Revisiting Experimental Models of Diabetic Nephropathy

 , and
, and
Abstract
:1. Introduction
2. Classical and New Models of Experimental DN
2.1. Diabetes Susceptible Background and Strain Crossing Models
2.2. Knockout Diabetic Models
2.3. Transgenic Diabetic Models
3. Genetic Background
4. Assessment of Renal Function in Experimental Models
4.1. Glomerular Filtration Rate Assessment
4.1.1. Creatinine Clearance
4.1.2. Anesthetized Inulin Clearance
4.1.3. Steady State Inulin Clearance
4.1.4. Non-Steady State Inulin Clearance
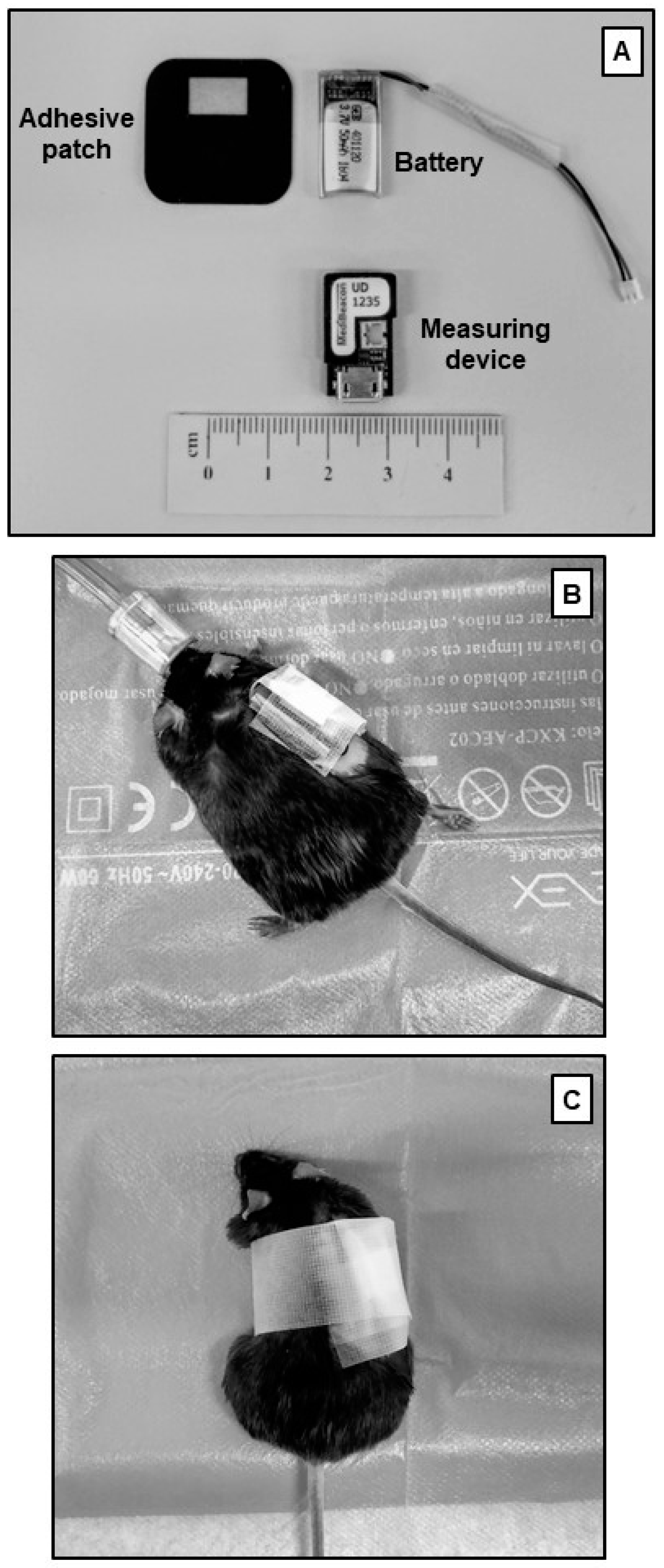
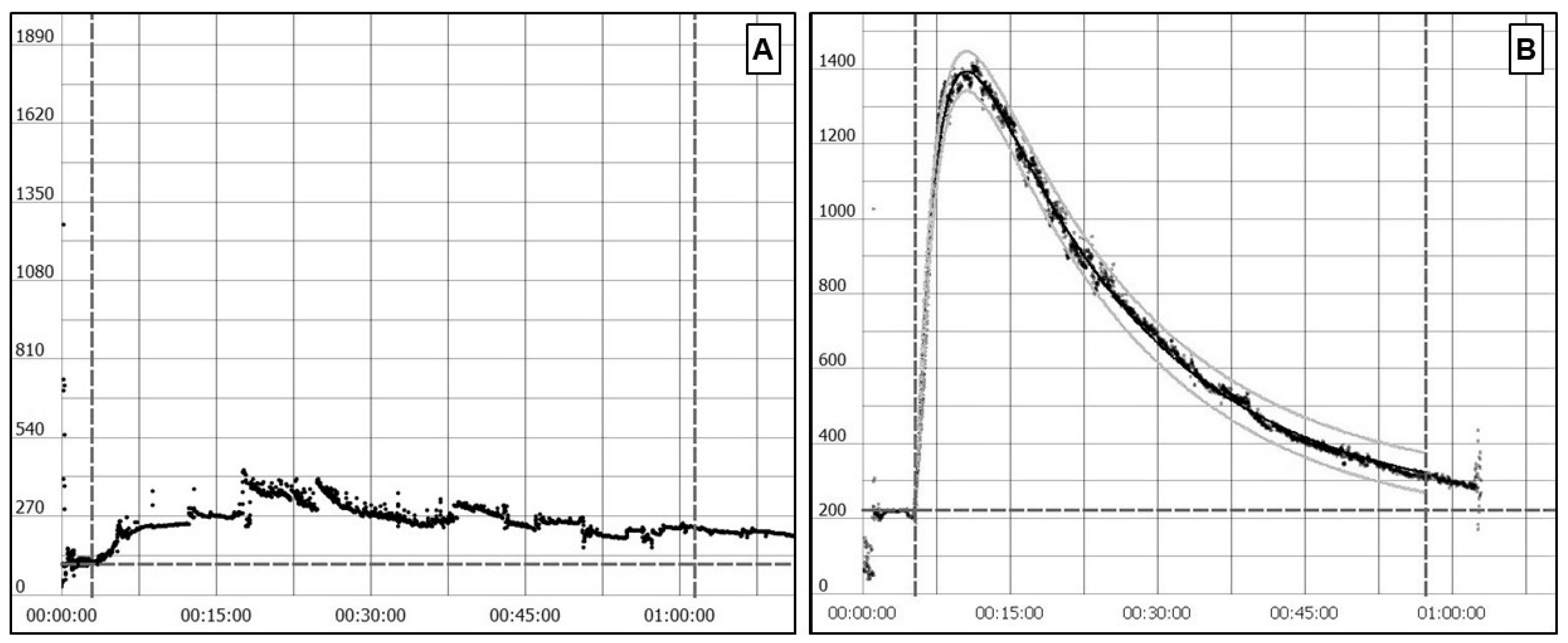
4.1.5. Transcutaneous Sinistrin Clearance Measurement
4.1.6. Iohexol Clearance
4.2. Albuminuria Measurement
5. Renal Lesions and Morphometry in Experimental Models of DN
5.1. Mesangial Matrix Expansion Measurement
5.2. Podocyte Number and Density
5.3. Glomerular Basement Membrane Thickness Measurement by Electron Microscopy
5.4. Tubular Injury
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Thomas, M.C.; Cooper, M.E.; Zimmet, P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.M.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: Implications for albuminuria in diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, A.F.; Bamagous, G.A.; Al-Harizy, R.M.; ElSawy, N.A.; Shahzad, N.; Ibrahim, I.A.; Ghamdi, S.S.A. Renal protective effect of SGLT2 inhibitor dapagliflozin alone and in combination with irbesartan in a rat model of diabetic nephropathy. Biomed. Pharmacother. 