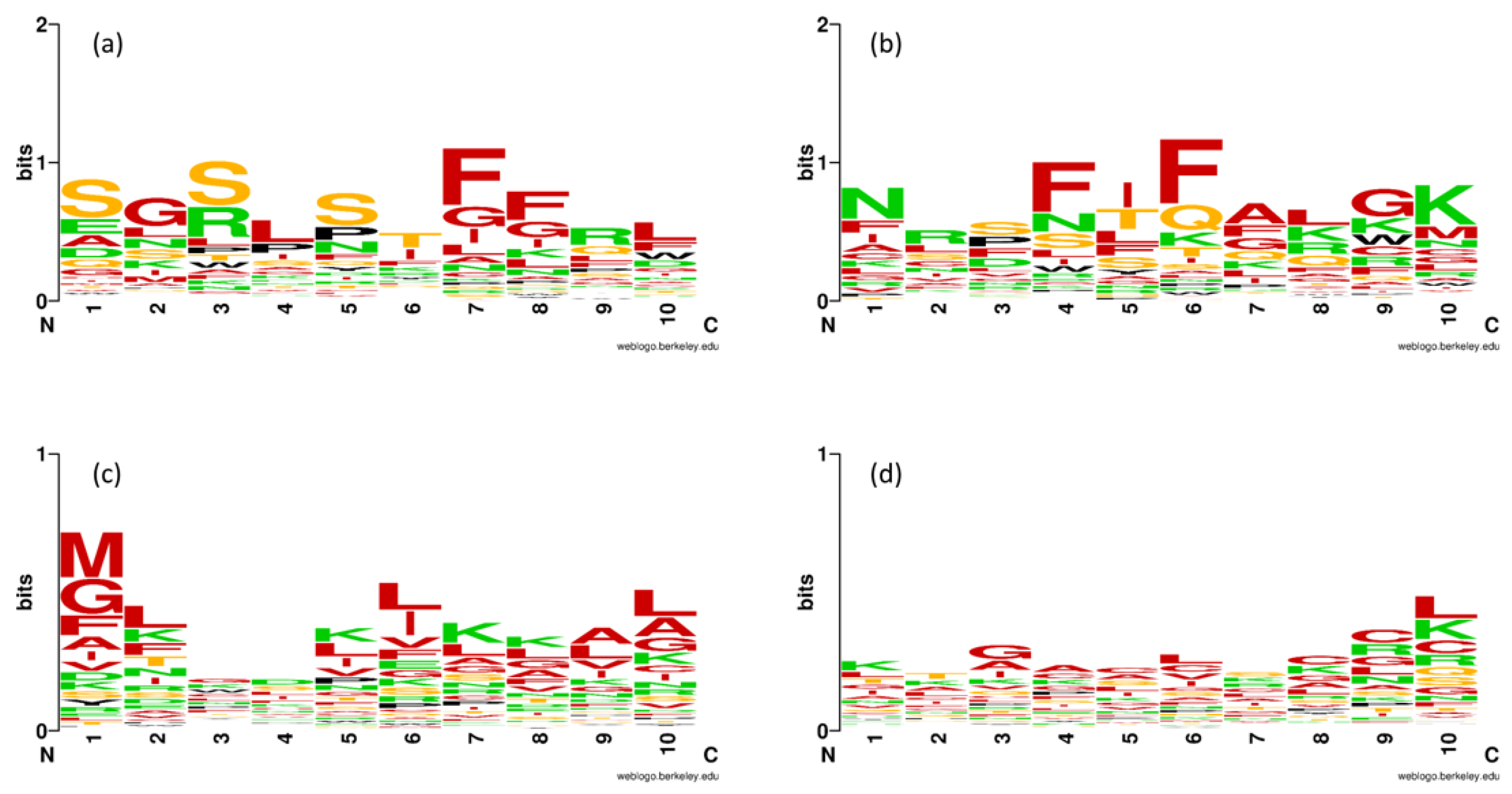
2.1. Composition Analysis
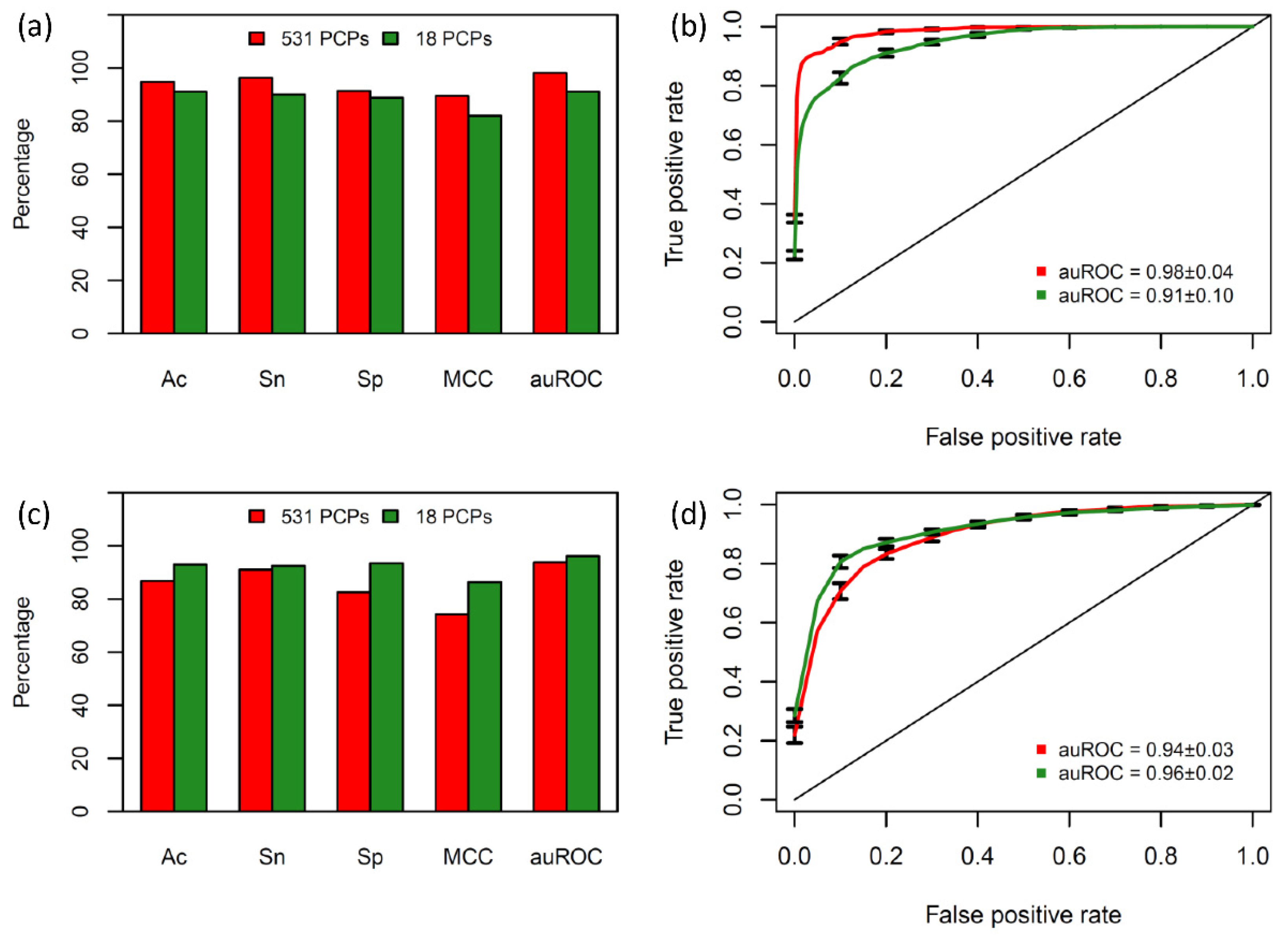
2.2. Prediction Capabilities of the Different Subset of Physicochemical Properties
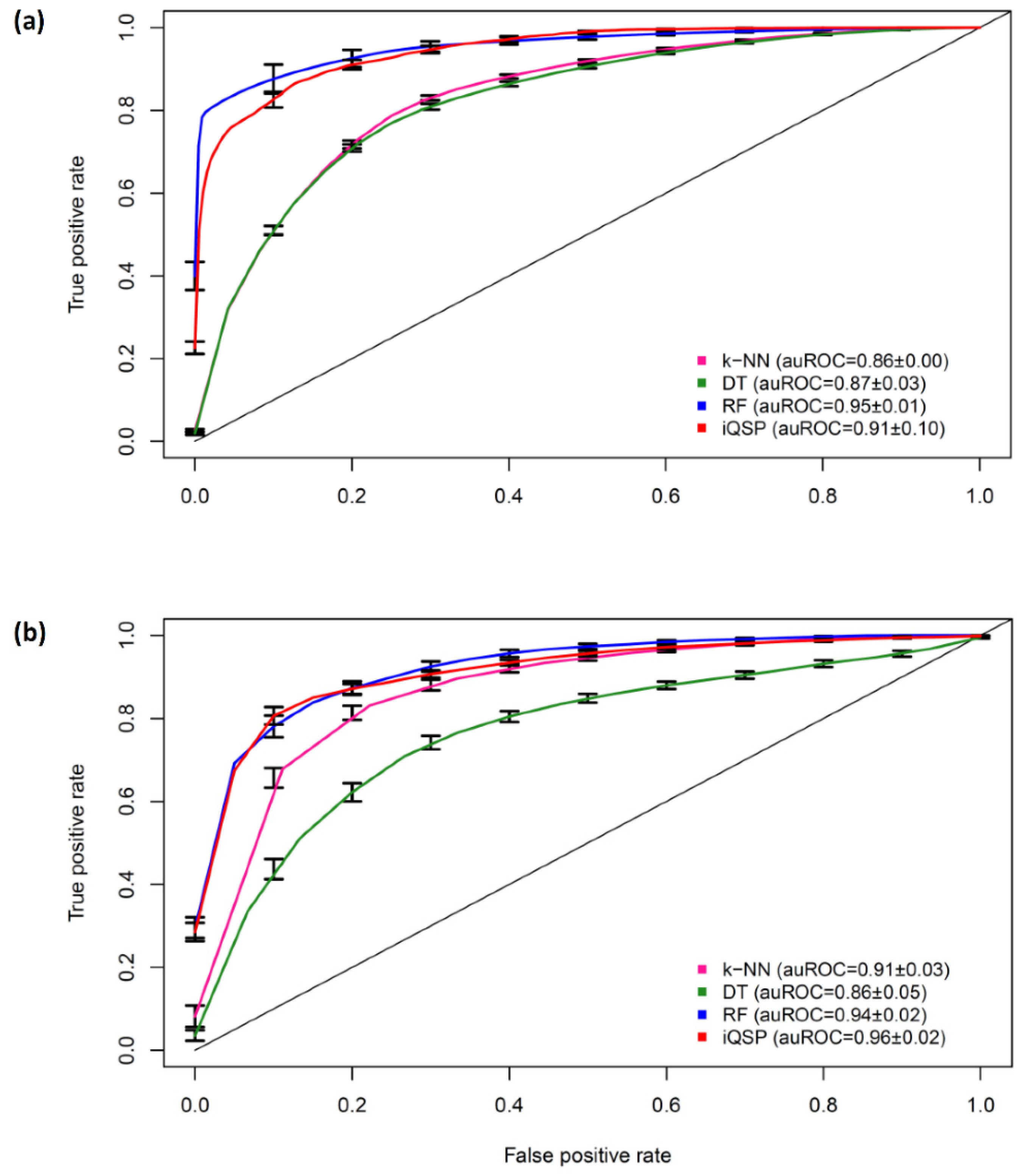
2.3. Comparison with Existing Methods
2.4. Feature Contribution Analysis
2.5. Interpretable Rules Acquisition
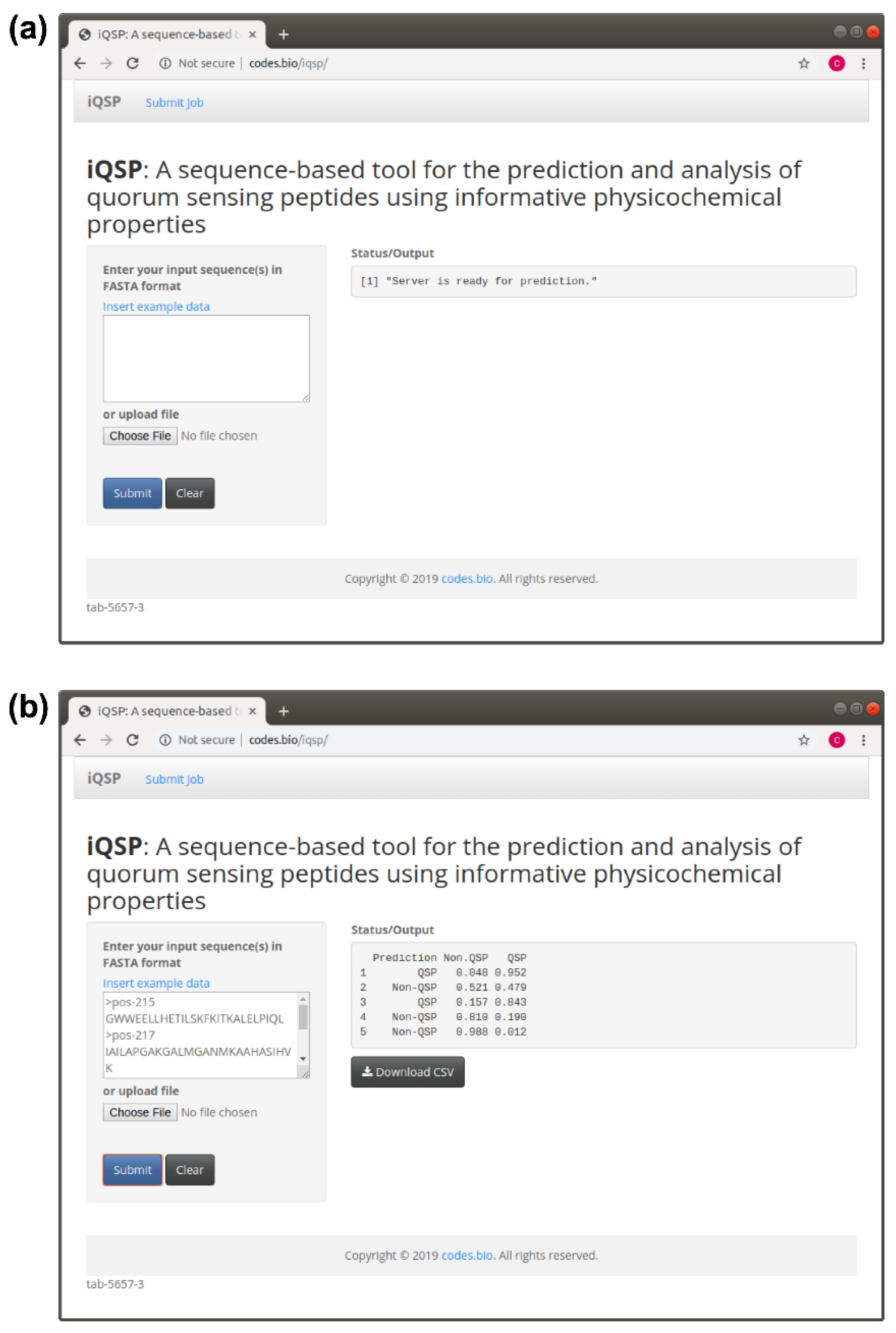
2.6. iQSP Web Server
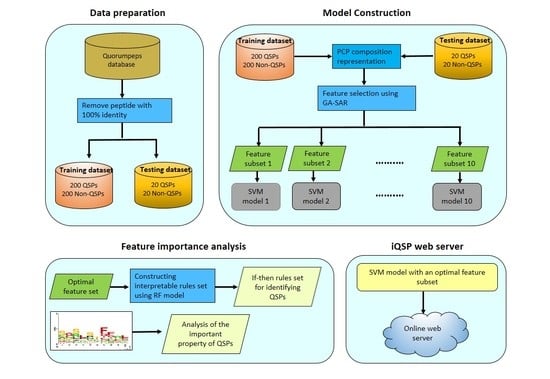
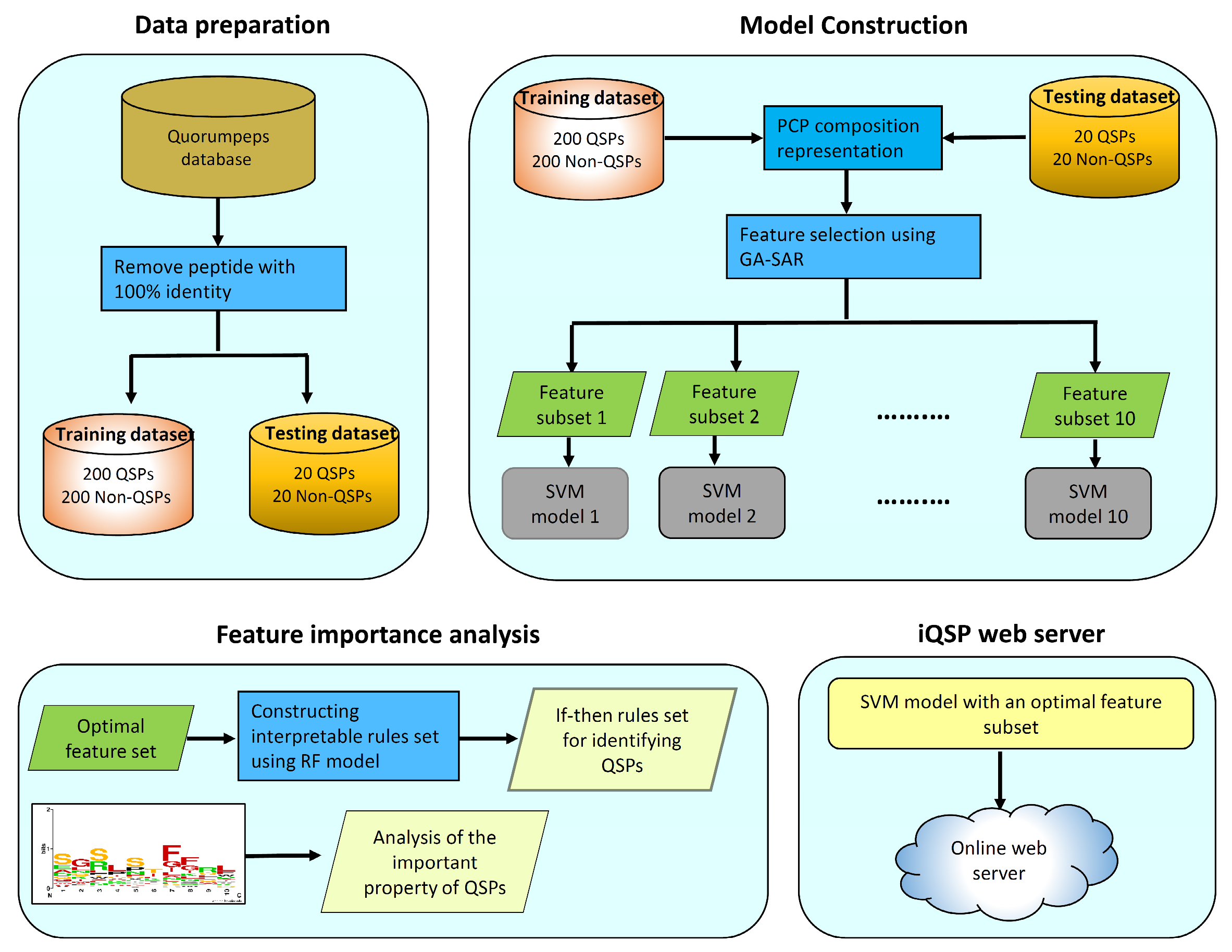
3. Materials and Methods
3.1. Benchmark Dataset
3.2. Feature Representation
3.3. Support Vector Machine
3.4. Performance Evaluation
3.5. Identification of Informative Physicochemical Properties
3.6. Construction of Interpretable Rules
3.7. Construction of the iQSP Web Server
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subset | # Feature | AAindex |
|---|---|---|
| 1 | 14 | FASG760103, ISOY800102, KYTJ820101, LIFS790103, MEIH800102, OOBM850101, PALJ810113, QIAN880137, ROSG850101, TANS770101, AURR980116, VINM940104, NADH010107, FUKS010106 |
| 2 | 17 | ANDN920101, GRAR740102, JANJ780103, LIFS790101, NAGK730102, PALJ810102, QIAN880137, RACS820102, ROBB760109, ROBB760111, SUEM840102, TANS770104, TANS770108, AURR980115, VINM940103, BLAM930101, PUNT030102 |
| 3 | 16 | EISD860101, GEIM800105, HOPA770101, LIFS790101, MEIH800103, OOBM770101, PALJ810111, QIAN880128, RACS770101, ROBB760107, ROBB790101, AURR980110, VINM940101, KIMC930101, FUKS010106, TSAJ990102, |
| 4 | 17 | DAYM780101, ISOY800108, MEIH800102, NAKH900104, PALJ810111, PALJ810113, PRAM820102, RACS770101, ROBB760111, SNEP660104, AURR980118, VINM940101, PARS000101, NADH010106, FUKS010106, TSAJ990101, GEOR030107 |
| 5 | 17 | ANDN920101, FINA910102, GEIM800102, GRAR740102, ISOY800101, LIFS790102, NOZY710101, PALJ810106, QIAN880138, ROBB760107, ROBB760112, ZIMJ680101, AURR980113, MUNV940102, MUNV940105, MONM990201, FUKS010109 |
| 6 | 18 | DAYM780101, GEIM800101, GRAR740101, ISOY800107, MANP780101, PALJ810111, PONP800102, PRAM820101, PRAM900102, QIAN880137, ROBB760104, ROBB760113, AURR980102, MUNV940103, WIMW960101, KUMS000103, NADH010104, FUKS010106 |
| 7 | 15 | GRAR740101, ISOY800108, LIFS790103, MIYS850101, OOBM850105, PALJ810114, QIAN880137, RACS820109, ROBB760108, ROSG850101, TANS770107, AURR980116, VINM940104, PARS000102, GEOR030108 |
| 8 | 17 | ANDN920101, ARGP820103, BIGC670101, BULH740101, BUNA790102, CHOC760104, CHOP780211, CHOP780212, DESM900101, FASG760101, GRAR740101, NAKH900101, RADA880107, ROBB790101, TANS770109, GUOD860101, PONJ960101 |
| 9 | 18 | FAUJ880112, FINA910103, GRAR740101, JOND750102, NISK860101, PALJ810108, PONP800104, PRAM820101, QIAN880125, QIAN880134, ROBB760108, WERD780104, AURR980113, VINM940101, VINM940102, PARS000102, FUKS010108, WOLR790101 |
| 10 | 17 | ANDN920101, BROC820102, BUNA790101, CHAM830104, CHOP780207, CIDH920101, CIDH920104, DESM900102, FASG760101, FINA910102, JANJ780101, LEWP710101, PALJ810110, QIAN880125, VASM830101, AURR980108, COSI940101 |
| % Covered Samples | Rule | Prediction Result |
|---|---|---|
| 3.57 | GRAR740101 ≤ 0.9055 & MANP780101 > 0.7495 & PRAM900102 > 0.848 & QIAN880137 ≤ 0.237 | QSP |
| 14.03 | PONP800102 > −0.751 & PONP800102 ≤ 1.0025 & QIAN880137 ≤ −0.104 & ROBB760104 ≤ 0.3645 & ROBB760104 > −0.5205 | QSP |
| 4.08 | PALJ810111 ≤ 1.369 & QIAN880137 > −0.104 & QIAN880137 ≤ 0.417 & ROBB760113 ≤ 0.5975 & AURR980102 ≤ 0.6955 | QSP |
| 9.95 | GEIM800101 > −0.3135 & GRAR740101 > −0.176 & ISOY800107 ≤ 1.367 & MANP780101 > −0.3325 & PALJ810111 ≤ 1.0905 & QIAN880137 ≤ −0.0985 | QSP |
| 9.95 | PALJ810111 > −0.786 & QIAN880137 > 0.237 & QIAN880137 > 0.403 & ROBB760113 > 0.5975 & AURR980102 ≤ 0.811 & KUMS000103 ≤ 0.793 | QSP |
| 2.81 | GRAR740101 ≤ 0.341 & ISOY800107 ≤ −0.089 & PALJ810111 ≤ 1.2455 & QIAN880137 > −0.009 & ROBB760113 ≤ −0.0285 | QSP |
| 2.81 | GRAR740101 > −0.708 & PRAM900102 ≤ 1.2985 & QIAN880137 > −0.104 & QIAN880137 ≤ 1.105 & AURR980102 ≤ 0.6585 & KUMS000103 ≤ 0.974 | QSP |
