The Role of High-Density Lipoproteins in Endothelial Cell Metabolism and Diabetes-Impaired Angiogenesis

 and
and 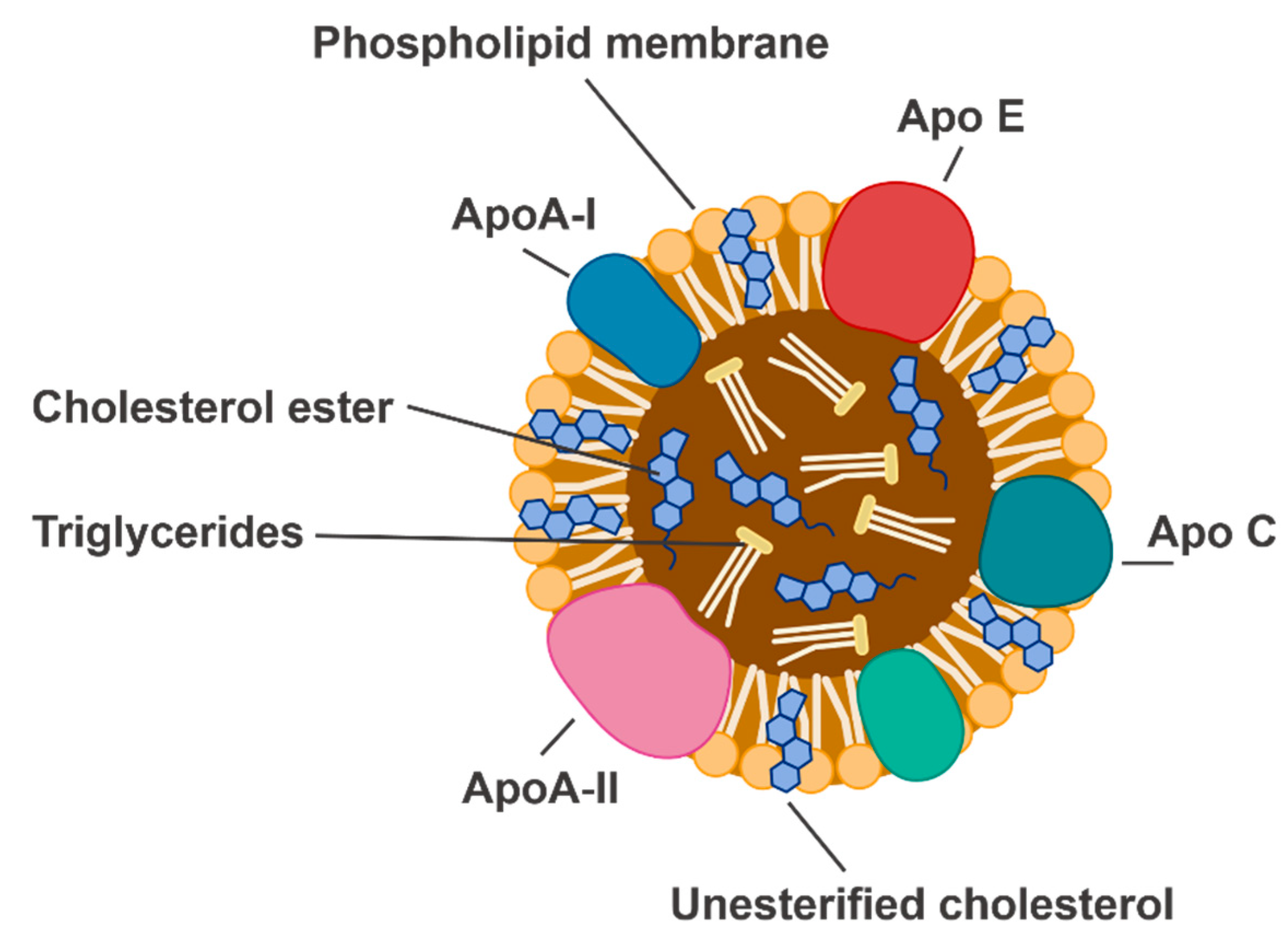{kind=link}
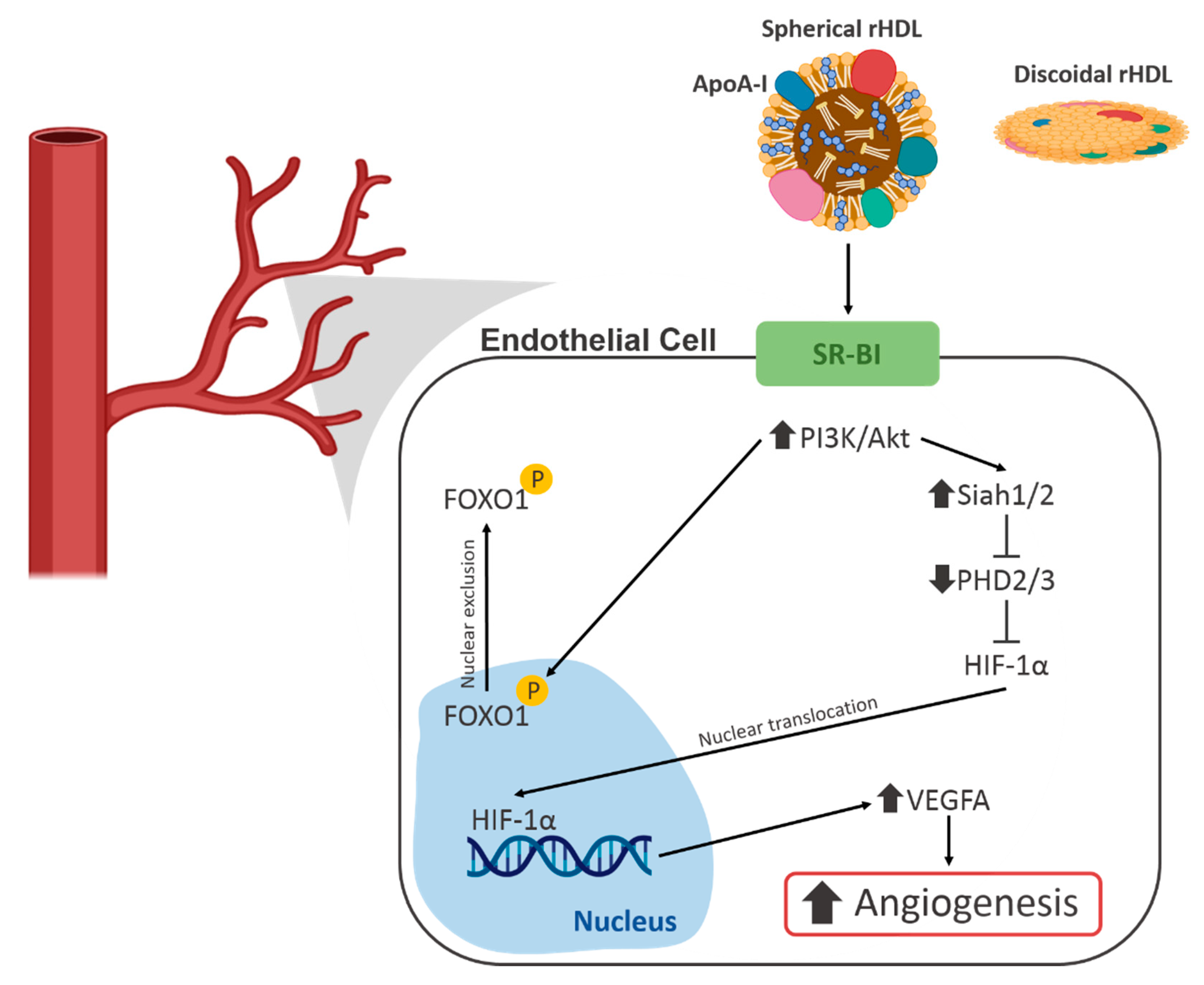{kind=link}
{kind=link}
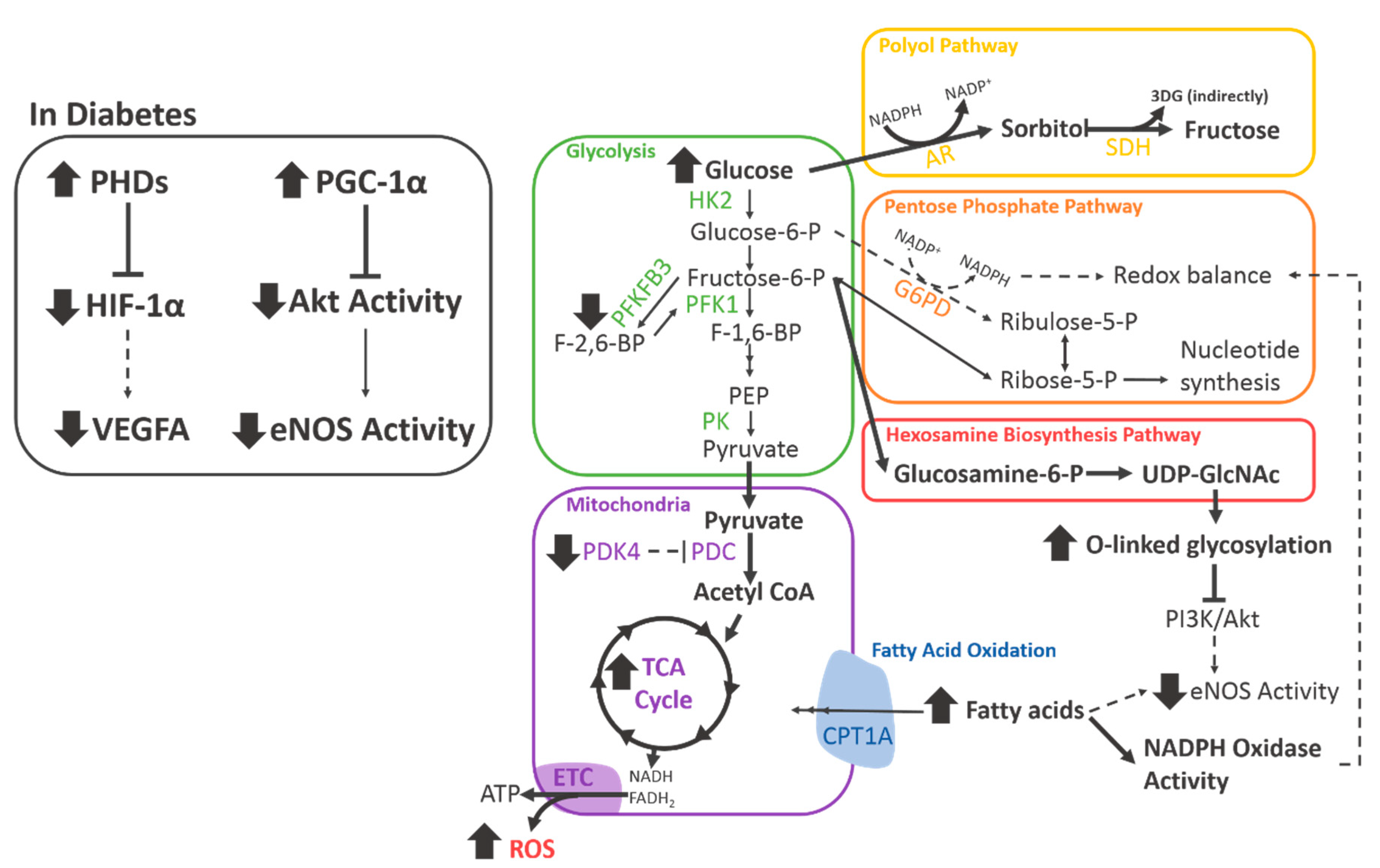{kind=link}
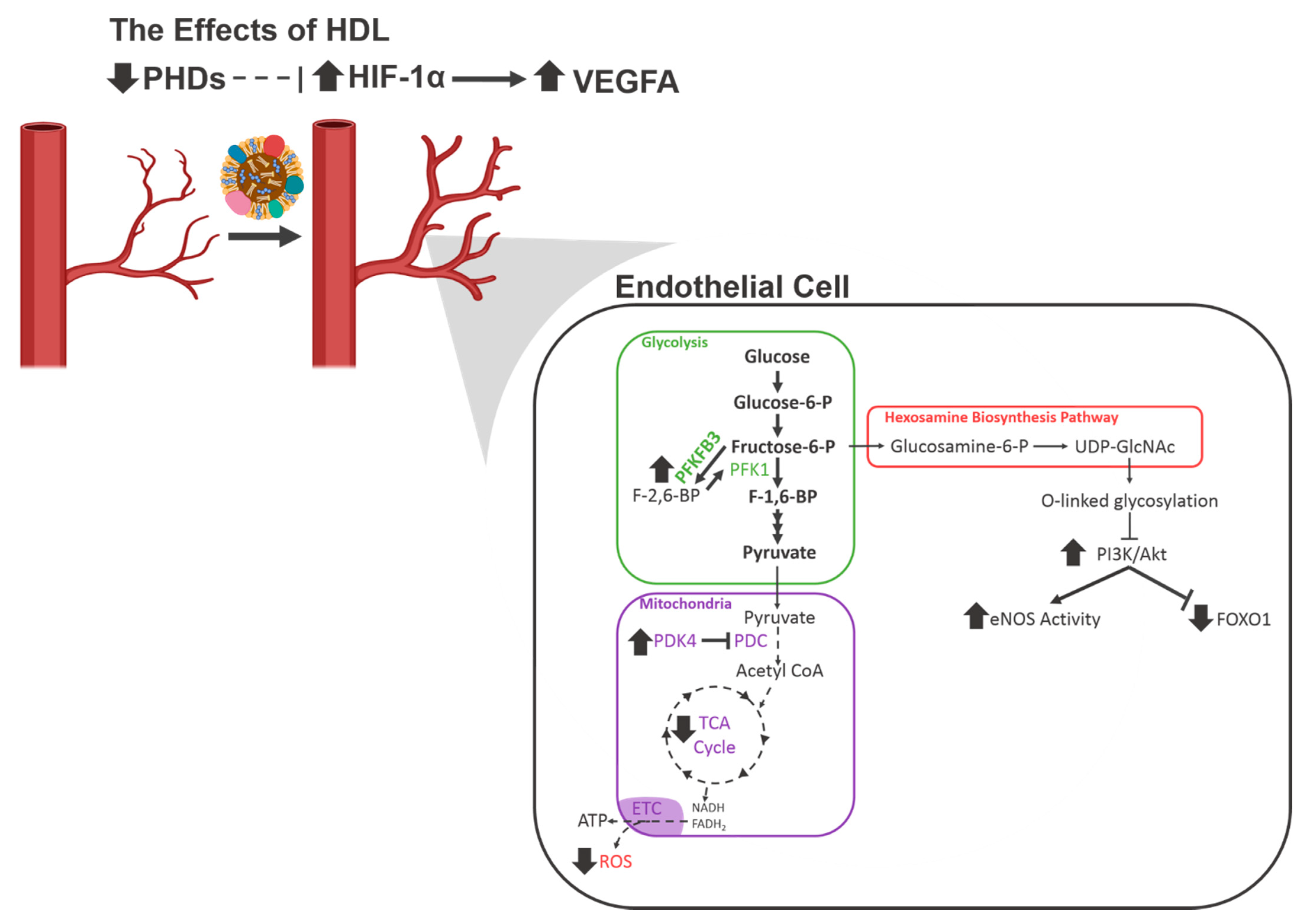{kind=link}
Abstract
:1. Introduction
2. High-Density Lipoproteins
3. Diabetes-Impaired Angiogenesis
3.1. Wound Healing
3.2. Recovery Following Myocardial Infarction
3.3. Dysregulated Angiogenesis in Diabetes
4. Physiological Angiogenesis and Its Regulation by HDL
5. Endothelial Cell Metabolism
5.1. Glycolysis
5.2. Mitochondrial Respiration and Hypoxia Tolerance
5.3. Fatty Acid Oxidation
5.4. The Pentose Phosphate Pathway