2018, 103, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Vergara, A.; Jacobs-Cachá, C.; Soler, M.J. Sodium-glucose cotransporter inhibitors: Beyond glycaemic control. Clin. Kidney J. 2019, 12, 322–325. [Google Scholar] [CrossRef] [Green Version]
- García-Carro, C.; Vergara, A.; Agraz, I.; Jacobs-Cachá, C.; Espinel, E.; Seron, D.; Soler, M.J. The New Era for Reno-Cardiovascular Treatment in Type 2 Diabetes. J. Clin. Med. 2019, 8, 864. [Google Scholar] [CrossRef] [Green Version]
- Azushima, K.; Gurley, S.B.; Coffman, T.M. Modelling diabetic nephropathy in mice. Nat. Rev. Nephrol. 2017, 14, 48–56. [Google Scholar] [CrossRef]
- Qi, Z.; Fujita, H.; Jin, J.; Davis, L.S.; Wang, Y.; Fogo, A.B.; Breyer, M.D. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes 2005, 54, 2628–2637. [Google Scholar] [CrossRef] [Green Version]
- Soler, M.J.; Riera, M.; Batlle, D. New experimental models of diabetic nephropathy in mice models of type 2 diabetes: Efforts to replicate human nephropathy. Exp. Diabetes Res. 2012, 2012, 616313. [Google Scholar] [CrossRef] [Green Version]
- Betz, B.; Conway, B.R. An Update on the Use of Animal Models in Diabetic Nephropathy Research. Curr. Diab. Rep. 2016, 16, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Animal Models of Diabetic Complications Consortium, (AMDCC). Available online: https://www.diacomp.org/shared/document.aspx?id=25&docType=Protocol (accessed on 16 May 2020).