| 1.53 | PONP800102 ≤ 1.1095 & PRAM900102 ≤ 0.8295 & QIAN880137 ≤ 0.2625 & QIAN880137 > −0.9055 & ROBB760113 > −0.5875 & ROBB760113 ≤ 1.031 | QSP |
| 11.73 | GEIM800101 > −0.547 & PONP800102 > 0.9625 & PRAM900102 ≤ 1.026 | Non-QSP |
| 3.57 | QIAN880137 > −0.0985 & QIAN880137 ≤ 0.482 & ROBB760113 > 0.5975 & AURR980102 > −0.1535 | Non-QSP |
| 2.30 | QIAN880137 > 0.257 & ROBB760113 > 0.613 & ROBB760113 > 0.7775 & AURR980102 > 0.584 | Non-QSP |
| 1.79 | DAYM780101 ≤ 0.1035 & MANP780101 > 0.5635 & QIAN880137 ≤ 0.2625 & ROBB760113 > 0.5215 | Non-QSP |
| 1.53 | GRAR740101 > −0.045 & PALJ810111 > 1.0905 & QIAN880137 ≤ −0.0985 | Non-QSP |
| 6.12 | MANP780101 > −0.5985 & ROBB760104 > −0.3545 & AURR980102 ≤ 0.5935 & AURR980102 ≤ 0.5125 & KUMS000103 > 0.663 | Non-QSP |
| 3.57 | PONP800102 > 0.5045 & QIAN880137 > −0.039 & ROBB760104 > −0.1345 & ROBB760113 ≤ 0.5975 & AURR980102 ≤ 1.22 & FUKS010106 ≤ 1.1985 | Non-QSP |
| 1.28 | QIAN880137 > 0.2625 & QIAN880137 ≤ 0.7795 & ROBB760104 > −0.3885 & AURR980102 ≤ 0.6585 & AURR980102 ≤ 0.515 & NADH010104 > −0.023 | Non-QSP |
| 3.06 | GEIM800101 ≤ 1.636 & PONP800102 ≤ 0.6385 & PRAM900102 > −0.017 & PRAM900102 > 0.627 & QIAN880137 > −0.009 | Non-QSP |
| 2.55 | PALJ810111 ≤ 0.436 & PRAM900102 > 0.1605 & QIAN880137 > −0.009 & AURR980102 ≤ 0.6525 | Non-QSP |
| 1.53 | GEIM800101 ≤ −0.3305 & GRAR740101 > −0.2425 & PONP800102 ≤ 0.98 & QIAN880137 ≤ 0.237 | Non-QSP |
| 12.24 | Else | Non-QSP |
References
- Fuqua, W.C.; Winans, S.C.; Greenberg, E.P. Quorum sensing in bacteria: The LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol. 1994, 176, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef]
- Remy, B.; Mion, S.; Plener, L.; Elias, M.; Chabriere, E.; Daude, D. Interference in bacterial quorum sensing: A biopharmaceutical perspective. Front. Pharm. 2018, 9, 203. [Google Scholar] [CrossRef]
- Monnet, V.; Juillard, V.; Gardan, R. Peptide conversations in Gram-positive bacteria. Crit. Rev. Microbiol. 2016, 42, 339–351. [Google Scholar] [CrossRef]
- Schuster, M.; Sexton, D.J.; Diggle, S.P.; Greenberg, E.P. Acyl-homoserine lactone quorum sensing: From evolution to application. Annu. Rev. Microbiol. 2013, 67, 43–63. [Google Scholar] [CrossRef]
- Hazan, R.; Que, Y.A.; Maura, D.; Strobel, B.; Majcherczyk, P.A.; Hopper, L.R.; Wilbur, D.J.; Hreha, T.N.; Barquera, B.; Rahme, L.G. Auto poisoning of the respiratory chain by a quorum-sensing-regulated molecule favors biofilm formation and antibiotic tolerance. Curr. Biol. 2016, 26, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Zhang, A.; Yin, H.; Chu, W. Bacillus sp. QSI-1 modulate quorum sensing signals reduce aeromonas hydrophila level and alter gut microbial community structure in fish. Front. Cell. Infect. Microbiol. 2016, 6, 184. [Google Scholar] [CrossRef] [Green Version]
- Tiaden, A.; Hilbi, H. alpha-Hydroxyketone synthesis and sensing by Legionella and Vibrio. Sensors 2012, 12, 2899–2919. [Google Scholar] [CrossRef] [Green Version]
- Kendall, M.M.; Sperandio, V. Quorum sensing by enteric pathogens. Curr. Opin. Gastroenterol. 2007, 23, 10–15. [Google Scholar] [CrossRef]
- Heeb, S.; Fletcher, M.P.; Chhabra, S.R.; Diggle, S.P.; Williams, P.; Camara, M. Quinolones: From antibiotics to autoinducers. FEMS Microbiol. Rev. 2011, 35, 247–274. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Schauder, S.; Potier, N.; Van Dorsselaer, A.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 2002, 415, 545–549. [Google Scholar] [CrossRef]
- Mukherjee, S.; Bassler, B.L. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef]
- Whiteley, M.; Diggle, S.P.; Greenberg, E.P. Progress in and promise of bacterial quorum sensing research. Nature 2017, 551, 313–320. [Google Scholar] [CrossRef]