6. Endothelial Cell Metabolism and Diabetes-Impaired Angiogenesis
6.1. Diabetes Impairs Central Metabolic Pathways
6.2. Glycolytic Side-Pathways
6.3. Diabetes and Fatty Acid Oxidation
7. Emerging Role of HDL in Mechanisms of Endothelial Cell Metabolic Reprogramming and Diabetes-Impaired Angiogenesis
7.1. HDL and Hypoxia Signalling
7.2. HDL and PI3K/Akt Signalling
8. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| 3DG | 3-deoxyglucosone |
| AGE | Advanced glycation end products |
| AMPK | AMP-activated protein kinase |
| ANGPTL4 | Angiopoietin-like 4 |
| Apo | Apolipoprotein |
| CoA | Coenzyme A |
| CPT1A | Carnitine Palmitoyltransferase 1A |
| CVD | Cardiovascular disease |
| DFU | Diabetic foot ulcer |
| DN | Diabetic nephropathy |
| DR | Diabetic retinopathy |
| eNOS | Endothelial nitric oxide synthase |
| EPC | Endothelial progenitor cell |
| ERRα | Oestrogen-related receptor α |
| F1,6P2 | Fructose-1,6-bisphosphate |
| F2,6P2 | Fructose-2,6-bisphosphate |
| F6P | Fructose-6-phosphate |
| FAO | Fatty acid oxidation |
| FFA | Free fatty acid |
| FGF | Fibroblast growth factor |
| FGFR1 | Fibroblast growth factor receptor 1 |
| FOXO | Forkhead box O |
| G6PD | Glucose-6-phosphate dehydrogenase |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GSH | Reduced glutathione |
| GSSG | Oxidised glutathione |
| HBP | Hexosamine biosynthesis pathway |
| HCAEC | Human coronary artery endothelial cell |
| HDL | High-density lipoprotein |
| HDL-C | High-density lipoprotein cholesterol |