- Brosius, F.C.; Alpers, C.E.; Bottinger, E.P.; Breyer, M.D.; Coffman, T.M.; Gurley, S.B.; Harris, R.C.; Kakoki, M.; Kretzler, M.; Leiter, E.H.; et al. Mouse models of diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 2503–2512. [Google Scholar] [CrossRef] [Green Version]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol. 2015, 70, 5.47.1–5.47.20. [Google Scholar] [CrossRef]
- Clotet-Freixas, S.; Soler, M.J.; Palau, V.; Anguiano, L.; Gimeno, J.; Konvalinka, A.; Pascual, J.; Riera, M. Sex dimorphism in ANGII-mediated crosstalk between ACE2 and ACE in diabetic nephropathy. Lab. Investig. 2018, 98, 1237–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, M.J.; Wysocki, J.; Ye, M.; Lloveras, J.; Kanwar, Y.; Batlle, D. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007, 72, 614–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Awar, A.; Kupai, K.; Veszelka, M.; Szucs, G.; Attieh, Z.; Murlasits, Z.; Török, S.; Pósa, A.; Varga, C. Experimental Diabetes Mellitus in Different Animal Models. J. Diabetes Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- McKnight, A.J.; Duffy, S.; Maxwell, A.P. Genetics of Diabetic Nephropathy: A Long Road of Discovery. Curr. Diab. Rep. 2015, 15. [Google Scholar] [CrossRef]
- Gurley, S.B.; Clare, S.E.; Snow, K.P.; Hu, A.; Meyer, T.W.; Coffman, T.M. Impact of genetic background on nephropathy in diabetic mice. Am. J. Physiol. Ren. Physiol. 2006, 290, 214–222. [Google Scholar] [CrossRef]
- Riera, M.; Márquez, E.; Clotet, S.; Gimeno, J.; Roca-Ho, H.; Lloreta, J.; Juanpere, N.; Batlle, D.; Pascual, J.; Soler, M.J. Effect of insulin on ACE2 activity and kidney function in the non-obese diabetic mouse. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Heydemann, A. An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus. J. Diabetes Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liang, K.; Zhen, J.; Zhou, M.; Wang, X.; Wang, Z.; Wei, X.; Zhang, Y.; Sun, Y.; Zhou, Z.; et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat. Commun. 2017, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Fan, B.; Han, H.; Zhen, J.; Shang, J.; Wang, X.; Li, X.; Shi, W.; Tang, W.; Bao, C.; et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int. 2013, 84, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskell, B.D.; Flurkey, K.; Duffy, T.M.; Sargent, E.E.; Leiter, E.H. The diabetes-prone NZO/HILt strain. I. Immunophenotypic comparison to the related NZB/BINJ and NZW/LacJ strains. Lab. Investig. 2002, 82, 833–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Son, C.; Aotani, D.; Nomura, H.; Hikida, T.; Hosoda, K.; Nakao, K. Role of leptin in conditioned place preference to high-fat diet in leptin-deficient ob/ob mice. Neurosci. Lett. 2017, 640, 60–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, S.J.; Batdorf, H.M.; Burk, D.H.; Noland, R.C.; Eder, A.E.; Boulos, M.S.; Karlstad, M.D.; Jason Collier, J. db/db Mice Exhibit Features of Human Type 2 Diabetes That Are Not Present in Weight-Matched C57BL/6J Mice Fed a Western Diet. J. Diabetes Res. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Meyer zum Gottesberge, A.M.; Massing, T.; Sasse, A.; Palma, S.; Hansen, S. Zucker diabetic fatty rats, a model for type 2 diabetes, develop an inner ear dysfunction that can be attenuated by losartan treatment. Cell Tissue Res. 2015, 362, 307–315. [Google Scholar] [CrossRef]
- Hempe, J.; Elvert, R.; Schmidts, H.L.; Kramer, W.; Herling, A.W. Appropriateness of the Zucker diabetic Fatty rat as a model for diabetic microvascular late complications. Lab. Anim. 2012, 46, 32–39. [Google Scholar] [CrossRef]
- Hudkins, K.L.; Pichaiwong, W.; Wietecha, T.; Kowalewska, J.; Banas, M.C.; Spencer, M.W.; Mühlfeld, A.; Koelling, M.; Pippin, J.W.; Shankland, S.J.; et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 1533–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, D.A.; Davis, D.B. Attention to background strain is essential for metabolic research: C57BL/6 and the international knockout mouse consortium. Diabetes 2016, 65, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Harbison, C.E.; Lipman, R.D.; Bronson, R.T. Strain- and Diet-Related Lesion Variability in Aging DBA/2, C57BL/6, and DBA/2xC57BL/6 F1 Mice. Vet. Pathol. 2016, 53, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Alpers, C.E.; Hudkins, K.L. Mouse models of diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2011, 20, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Clee, S.M.; Nadler, S.T.; Attie, A.D. Genetic and genomic studies of the BTBR ob/ob mouse model of type 2 diabetes. Am. J. Ther. 2005, 12, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiter, E.H.; Reifsnyder, P.C. Differential Levels of Diabetogenic Stress in Two New Mouse Models of Obesity and Type 2 Diabetes. Diabetes 2004, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Itoh, Y.; Yoshida, F.; Koh, N.; Tamai, H.; Fukatsu, A.; Matsuo, S.; Hotta, N.; Sakamoto, N. Unique glomerular lesion with spontaneous lipid deposition in glomerular capillary lumina in the NON strain of mice. Nephron 1991, 58, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Deeds, M.C.; Anderson, J.M.; Armstrong, A.S.; Gastineau, D.A.; Hiddinga, H.J.; Jahangir, A.; Eberhardt, N.L.; Kudva, Y.C. Single dose streptozotocin-induced diabetes: Considerations for study design in islet transplantation models. Lab. Anim. 2011, 45, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, F.; Ortsäter, H.; Hansell, P.; Liss, P.; Carlsson, P.O. Differentiating between effects of streptozotocin per se and subsequent hyperglycemia on renal function and metabolism in the streptozotocin-diabetic rat model. Diabetes. Metab. Res. Rev. 2004, 20, 452–459. [Google Scholar] [CrossRef]