- Jiang, Q.; Chen, J.; Yang, C.; Yin, Y.; Yao, K. Quorum Sensing: A prospective therapeutic target for bacterial diseases. Biomed. Res. Int. 2019, 2019, 2015978. [Google Scholar] [CrossRef] [Green Version]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef]
- Verbeke, F.; De Craemer, S.; Debunne, N.; Janssens, Y.; Wynendaele, E.; Van de Wiele, C.; De Spiegeleer, B. Peptides as quorum sensing molecules: Measurement techniques and obtained levels in vitro and in vivo. Front. Neurosci. 2017, 11, 183. [Google Scholar] [CrossRef] [Green Version]
- De Kievit, T.R.; Gillis, R.; Marx, S.; Brown, C.; Iglewski, B.H. Quorum-sensing genes in Pseudomonas aeruginosa biofilms: Their role and expression patterns. Appl. Environ. Microbiol. 2001, 67, 1865–1873. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhang, L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 2015, 6, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Eickhoff, M.J.; Bassler, B.L. SnapShot: Bacterial quorum sensing. Cell 2018, 174, 1328–1328.e1. [Google Scholar] [CrossRef]
- Gallardo-Garcia, M.M.; Sanchez-Espin, G.; Ivanova-Georgieva, R.; Ruiz-Morales, J.; Rodriguez-Bailon, I.; Vinuela Gonzalez, V.; Garcia-Lopez, M.V. Relationship between pathogenic, clinical, and virulence factors of Staphylococcus aureus in infective endocarditis versus uncomplicated bacteremia: A case-control study. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 821–828. [Google Scholar] [CrossRef]
- Liu, J.; Yu, H.; Huang, Y.; Yu, Z.; Fan, G.; Jin, J.; Liu, X.; Wang, G. Complete genome sequence of a novel bacteriophage infecting Bradyrhizobium diazoefficiens USDA110. Sci. China Life Sci. 2018, 61, 118–121. [Google Scholar] [CrossRef]
- Han, M.; Gu, J.; Gao, G.F.; Liu, W.J. China in action: National strategies to combat against emerging infectious diseases. Sci. China Life Sci. 2017, 60, 1383–1385. [Google Scholar] [CrossRef]
- Vadekeetil, A.; Saini, H.; Chhibber, S.; Harjai, K. Exploiting the antivirulence efficacy of an ajoene-ciprofloxacin combination against Pseudomonas aeruginosa biofilm associated murine acute pyelonephritis. Biofouling 2016, 32, 371–382. [Google Scholar] [CrossRef]
- Bahari, S.; Zeighami, H.; Mirshahabi, H.; Roudashti, S.; Haghi, F. Inhibition of Pseudomonas aeruginosa quorum sensing by subinhibitory concentrations of curcumin with gentamicin and azithromycin. J. Glob. Antimicrob. Resist. 2017, 10, 21–28. [Google Scholar] [CrossRef]
- Furiga, A.; Lajoie, B.; El Hage, S.; Baziard, G.; Roques, C. Impairment of pseudomonas aeruginosa biofilm resistance to antibiotics by combining the drugs with a new quorum-sensing inhibitor. Antimicrob. Agents Chemother. 2015, 60, 1676–1686. [Google Scholar] [CrossRef] [Green Version]
- Brackman, G.; Cos, P.; Maes, L.; Nelis, H.J.; Coenye, T. Quorum sensing inhibitors increase the susceptibility of bacterial biofilms to antibiotics in vitro and in vivo. Antimicrob. Agents Chemother. 2011, 55, 2655–2661. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Hu, J.; Li, F.; Song, J.; Su, R.; Zou, Q. Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief. Bioinform. 2018, 10, 1–14. [Google Scholar] [CrossRef]
- Rajput, A.; Gupta, A.K.; Kumar, M. Prediction and analysis of quorum sensing peptides based on sequence features. PLoS ONE 2015, 10, e0120066. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.-C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895. [Google Scholar] [CrossRef] [Green Version]
- Raiko, T.; Ilin, A.; Karhunen, J. Principal component analysis for large scale problems with lots of missing values. In Proceedings of European Conference on Machine Learning; Springer: Berlin/Heidelberg, Germany, 2007; pp. 691–698. [Google Scholar]
- Subramanian, J.; Simon, R. Overfitting in prediction models—Is it a problem only in high dimensions? Contemp. Clin. Trials 2013, 36, 636–641. [Google Scholar] [CrossRef]
- Xing, E.P.; Jordan, M.I.; Karp, R.M. Feature selection for high-dimensional genomic microarray data. ICML 2001, 1, 601–608. [Google Scholar]