- Danda, R.S.; Habiba, N.M.; Rincon-Choles, H.; Bhandari, B.K.; Barnes, J.L.; Abboud, H.E.; Pergola, P.E. Kidney involvement in a nongenetic rat model of type 2 diabetes. Kidney Int. 2005, 68, 2562–2571. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Lane, P.H.; Lane, J.T.; Padanilam, B.J.; Sansom, S.C. Glomerular structural and functional changes in a high-fat diet mouse model of early-stage Type 2 diabetes. Diabetologia 2004, 47, 1541–1549. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.M.; Dang, H.; Kamat, A.; Yeh, C.K.; Zhang, B.X. Geldanamycin derivative ameliorates high fat diet-induced renal failure in diabetes. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Glastras, S.J.; Chen, H.; Teh, R.; McGrath, R.T.; Chen, J.; Pollock, C.A.; Wong, M.G.; Saad, S. Mouse models of diabetes, obesity and related kidney disease. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Xu, H.; Ma, Z.; Lu, S.; Li, R.; Lyu, L.; Ding, L.; Lu, Q. Renal resistive index as a novel indicator for renal complications in high-fat diet-fed mice. Kidney Blood Press. Res. 2017, 42, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Fan, T.T.; Ji, Y.S.; Yu, J.Y.; Wu, S.; Zhang, L. Spironolactone alleviates diabetic nephropathy through promoting autophagy in podocytes. Int. Urol. Nephrol. 2019, 51, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Choi, B.H.; Ku, S.K.; Park, J.H.; Oh, E.; Kwak, M.K.K. Beneficial effects of sarpogrelate and rosuvastatin in high fat diet/streptozotocin-induced nephropathy in mice. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurley, S.B.; Mach, C.L.; Stegbauer, J.; Yang, J.; Snow, K.P.; Hu, A.; Meyer, T.W.; Coffman, T.M. Influence of genetic background on albuminuria and kidney injury in Ins2+/C96Y(Akita) mice. Am. J. Physiol. Ren. Physiol. 2010, 298, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Bivona, B.J.; Park, S.; Harrison-Bernard, L.M. Glomerular filtration rate determinations in conscious type II diabetic mice. Am. J. Physiol. Ren. Physiol. 2011, 300, 618–625. [Google Scholar] [CrossRef]
- Ericsson, A.; Tonelius, P.; Lal, M.; Sabirsh, A.; Böttcher, G.; William-Olsson, L.; Strömstedt, M.; Johansson, C.; Hyberg, G.; Tapani, S.; et al. The effects of dual PPARα/γ agonism compared with ACE inhibition in the BTBRob/ob mouse model of diabetes and diabetic nephropathy. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Kanetsuna, Y.; Takahashi, K.; Nagata, M.; Gannon, M.A.; Breyer, M.D.; Harris, R.C.; Takahashi, T. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am. J. Pathol. 2007, 170, 1473–1484. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Sato, W.; Glushakova, O.; Heinig, M.; Clarke, T.; Campbell-Thompson, M.; Yuzawa, Y.; Atkinson, M.A.; Johnson, R.J.; Croker, B. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J. Am. Soc. Nephrol. 2007, 18, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.J.; Wang, S.; Cheng, H.; Zhang, M.; Takahashi, T.; Fogo, A.B.; Breyer, M.D.; Harris, R.C. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J. Am. Soc. Nephrol. 2006, 17, 2664–2669. [Google Scholar] [CrossRef]
- Kakoki, M.; Takahashi, N.; Jennette, J.C.; Smithies, O. Diabetic nephropathy is markedly enhanced in mice lacking the bradykinin B2 receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 13302–13305. [Google Scholar] [CrossRef] [Green Version]
- Kakoki, M.; Sullivan, K.A.; Backus, C.; Hayes, J.M.; Oh, S.S.; Hua, K.; Gasim, A.M.H.; Tomita, H.; Grant, R.; Nossov, S.B.; et al. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.J.; Qiu, G.; Usui, H.K.; Dunn, S.R.; McCue, P.; Bottinger, E.; Iozzo, R.V.; Sharma, K. Decorin deficiency enhances progressive nephropathy in diabetic mice. Am. J. Pathol. 2007, 171, 1441–1450. [Google Scholar] [CrossRef] [Green Version]
- Thibodeau, J.F.; Holterman, C.E.; Burger, D.; Read, N.C.; Reudelhuber, T.L.; Kennedy, C.R.J. A novel mouse model of advanced diabetic kidney disease. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Conway, B.R.; Rennie, J.; Bailey, M.A.; Dunbar, D.R.; Manning, J.R.; Bellamy, C.O.; Hughes, J.; Mullins, J.J. Hyperglycemia and renin-dependent hypertension synergize to model diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Conway, B.R.; Betz, B.; Sheldrake, T.A.; Manning, J.R.; Dunbar, D.R.; Dobyns, A.; Hughes, J.; Mullins, J.J. Tight blood glycaemic and blood pressure control in experimental diabetic nephropathy reduces extracellular matrix production without regression of fibrosis. Nephrology 2014, 19, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Herbach, N.; Schairer, I.; Blutke, A.; Kautz, S.; Siebert, A.; Göke, B.; Wolf, E.; Wanke, R. Diabetic kidney lesions of GIPRdn transgenic mice: Podocyte hypertrophy and thickening of the GBM precede glomerular hypertrophy and glomerulosclerosis. Am. J. Physiol. Physiol. 2009, 296, F819–F829. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Heilig, K.; Saunders, T.; Minto, A.; Deb, D.K.; Chang, A.; Brosius, F.; Monteiro, C.; Heilig, C.W. Transgenic overexpression of GLUT1 in mouse glomeruli produces renal disease resembling diabetic glomerulosclerosis. Am. J. Physiol. Physiol. 2010, 299, F99–F111. [Google Scholar] [CrossRef] [Green Version]
- Betz, B.; Conway, B.R. Recent advances in animal models of diabetic nephropathy. Nephron Exp. Nephrol. 2014, 126, 191–195. [Google Scholar] [CrossRef]
- Manolis, A.J.; Marketou, M.E.; Gavras, I.; Gavras, H. Cardioprotective properties of bradykinin: Role of the B 2 receptor. Hypertens. Res. 2010, 33, 772–777. [Google Scholar] [CrossRef] [Green Version]