- Chou, K.-C. Advance in predicting subcellular localization of multi-label proteins and its implication for developing multi-target drugs. Curr. Med. Chem. 2019, 26, 4918–4943. [Google Scholar] [CrossRef]
- Chou, K.-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. ACPred: A computational tool for the prediction and analysis of anticancer peptides. Molecules 2019, 24, 1973. [Google Scholar] [CrossRef] [Green Version]
- Simeon, S.; Li, H.; Win, T.S.; Malik, A.A.; Kandhro, A.H.; Piacham, T.; Shoombuatong, W.; Nuchnoi, P.; Wikberg, J.E.; Gleeson, M.P. PepBio: Predicting the bioactivity of host defense peptides. RSC Adv. 2017, 7, 35119–35134. [Google Scholar] [CrossRef] [Green Version]
- Simeon, S.; Shoombuatong, W.; Anuwongcharoen, N.; Preeyanon, L.; Prachayasittikul, V.; Wikberg, J.E.; Nantasenamat, C. osFP: A web server for predicting the oligomeric states of fluorescent proteins. J. Cheminform. 2016, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Win, T.S.; Schaduangrat, N.; Prachayasittikul, V.; Nantasenamat, C.; Shoombuatong, W. PAAP: A web server for predicting antihypertensive activity of peptides. Future Med. Chem. 2018, 10, 1749–1767. [Google Scholar] [CrossRef]
- Laengsri, V.; Nantasenamat, C.; Schaduangrat, N.; Nuchnoi, P.; Prachayasittikul, V.; Shoombuatong, W. TargetAntiAngio: A sequence-based tool for the prediction and analysis of anti-angiogenic peptides. Int. J. Mol. Sci. 2019, 20, 2950. [Google Scholar] [CrossRef] [Green Version]
- Shoombuatong, W.; Schaduangrat, N.; Pratiwi, R.; Nantasenamat, C. THPep: A machine learning-based approach for predicting tumor homing peptides. Comput. Biol. Chem. 2019, 80, 441–451. [Google Scholar] [CrossRef]
- Pratiwi, R.; Malik, A.A.; Schaduangrat, N.; Prachayasittikul, V.; Wikberg, J.E.; Nantasenamat, C.; Shoombuatong, W. CryoProtect: A web server for classifying antifreeze proteins from nonantifreeze proteins. J. Chem. 2017, 2017, 15. [Google Scholar] [CrossRef]
- Hongjaisee, S.; Nantasenamat, C.; Carraway, T.S.; Shoombuatong, W. HIVCoR: A sequence-based tool for predicting HIV-1 CRF01_AE coreceptor usage. Comput. Biol. Chem. 2019, 80, 419–432. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Shoombuatong, W.; Lee, H.-C.; Chaijaruwanich, J.; Huang, H.-L.; Ho, S.-Y. SCMCRYS: Predicting protein crystallization using an ensemble scoring card method with estimating propensity scores of P-collocated amino acid pairs. PLoS ONE 2013, 8, e72368. [Google Scholar] [CrossRef]
- Win, T.S.; Malik, A.A.; Prachayasittikul, V.S.; Wikberg, J.E.; Nantasenamat, C.; Shoombuatong, W. HemoPred: A web server for predicting the hemolytic activity of peptides. Future Med. Chem. 2017, 9, 275–291. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Syvitski, R.T.; Tian, X.-L.; Sampara, K.; Salman, A.; Lee, S.F.; Jakeman, D.L.; Li, Y.-H. Structure-activity analysis of quorum-sensing signaling peptides from Streptococcus mutans. J. Bacteriol. 2007, 189, 1441–1450. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Zhou, C.; Su, R.; Zou, Q. PEPred-Suite: Improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics 2019, 35, 4272–4280. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Towards understanding aromatase inhibitory activity via QSAR modeling. EXCLI J. 2018, 17, 688. [Google Scholar]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Unraveling the bioactivity of anticancer peptides as deduced from machine learning. EXCLI J. 2018, 17, 734. [Google Scholar]
- Shoombuatong, W.; Prachayasittikul, V.; Prachayasittikul, V.; Nantasenamat, C. Prediction of aromatase inhibitory activity using the efficient linear method (ELM). EXCLI J. 2015, 14, 452. [Google Scholar]