- Kuoppala, A.; Shiota, N.; Lindstedt, K.A.; Rysä, J.; Leskinen, H.K.; Luodonpää, M.; Liesmaa, I.; Ruskoaho, H.; Kaaja, R.; Kovanen, P.T.; et al. Expression of bradykinin receptors in the left ventricles of rats with pressure overload hypertrophy and heart failure. J. Hypertens. 2003, 21, 1729–1736. [Google Scholar] [CrossRef]
- Loke, K.E.; Curran, C.M.; Messina, E.J.; Laycock, S.K.; Shesely, E.G.; Carretero, O.A.; Hintze, T.H. Role of nitric oxide in the control of cardiac oxygen consumption in B(2)-kinin receptor knockout mice. Hypertension 1999, 34, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönherr, E.; Sunderkötter, C.; Iozzo, R.V.; Schaefer, L. Decorin, a novel player in the insulin-like growth factor system. J. Biol. Chem. 2005, 280, 15767–15772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönherr, E.; Sunderkötter, C.; Schaefer, L.; Thanos, S.; Grässel, S.; Oldberg, Å.; Iozzo, R.V.; Young, M.F.; Kresse, H. Decorin deficiency leads to impaired angiogenesis in injured mouse cornea. J. Vasc. Res. 2004, 41, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Kodama, Y.; Matsumoto, T. Bone matrix decorin binds transforming growth factor-beta and enhances its bioactivity. J. Biol. Chem. 1994, 269, 32634–32638. [Google Scholar]
- Marshall, S.M. Natural history and clinical characteristics of CKD in type 1 and type 2 diabetes mellitus. Adv. Chronic Kidney Dis. 2014, 21, 267–272. [Google Scholar] [CrossRef]
- Flier, J.S.; Underhill, L.H.; Krolewski, A.S.; Warram, J.H.; Rand, L.I.; Kahn, C.R. Epidemiologic Approach to the Etiology of Type I Diabetes Mellitus and Its Complications. N. Engl. J. Med. 1987, 317, 1390–1398. [Google Scholar] [CrossRef]
- Quinn, M.; Angelico, M.C.; Warram, J.H.; Krolewski, A.S. Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia 1996, 39, 940–945. [Google Scholar] [CrossRef]
- Afkarian, M.; Zelnick, L.R.; Hall, Y.N.; Heagerty, P.J.; Tuttle, K.; Weiss, N.S.; De Boer, I.H. Clinical manifestations of kidney disease among US adults with diabetes, 1988-2014. JAMA J. Am. Med. Assoc. 2016, 316, 602–610. [Google Scholar] [CrossRef]
- Gu, T.; Gu, H.F.; Hilding, A.; Sjöholm, L.K.; Östenson, C.-G.; Ekström, T.J.; Brismar, K. Increased DNA methylation levels of the insulin-like growth factor binding protein 1 gene are associated with type 2 diabetes in Swedish men. Clin. Epigenetics 2013, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Coresh, J.; Turin, T.C.; Matsushita, K.; Sang, Y.; Ballew, S.H.; Appel, L.J.; Arima, H.; Chadban, S.J.; Cirillo, M.; Djurdjev, O.; et al. Decline in estimated glomerular filtration rate and subsequent risk of end-stage renal disease and mortality. JAMA J. Am. Med. Assoc. 2014, 311, 2518–2531. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.M. Diabetic nephropathy in type 1 diabetes: Has the outlook improved since the 1980s? Diabetologia 2012, 55, 2301–2306. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, C.E.; Keane, W.F.; Bennett, P.H.; Jerums, G.; Parving, H.H.; Passa, P.; Steffes, M.W.; Striker, G.E.; Viberti, G.C. Prevention of diabetic renal disease with special reference to microalbuminuria. Lancet 1995, 346, 1080–1084. [Google Scholar] [CrossRef]
- Perkins, B.A.; Ficociello, L.H.; Roshan, B.; Warram, J.H.; Krolewski, A.S. In patients with type 1 diabetes and new-onset microalbuminuria the development of advanced chronic kidney disease may not require progression to proteinuria. Kidney Int. 2010, 77, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 1Sharma, K.; McCue, P.; Dunn, S.R. Diabetic kidney disease in the db/dbmouse. Am. J. Physiol. Physiol. 2003, 284, F1138–F1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, A.; Shulhevich, Y.; Geraci, S.; Hesser, J.; Stsepankou, D.; Neudecker, S.; Koenig, S.; Heinrich, R.; Hoecklin, F.; Pill, J.; et al. Transcutaneous measurement of renal function in conscious mice. Am. J. Physiol. Ren. Physiol. 2012, 303, F783–F788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, M.T.; Nishida, S.K.; Câmara, N.O.S.; Shimizu, M.H.; Mastroianni-Kirsztajn, G. Iohexol clearance for determination of glomerular filtration rate in rats induced to acute renal failure. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Cağlar, Y.; Mete, U.O.; Kaya, M. Ultrastructural evaluation of the effects of the contrast media on the rat kidney. J. Submicrosc. Cytol. Pathol. 2001, 33, 443–451. [Google Scholar]
- Sharma, K. Assay for Creatinine Clearance by HPLC. Animal Models of Diabetic Complications Consortium (AMDCC) 2003. Available online: https://www.diacomp.org/shared/document.aspx?id=21&docType=Protocol (accessed on 16 May 2020).
- Stevens, L.A.; Coresh, J.; Greene, T.; Levey, A.S. Assessing kidney function Measured and estimated glomerular filtration rate. N. Engl. J. Med. 2006, 354, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Carrara, F.; Azzollini, N.; Nattino, G.; Corna, D.; Villa, S.; Cerullo, D.; Zoja, C.; Abrante, B.; Luis-Lima, S.; Porrini, E.; et al. Simplified method to measure glomerular filtration rate by Iohexol plasma clearance in conscious rats. Nephron 2016, 133, 62–70. [Google Scholar] [CrossRef]
- Viswanathan, G.; Upadhyay, A. Assessment of Proteinuria. Adv. Chronic Kidney Dis. 2011, 18, 243–248. [Google Scholar] [CrossRef]
- Burke, K.; Reifsnyder, P. Protocol for Albuwell M kit: Murine Microalbuminuria ELISA by Exocell Inc., 2008 Animal Models of Diabetic Complications Consortium (AMDCC). Available online: https://www.diacomp.org/shared/document.aspx?id=22&docType=Protocol (accessed on 16 May 2020).