- Shoombuatong, W.; Mekha, P.; Chaijaruwanich, J. Sequence based human leukocyte antigen gene prediction using informative physicochemical properties. Int. J. Data Min. Bioinform. 2015, 13, 211–224. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Huang, H.-L.; Chaijaruwanich, J.; Charoenkwan, P.; Lee, H.-C.; Ho, S.-Y. Predicting protein crystallization using a simple scoring card method. In Proceedings of the IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Singapore, 16–19 April 2013; pp. 23–30. [Google Scholar]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Anuwongcharoen, N.; Shoombuatong, W.; Tantimongcolwat, T.; Prachayasittikul, V.; Nantasenamat, C. Exploring the chemical space of influenza neuraminidase inhibitors. PeerJ 2016, 4, e1958. [Google Scholar] [CrossRef] [Green Version]
- Prachayasittikul, V.; Worachartcheewan, A.; Shoombuatong, W.; Prachayasittikul, V.; Nantasenamat, C. Classification of P-glycoprotein-interacting compounds using machine learning methods. EXCLI J. 2015, 14, 958. [Google Scholar]
- Shoombuatong, W.; Mekha, P.; Waiyamai, K.; Cheevadhanarak, S.; Chaijaruwanicha, J. Prediction of human leukocyte antigen gene using k-nearest neighbour classifier based on spectrum kernel. Sci. Asia 2013, 39, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Shoombuatong, W.; Prachayasittikul, V.; Anuwongcharoen, N.; Songtawee, N.; Monnor, T.; Prachayasittikul, S.; Prachayasittikul, V.; Nantasenamat, C. Navigating the chemical space of dipeptidyl peptidase-4 inhibitors. Drug Des. Dev. Ther. 2015, 9, 4515. [Google Scholar]
- Shoombuatong, W.; Prathipati, P.; Prachayasittikul, V.; Schaduangrat, N.; Ahmad Malik, A.; Pratiwi, R.; Wanwimolruk, S.; ES Wikberg, J.; Paul Gleeson, M.; Spjuth, O. Towards predicting the cytochrome P450 modulation: From QSAR to proteochemometric modeling. Curr. Drug Metab. 2017, 18, 540–555. [Google Scholar] [CrossRef]
- Worachartcheewan, A.; Prachayasittikul, V.; Anuwongcharoen, N.; Shoombuatong, W.; Prachayasittikul, V.; Nantasenamat, C. On the origins of hepatitis C virus NS5B polymerase inhibitory activity using machine learning approaches. Curr. Top. Med. Chem. 2015, 15, 1814–1826. [Google Scholar] [CrossRef]
- Worachartcheewan, A.; Shoombuatong, W.; Pidetcha, P.; Nopnithipat, W.; Prachayasittikul, V.; Nantasenamat, C. Predicting metabolic syndrome using the random forest method. Sci. World J. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [Green Version]
- Shoombuatong, W.; Nabu, S.; Simeon, S.; Prachayasittikul, V.; Lapins, M.; Wikberg, J.E.; Nantasenamat, C. Extending proteochemometric modeling for unraveling the sorption behavior of compound–soil interaction. Chemom. Intell. Lab. Syst. 2016, 151, 219–227. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Prathipati, P.; Owasirikul, W.; Worachartcheewan, A.; Simeon, S.; Anuwongcharoen, N.; Wikberg, J.E.; Nantasenamat, C. Towards the revival of interpretable QSAR models. In Advances in QSAR Modeling; Springer: Cham, Switzerland, 2017; pp. 3–55. [Google Scholar]
- Liu, K.; Chen, W.; Lin, H. XG-PseU: An eXtreme Gradient Boosting based method for identifying pseudouridine sites. Mol. Genet. Genom. 2019. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [Green Version]
- Awais, M.; Hussain, W.; Khan, Y.D.; Rasool, N.; Khan, S.A.; Chou, K.-C. iPhosH-PseAAC: Identify phosphohistidine sites in proteins by blending statistical moments and position relative features according to the Chou’s 5-step rule and general pseudo amino acid composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef]