- Jaffe, M. Ueber den Niederschlag, welchen Pikrinsäure im normalen Harn erzeugt, und über eine neue Reaction des Kreatinins. Z. Physio.l Chem. 1886, 10, 391–400. [Google Scholar]
- Tervaert, T.W.C.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; TerenceCook, H.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; De Heer, E.; et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-De La Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayo, S.H.; Radnik, R.A.; Glass, W.F.; Garoni, J.A.; Rampt, E.R.; Appling, D.R.; Kreisberg, J.I. Increased extracellular matrix synthesis and mRNA in mesangial cells grown in high-glucose medium. Am. J. Physiol. 1991, 260, F185–F191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, M.; Oltean, S. Assessment of kidney function in mouse models of glomerular disease. J. Vis. Exp. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, S.M.; Korstanje, R. Automatic glomerular identification and quantification of histological phenotypes using image analysis and machine learning. Am. J. Physiol. Ren. Physiol. 2018, 315, F1644–F1651. [Google Scholar] [CrossRef] [Green Version]
- Rangan, G.K.; Tesch, G.H. Quantification of renal pathology by image analysis (Methods in Renal Research). Nephrology 2007, 12, 553–558. [Google Scholar] [CrossRef]
- Cohen, M.P.; Sharma, K.; Yulin Jin, I.; Hud, E.; Wu, V.-Y.; Tomaszewski, J.; Ziyadehtt, F.N. Prevention of Diabetic Nephropathy in dbldb Mice with Glycated Albumin Antagonists A Novel Treatment Strategy. J. Clin. Investig. 1995, 95, 2338–2345. [Google Scholar] [CrossRef]
- Venkatareddy, M.; Wang, S.; Yang, Y.; Patel, S.; Wickman, L.; Nishizono, R.; Chowdhury, M.; Hodgin, J.; Wiggins, P.A.; Wiggins, R.C. Estimating podocyte number and density using a single histologic section. J. Am. Soc. Nephrol. 2014, 25, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Clotet, S.; Soler, M.J.; Rebull, M.; Gimeno, J.; Gurley, S.B.; Pascual, J.; Riera, M. Gonadectomy prevents the increase in blood pressure and glomerular injury in angiotensin-converting enzyme 2 knockout diabetic male mice. Effects on renin–angiotensin system. J. Hypertens. 2016, 34, 1752–1765. [Google Scholar] [CrossRef] [Green Version]
- Sanden, S.K.; Wiggins, J.E.; Goyal, M.; Riggs, L.K.; Wiggins, R.C. Evaluation of a thick and thin section method for estimation of podocyte number, glomerular volume, and glomerular volume per podocyte in rat kidney with Wilms’ tumor-1 protein used as a podocyte nuclear marker. J. Am. Soc. Nephrol. 2003, 14, 2484–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M. Thin glomerular basement membrane nephropathy: Incidence in 3471 consecutive renal biopsies examined by electron microscopy. Arch. Pathol. Lab. Med. 2006, 130, 699–706. [Google Scholar] [PubMed]
- Hirose, K.; Østerby, R.; Nozawa, M.; Gundersen, H.J.G. Development of glomerular lesions in experimental long-term diabetes in the rat. Kidney Int. 1982, 21, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letts, R.F.R.; Zhai, X.Y.; Bhikha, C.; Grann, B.L.; Blom, N.B.; Thomsen, J.S.; Rubin, D.M.; Christensen, E.I.; Andreasen, A. Nephron morphometry in mice and rats using tomographic microscopy. Am. J. Physiol. Ren. Physiol. 2017, 312, F210–F229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, J.M.; White, K.E.; Tate, H.; Bilous, R.W. Efficient morphometric analysis of glomerular mesangium in insulin-dependent diabetic patients with early nephropathy. Kidney Int. 1997, 51, 1624–1628. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.W.; Isono, M.; Chen, S.; Iglesias-De La Cruz, M.C.; Han, D.C.; Ziyadeh, F.N. Increased glomerular and tubular expression of transforming growth factor-β1, its type II receptor, and activation of the smad signaling pathway in the db/db mouse. Am. J. Pathol. 2001, 158, 1653–1663. [Google Scholar] [CrossRef]
- Jensen, E.B.; Gundersen, H.J.G.; Østerby, R. Determination of membrane thickness distribution from orthogonal intercepts. J. Microsc. 1979, 115, 19–33. [Google Scholar] [CrossRef]
- Rangan, G.K.; Wang, Y.; Tay, Y.C.; Harris, D.C.H. Inhibition of nuclear factor-κB activation reduces cortical tubulointerstitial injury in proteinuric rats. Kidney Int. 1999, 56, 118–134. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Clinical features |
|---|
| -Greater than 50% decline in GFR over the lifetime of the animal. -Greater than 10-fold increase in albuminuria compared with not affected controls for that strain at the same age and gender. |
| Pathological features |
| -Mesangial matrix expansion. More severe models may show nodular sclerosis. -Any degree of arteriolar hyalinosis. -Glomerular basement membrane thickening by more than 50% over baseline. -Tubulointerstitial fibrosis (usually seen in severe DN models). |
| Type | Name | Strain | Diabetic Model | Diabetes Type | Systemic Metabolic Features | Albuminuria Increase (Age/Grade) | eGFR 50% Decline (Age) * | Kidney Features | References |
|---|---|---|---|---|---|---|---|---|---|
| Induced | Streptozotocin (STZ) | Any (mice/rats) | STZ-induced | T1DM | Hyperglycaemia in 90% of the STZ-induced animals. In mice, C57BL/6 and DBA/2 are high responders to STZ. | Mice: - BALB/c, DBA/2 and KK/HIJ strains: at 16 weeks of DM onset/10-fold - C57BL/6, MRL/Mp, 129/SvEv and A/J strains: minor or no changes at 16 weeks. | No decline of eGFR at 15 weeks of DM onset in C57BL/6J, DBA/2J, FVB/NJ, MRL/MpJ, A/J, and KK/HlJ strains (Hyperfiltration, except for A/J strain) | Mesangial expansion (more important in DBA/2 and KK/HIJ strain) | Zhonghua et al. 2005 [9] Gurley et al. 2006 [19] Deeds et al. 2011 [36] |
| Rats -Wistar-Furth: 1 week post-STZ/10-fold and 4 weeks post-STZ/20-fold - Sprague-Dawley: 6 weeks post-STZ/400-fold | Wistar-Furth: no eGFR decline at 4 weeks after STZ (hyperfiltration). | Sprague-Dawley: mild mesangial expansion. Increase of fibronectin expression. | Palm et al. 2004 [37] Danda et al. 2005 [38] | ||||||
| High Fat Diet (HFD) | Any (mice/rats) | Diet-induced | T2DM | Obesity, dyslipidemia, hypertension, hyperglycemia. Increases diabetic features and DN severity in most models. | Mice - C57BL/6 at 32 weeks/1.5 fold - db/db at 16 weeks (2 weeks of HFD)/3-fold. (vs. same model on chow diet). | No decline after 22 weeks (early hyperfiltration). | C56BL/6: 2 to 4 weeks of HFD: mesangial matrix expansion. 8 weeks of HFD: increased inflammation. 16 weeks of HFD: glomerular fibrosis. | Wei et al. 2004 [39] Zhang et al. 2012 [40] Glastras et al. 2016 [41] Xu et al. 2017 [42] | |
| Rats Sprague-Dawley: after 4 weeks of HFD/4-fold (vs. Chow diet) | Sprague-Dawley: No decline after 14 weeks of DM induction. | Sprague-Dawley: GBM thickening and mesangial matrix expansion. Not tubular atrophy. | Dong et al. 2019 [43] Danda et al. 2005 [38] | ||||||
| STZ + HFD | Any (mice/rats) | Diet and STZ-induced | T2DM | Add-on effects vs. STZ or HFD models | Mice: -C57BL/6J: DM induction with 7 weeks of HFD + single dose STZ. After 15 weeks/3-fold (vs. Chow diet). -C57BL/6J: DM induction with 5 weeks of HFD + single dose STZ. After 24 weeks/2-fold (vs. Chow diet) and mild increase (vs. HFD). | C57BL/6J: DM induction with 5 weeks of HFD + single dose STZ. After 24 weeks/Mild increase in serum creatinine levels (vs. chow diet but not vs. HFD). | C57BL/6J: mesangial expansion and tubular vacuolization. Lipid deposition. | Kim et al. 2016 [44] Glastras et al. 2016 [41] | |
| Rats Sprague-Dawley: DM induction with 5 weeks of HFD + single dose STZ. After 6 weeks/400-fold | Sprague-Dawley: No decline after 14 weeks of DM induction. | Sprague-Dawley: Increased mesangial matrix expansion. Increased fibronectin and collagen expression. (vs. STZ and HFD) | Danda et al. 2005 [38] | ||||||
| Spontaneous mutations | Akita mice | DBA/2, 129/SvEv, C57BL6 | Akita | T1DM | Hyperglycaemia, hypoinsulinemia, polydipsia, and polyuria at 3–4 weeks of age | Depending on the strain, at 6 months of age/8-fold (DBA/2 x C57BL/6), 2-fold (DBA/2 and 129/SvEv). No change in C57BL/6. | No decline at 6 months of age (hyperfiltration) | Mesangial matrix expansion | Gurley et al. 2010 [45] |
| Non-obese diabetic (NOD) mice | NOD | T1DM | Autoimmune diabetes onset at 12–13 weeks of age hyperglycaemia | 21 days of diabetes onset/10-fold 40 days of diabetes onset/20-fold (vs. NOR mice) | No decline after 40 days of diabetes onset (hyperfiltration) | Early kidney disease | Riera et al. 2014 [20] | ||
| ob/ob mice (leptin deficient) | C57BL/6 | ob/ob | T2DM | Obesity, hyperglycaemia, insulin resistance | 22 weeks/4-fold (vs. WT) | - | Mesangial matrix expansion | Hudkins et al. 2010 [29] | |
| db/db mice (leptin receptor deficient) | C57BL/6 C57BLKS | db/db | T2DM | Hyperglycaemia (C57BLKS more susceptible), insulin resistance, hypertension, hyperphagia, obesity | 18 weeks/6-fold (vs. db/m) | 28 weeks (in aprox. 40% of the cases, vs. db/m) | Mesangial matrix expansion | Bivona et al. 2011 [46] | |
| Zucker diabetic Rats (leptin receptor deficient) | Merck M crossed with Sherman rats | T2DM | Hyperphagic and hyperinsulinemic, hyperglycaemia, hyperlipidemia | 16 weeks/200-fold 26 weeks/1000-fold (vs. Lean) | No decline after 26 weeks (hyperfiltration) | Glomerular and tubular damage | Hempe et al. 2012 [28] | ||