- Ju, Z.; Wang, S.-Y. Prediction of lysine formylation sites using the composition of k-spaced amino acid pairs via Chou’s 5-steps rule and general pseudo components. Genomics 2019. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Yapp, E.K.Y.; Ho, Q.-T.; Nagasundaram, N.; Ou, Y.-Y.; Yeh, H.-Y. iEnhancer-5Step: Identifying enhancers using hidden information of DNA sequences via Chou’s 5-step rule and word embedding. Anal. Biochem. 2019, 571, 53–61. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Yapp, E.K.Y.; Ou, Y.-Y.; Yeh, H.-Y. iMotor-CNN: Identifying molecular functions of cytoskeleton motor proteins using 2D convolutional neural network via Chou’s 5-step rule. Anal. Biochem. 2019, 575, 17–26. [Google Scholar] [CrossRef]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A support vector machine-based meta-predictor for identification of anticancer peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. mAHTPred: A sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 2018, 35, 2757–2765. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. Meta-4mCpred: A Sequence-Based Meta-Predictor for Accurate DNA 4mC Site Prediction Using Effective Feature Representation. Mol. Ther. Nucleic Acids 2019, 16, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A new ensemble learning framework for improved linear B-cell epitope prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. PIP-EL: A new ensemble learning method for improved proinflammatory peptide predictions. Front. Immunol. 2018, 9, 1783. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Hongjaisee, S.; Barin, F.; Chaijaruwanich, J.; Samleerat, T. HIV-1 CRF01_AE coreceptor usage prediction using kernel methods based logistic model trees. Comput. Biol. Med. 2012, 42, 885–889. [Google Scholar] [CrossRef]
- Srinivasulu, Y.S.; Wang, J.-R.; Hsu, K.-T.; Tsai, M.-J.; Charoenkwan, P.; Huang, W.-L.; Huang, H.-L.; Ho, S.-Y. Characterizing informative sequence descriptors and predicting binding affinities of heterodimeric protein complexes. BMC Bioinform. 2015, 16, S14. [Google Scholar] [CrossRef] [Green Version]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. Meta-iAVP: A sequence-based meta-predictor for improving the prediction of antiviral peptides using effective feature representation. Int. J. Mol. Sci. 2019, 20, 5743. [Google Scholar] [CrossRef] [Green Version]
- Vapnik, V. The Nature of Statistical Learning Theory; Springer Science & Business Media: Cham, Switzerland, 2013. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Vapnik, V.N. An overview of statistical learning theory. IEEE Trans. Neural Netw. 1999, 10, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Karatzoglou, A.; Smola, A.; Hornik, K.; Zeileis, A. kernlab-an S4 package for kernel methods in R. J. Stat. Softw. 2004, 11, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.-Y.; Chen, J.-H.; Huang, M.-H. Inheritable genetic algorithm for biobjective 0/1 combinatorial optimization problems and its applications. IEEE Trans. Syst. ManCybern. Part B (Cybern.) 2004, 34, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.A. Correlation-Based Feature Selection for Machine Learning; University of Waikato: Hamilton, New Zealand, 1999. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.M.; Yang, S.; Zhou, Y.; Mollah, M.N.H. SuccinSite: A computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol. Biosyst. 2016, 12, 786–795. [Google Scholar] [CrossRef]
- Breiman, L. Classification and Regression Trees; Routledge: London, UK, 2017. [Google Scholar]
- Laengsri, V.; Shoombuatong, W.; Adirojananon, W.; Nantasenamart, C.; Prachayasittikul, V.; Nuchnoi, P. ThalPred: A web-based prediction tool for discriminating thalassemia trait and iron deficiency anemia. BMC Med. Inform. Decis. Mak. 2019, 19, 212. [Google Scholar]






| Method | Classifier a | Sequence Feature b | No. of Features | Independent Test |
|---|---|---|---|---|
| QSPpred | SVM | AAC, DPC, N5C5Bin, PCP | 630 | Yes |
| QSPpred-FL | RF | GDP, OVP, CTD, ASDC | 913 | No |
| iQSP (this study) | SVM | PCP | 18 | Yes |
| Amino Acid | QSP (%) | Non-QSP (%) | Difference | p-Value | MDGI |
|---|---|---|---|---|---|
| F | 0.109 | 0.049 | 0.059 (1) | 0.000 | 41.94 (1) |
| K | 0.047 | 0.093 | −0.045 (20) | 0.000 | 19.37 (2) |
| L | 0.076 | 0.106 | −0.030 (18) | 0.002 | 17.57 (3) |
| V | 0.043 | 0.054 | −0.011 (14) | 0.114 | 15.56 (4) |
| W | 0.034 | 0.015 | 0.019 (4) | 0.000 | 10.99 (5) |
| I | 0.063 | 0.067 | −0.004 (11) | 0.629 | 10.15 (6) |
| A | 0.053 | 0.084 | −0.032 (19) | 0.000 | 9.15 (7) |
| Y | 0.042 | 0.020 | 0.022 (3) | 0.000 | 8.69 (8) |
| R | 0.039 | 0.050 | −0.011 (15) | 0.128 | 7.99 (9) |
| C | 0.049 | 0.061 | −0.013 (16) | 0.091 | 7.82 (10) |
| S | 0.079 | 0.062 | 0.017 (5) | 0.023 | 6.70 (11) |
| G | 0.078 | 0.094 | −0.016 (17) | 0.032 | 6.56 (12) |
| P | 0.041 | 0.043 | −0.001 (10) | 0.846 | 6.25 (13) |
| N | 0.070 | 0.043 | 0.026 (2) | 0.009 | 5.24 (14) |
| T | 0.051 | 0.041 | 0.010 (7) | 0.098 | 4.38 (15) |
| D | 0.033 | 0.029 | 0.004 (8) | 0.411 | 4.37 (16) |
| E | 0.026 | 0.030 | −0.004 (12) | 0.423 | 3.64 (17) |
| M | 0.028 | 0.016 | 0.012 (6) | 0.013 | 2.84 (18) |
| H | 0.010 | 0.017 | −0.007 (13) | 0.048 | 2.71 (19) |
| Q | 0.029 | 0.026 | 0.003 (9) | 0.484 | 2.49 (20) |
| Subset | # Feature | Ac (%) | Sn (%) | Sp (%) | MCC | AUC |
|---|---|---|---|---|---|---|
| 1 | 14 | 91.23 ± 1.31 | 91.17 ± 2.65 | 88.23 ± 3.03 | 0.82 ± 0.03 | 0.95 ± 0.05 |
| 2 | 17 | 90.69 ± 1.26 | 92.08 ± 2.82 | 87.04 ± 2.89 | 0.82 ± 0.03 | 0.95 ± 0.05 |
| 3 | 16 | 91.58 ± 1.77 | 92.79 ± 2.73 | 88.45 ± 2.97 | 0.83 ± 0.04 | 0.94 ± 0.05 |
| 4 | 17 | 91.63 ± 1.75 | 91.49 ± 2.73 | 89.55 ± 3.71 | 0.84 ± 0.04 | 0.92 ± 0.05 |
| 5 | 17 | 92.01 ± 1.40 | 91.43 ± 3.64 | 90.27 ± 2.39 | 0.84 ± 0.03 | 0.92 ± 0.08 |
| 6 | 18 | 91.07 ± 1.77 | 90.06 ± 2.69 | 88.79 ± 3.40 | 0.82 ± 0.04 | 0.91 ± 0.10 |
| 7 | 15 | 89.28 ± 1.99 | 88.78 ± 3.88 | 86.44 ± 2.67 | 0.79 ± 0.04 | 0.93 ± 0.07 |
| 8 | 17 | 88.24 ± 2.15 | 85.04 ± 3.61 | 86.81 ± 3.86 | 0.76 ± 0.05 | 0.92 ± 0.07 |
| 9 | 18 | 90.54 ± 1.26 | 90.60 ± 4.12 | 87.57 ± 2.68 | 0.81 ± 0.03 | 0.93 ± 0.09 |
| 10 | 17 | 92.19 ± 1.09 | 90.12 ± 2.98 | 92.16 ± 1.96 | 0.84 ± 0.02 | 0.93 ± 0.07 |
| Subset | # Feature | Ac (%) | Sn (%) | Sp (%) | MCC | AUC |
|---|---|---|---|---|---|---|
| 1 | 14 | 92.50 ± 1.67 | 95.50 ± 3.69 | 89.50 ± 4.97 | 0.85 ± 0.03 | 0.95 ± 0.03 |
| 2 | 17 | 91.50 ± 3.58 | 98.00 ± 3.50 | 85.00 ± 7.07 | 0.84 ± 0.07 | 0.96 ± 0.03 |
| 3 | 16 | 92.00 ± 2.58 | 92.50 ± 5.40 | 91.50 ± 5.80 | 0.84 ± 0.05 | 0.95 ± 0.02 |
| 4 | 17 | 92.25 ± 2.19 | 93.00 ± 4.83 | 91.50 ± 4.12 | 0.85 ± 0.04 | 0.97 ± 0.01 |
| 5 | 17 | 92.50 ± 2.36 | 94.00 ± 5.68 | 91.00 ± 6.15 | 0.86 ± 0.05 | 0.96 ± 0.02 |
| 6 | 18 | 93.00 ± 1.97 | 92.50 ± 5.40 | 93.50 ± 4.12 | 0.86 ± 0.04 | 0.96 ± 0.02 |
| 7 | 15 | 92.00 ± 1.97 | 94.00 ± 5.16 | 90.00 ± 7.45 | 0.85 ± 0.04 | 0.96 ± 0.02 |
| 8 | 17 | 91.75 ± 2.90 | 94.00 ± 5.16 | 89.50 ± 6.43 | 0.84 ± 0.06 | 0.95 ± 0.04 |
| 9 | 18 | 91.50 ± 3.38 | 90.50 ± 7.62 | 92.50 ± 7.91 | 0.84 ± 0.06 | 0.97 ± 0.04 |
| 10 | 17 | 92.50 ± 1.67 | 95.00 ± 3.33 | 90.00 ± 4.08 | 0.85 ± 0.03 | 0.95 ± 0.04 |
| Method | # Feature | 10-Fold CV | Independent Test | ||||
|---|---|---|---|---|---|---|---|
| Ac (%) | MCC | auROC | Ac (%) | MCC | auROC | ||
| QSPpred a | 430 | 91.25 | 0.830 | 0.960 | 90.00 | 0.800 | 0.950 |
| QSPpred-FL b | 913 | 94.30 | 0.885 | N/A | 92.50 | 0.860 | 0.962 |
| iQSP | 18 | 91.07 ± 1.77 | 0.82 ± 0.04 | 0.91 ± 0.10 | 93.00 ± 1.97 | 0.86 ± 0.04 | 0.96 ± 0.02 |
| Classifier | 10-Fold CV | Independent Test | ||||
|---|---|---|---|---|---|---|
| Ac (%) | MCC | auROC | Ac (%) | MCC | auROC | |
| k-NN | 85.13 ± 0.27 | 0.72 ± 0.01 | 0.86 ± 0.00 | 85.75 ± 1.21 | 0.73 ± 0.02 | 0.91 ± 0.03 |
| DT | 83.57 ± 2.74 | 0.67 ± 0.06 | 0.87 ± 0.03 | 83.75 ± 3.39 | 0.68 ± 0.07 | 0.86 ± 0.05 |
| RF | 87.93 ± 0.48 | 0.76 ± 0.01 | 0.95 ± 0.01 | 91.00 ± 3.16 | 0.82 ± 0.06 | 0.94 ± 0.02 |
| iQSP | 91.07 ± 1.77 | 0.82 ± 0.04 | 0.91 ± 0.10 | 93.00 ± 1.97 | 0.86 ± 0.04 | 0.96 ± 0.02 |
| Rank | AAindex ID | MDGI | Description |
|---|---|---|---|
| 1 | QIAN880137 | 32.55 | Weights for coil at the window position of 4 (Qian-Sejnowski, 1988) |
| 2 | AURR980102 | 16.62 | Normalized positional residue frequency at helix termini N’ (Aurora-Rose, 1998) |