| Polygenic | Non-obese Non-diabetic (NON) mice | NON | T2DM | Impaired glucose tolerance (in 50% of the cases) | UK | Increased serum creatinine at 6 months | Glomerular lesions not resembling DN | Watanabe et al. 1991 [35] | |
| New Zeland Obese (NZO)/HlLt mice | NZO/H1Lt | T2DM | Obesity and 50% are diabetic (hyperinsulinemia, insulin resistance, glucose intolerance) | UK | UK | UK | Haskell et al. 2002 [24] | ||
| Backcrossing models | BTBR-ob/ob mice | BTBR crossed with C57BL/6 | ob/ob | T2DM | Insulin resistance, hyperglycaemia, hypertension, hyperphagia, obesity | 8 weeks/2-fold and 20 weeks/10-fold (vs. BTBR WT) | No decline at 24 weeks (hyperfiltration) | Progressive renal damage, hypertrophy and accumulation of mesangial matrix (8 w), glomerular lesions (20 w). | Clee et al. 2005 [33] Hudkins et al. 2010 [29] Ericsson el al 2017 [47] |
| NONcNZO10/LtJ mice | NON/LtJ + NZO/H1Lt | - | T2DM | Insulin resistance, maturity-onset hyperglycaemia, visceral obesity, dyslipidemia | Albuminuria - | UK | Glomerulosclerosis, intraglomerular capillary thrombi and lipid deposition, nephritis, and Ig deposition. | Leiter et al. 2004 [34] Soler et al. 2012 [10] | |
| Knockout models | eNOS deficiency mice | C57BL/6 | STZ-induced | T1DM | Hypertension, hyperglycaemia | 15 weeks/3-fold 30 weeks/up to 30-fold (vs. WT-STZ) | 26–28 weeks (vs. WT-STZ) | Glomerular and tubulointerstitial damage. | Kanetsuna et al. 2007 [48] Nakagawa et al. 2007 [49] |
| eNOS deficiency mice | C57BLKS | db/db | T2DM | Hypertension, hyperglycaemia | 24–26 weeks/30-fold (vs. db/db) | 24–26 weeks (vs. db/db) | Glomerular and tubulointerstitial damage | Zhao et al. 2006 [50] | |
| B2R deficiency mice | C57BL/6 | Akita | T1DM | Hyperglycaemia, hypoinsulinemia, hypertension | 6 months/4-fold (vs. Akita) | Hyperfiltration (12 weeks) | Glomerular and tubulointerstitial damage | Kakoki et al. 2004 [51] Kakoki et al. 2010 [52] | |
| Decorin deficiency mice | C57BL/6 | STZ-induced | T1DM | Hypoinsulinemia, hyperglycaemia | 6 months/2 fold (vs. Decorin +/+ STZ) | 10 months (vs. Decorin +/+ STZ) | Mesangial matrix expansions and kidney inflammation | Williams et al. 2007 [53] | |
| Transgenic models | TTRhRen mice | FVB/NJ | STZ-induced | T1DM | Hypertension, hypoinsulinemia, hyperglycaemia | 20 weeks/3-fold (vs. WT-STZ) | 18 weeks (vs. WT-STZ) | Mesangial expansion. tubulointerstitial fibrosis | Thibodeau et al. 2014 [54] |
| OVE26-TTRhRen mice | FVB/NJ | Crossed with OVE26 T1DM | T1DM | Hypertension, hypoinsulinemia, hyperglycaemia | 20 weeks/40-fold (vs. OVE-26) | 20 weeks (vs. OVE-26) | Mesangial expansion. tubulointerstitial fibrosis | ||
| CYP1a1mRen2 rat (also found in literature as CYP1a1Ren2 rat) | Fisher rat | STZ-induced | T1DM | Hypertension, hypoinsulinemia, hyperglycaemia | Onset at 16 weeks. 28 weeks/10-fold (non STZ-induced vs. WT) 500-fold (STZ-induced vs. WT) | No decline at 28 weeks (vs. WT). | Glomerulosclerosis and tubulointerstitial fibrosis | Conway et al. 2012, 2014 [55,56] | |
| GIPRdn mice | CD1 | - | T1DM | Hypoinsulinemia, hyperglycaemia | 20 weeks (50% of the animals) and 28 weeks (all the animals)/5-fold (vs. WT) | NA | Progressive kidney disfunction | Herbach et al. 2009 [57] | |
| GLUT-1 mice | C57BL/6 | - | Non- diabetic | - | 26 weeks/2.4-fold (vs. WT) | Unknown (25% serum creatinine increase at 26 weeks, vs. WT) | Slow kidney disease progression, increased glomerular matrix, thickened GBM and glomerulosclerosis | Wang et al. 2010 [58] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giralt-López, A.; Molina-Van den Bosch, M.; Vergara, A.; García-Carro, C.; Seron, D.; Jacobs-Cachá, C.; Soler, M.J. Revisiting Experimental Models of Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 3587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103587
Giralt-López A, Molina-Van den Bosch M, Vergara A, García-Carro C, Seron D, Jacobs-Cachá C, Soler MJ. Revisiting Experimental Models of Diabetic Nephropathy. International Journal of Molecular Sciences. 2020; 21(10):3587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103587
Chicago/Turabian StyleGiralt-López, Anna, Mireia Molina-Van den Bosch, Ander Vergara, Clara García-Carro, Daniel Seron, Conxita Jacobs-Cachá, and Maria José Soler. 2020. "Revisiting Experimental Models of Diabetic Nephropathy" International Journal of Molecular Sciences 21, no. 10: 3587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103587