| 3 | ROBB760113 | 13.56 | Information measure for loop (Robson-Suzuki, 1976) |
| 4 | PRAM820101 | 12.62 | Intercept in regression analysis (Prabhakaran-Ponnuswamy, 1982) |
| 5 | GRAR740101 | 12.26 | Composition (Grantham, 1974) |
| 6 | PALJ810111 | 11.71 | Normalized frequency of beta-sheet in alpha + beta class (Palau et al., 1981) |
| 7 | PONP800102 | 10.96 | Average gain in surrounding hydrophobicity (Ponnuswamy et al., 1980) |
| 8 | MUNV940103 | 9.07 | Free energy in beta-strand conformation (Munoz-Serrano, 1994) |
| 9 | DAYM780101 | 8.64 | Amino acid composition (Dayhoff et al., 1978a) |
| 10 | MANP780101 | 8.36 | Average surrounding hydrophobicity (Manavalan-Ponnuswamy, 1978) |
| 11 | KUMS000103 | 8.23 | Distribution of amino acid residues in the alpha-helices in thermophilic proteins (Kumar et al., 2000) |
| 12 | ROBB760104 | 8.18 | Information measure for C-terminal helix (Robson-Suzuki, 1976) |
| 13 | ISOY800107 | 8.09 | Normalized relative frequency of double bend (Isogai et al., 1980) |
| 14 | GEIM800101 | 7.80 | Alpha-helix indices (Geisow-Roberts, 1980) |
| 15 | PRAM900102 | 7.59 | Relative frequency in alpha-helix (Prabhakaran, 1990) |
| 16 | NADH010104 | 7.20 | Hydropathy scale based on self-information values in the two-state model (20% accessibility) (Naderi-Manesh et al., 2001) |
| 17 | FUKS010106 | 6.47 | Interior composition of amino acids in intracellular proteins of mesophiles (percent) (Fukuchi-Nishikawa, 2001) |
| 18 | WIMW960101 | 5.54 | Free energies of transfer of AcWl-X-LL peptides from bilayer interface to water (Wimley-White, 1996) |
| No. | Rule | Cover Samples | Misclassified Samples | Ac (%) |
|---|---|---|---|---|
| 1 | GRAR740101 ≤ 0.9055 & MANP780101 > 0.7495 & PRAM900102 > 0.848 & QIAN880137 ≤ 0.237 | 10 | 0 | 100.00 |
| 2 | PONP800102 > −0.751 & PONP800102 ≤ 1.0025 & QIAN880137 ≤ −0.104 & ROBB760104 ≤ 0.3645 & ROBB760104 > −0.5205 | 61 | 1 | 98.36 |
| 3 | PALJ810111 ≤ 1.369 & QIAN880137 > −0.104 & QIAN880137 ≤ 0.417 & ROBB760113 ≤ 0.5975 & AURR980102 ≤ 0.6955 | 21 | 2 | 90.48 |
| 4 | GEIM800101 > −0.3135 & GRAR740101 > −0.176 & ISOY800107 ≤ 1.367 & MANP780101 > −0.3325 & PALJ810111 ≤ 1.0905 & QIAN880137 ≤ −0.0985 | 94 | 6 | 93.62 |
| 5 | PALJ810111 > −0.786 & QIAN880137 > 0.237 & QIAN880137 > 0.403 & ROBB760113 > 0.5975 & AURR980102 ≤ 0.811 & KUMS000103 ≤ 0.793 | 45 | 7 | 84.44 |
| 6 | GRAR740101 ≤ 0.341 & ISOY800107 ≤ −0.089 & PALJ810111 ≤ 1.2455 & QIAN880137 > −0.009 & ROBB760113 ≤ −0.0285 | 17 | 3 | 82.35 |
| 7 | GRAR740101 > −0.708 & PRAM900102 ≤ 1.2985 & QIAN880137 > −0.104 & QIAN880137 ≤ 1.105 & AURR980102 ≤ 0.6585 & KUMS000103 ≤ 0.974 | 94 | 28 | 70.21 |
| 8 | PONP800102 ≤ 1.1095 & PRAM900102 ≤ 0.8295 & QIAN880137 ≤ 0.2625 & QIAN880137 > −0.9055 & ROBB760113 > −0.5875 & ROBB760113 ≤ 1.031 | 121 | 15 | 87.60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charoenkwan, P.; Schaduangrat, N.; Nantasenamat, C.; Piacham, T.; Shoombuatong, W. iQSP: A Sequence-Based Tool for the Prediction and Analysis of Quorum Sensing Peptides Using Informative Physicochemical Properties. Int. J. Mol. Sci. 2020, 21, 75. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010075
Charoenkwan P, Schaduangrat N, Nantasenamat C, Piacham T, Shoombuatong W. iQSP: A Sequence-Based Tool for the Prediction and Analysis of Quorum Sensing Peptides Using Informative Physicochemical Properties. International Journal of Molecular Sciences. 2020; 21(1):75. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010075
Chicago/Turabian StyleCharoenkwan, Phasit, Nalini Schaduangrat, Chanin Nantasenamat, Theeraphon Piacham, and Watshara Shoombuatong. 2020. "iQSP: A Sequence-Based Tool for the Prediction and Analysis of Quorum Sensing Peptides Using Informative Physicochemical Properties" International Journal of Molecular Sciences 21, no. 1: 75. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010075