| HIF | Hypoxia-inducible factor |
| HIF-1α | Hypoxia-inducible factor 1α |
| HK2 | Hexokinase 2 |
| HUVEC | Human umbilical vein endothelial cell |
| LDL | Low-density lipoprotein |
| LDL-C | Low-density lipoprotein cholesterol |
| MAPK | Mitogen-activated protein kinase |
| MI | Myocardial infarction |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| PARP1 | Poly (ADP-Ribose) Polymerase 1 |
| PDC | Pyruvate dehydrogenase complex |
| PDK | Pyruvate dehydrogenase kinase |
| PDK4 | Pyruvate dehydrogenase kinase 4 |
| PDR | Proliferative diabetic retinopathy |
| PFK-1 | Phosphofructokinase-1 |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma co-activator 1α |
| PHD | Prolyl hydroxylase domain |
| PI3K | Phosphoinositide 3-kinases |
| PK | Pyruvate kinase |
| PPP | Pentose phosphate pathway |
| R5P | Ribose-5-phosphate |
| RA | Rheumatoid arthritis |
| rHDL | Reconstitute high-density lipoprotein |
| ROS | Reactive oxygen species |
| shRNA | Short hairpin RNA |
| siRNA | Small interfering RNA |
| SR-BI | Scavenger receptor BI |
| TCA | Tricarboxylic acid |
| UDP-GlcNAc | Uridine diphosphate N-acetylglucosamine |
| VEGFA | Vascular endothelial growth factor A |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| VHL | Von Hippel-Lindau |
References
- IDF. IDF Diabetes Atlas, 8th ed.; IDF: Brussels, Belgium, 2017. [Google Scholar]
- Tan, J.T.M.; Prosser, H.C.; Vanags, L.Z.; Monger, S.A.; Ng, M.K.C.; Bursill, C.A. High-density lipoproteins augment hypoxia-induced angiogenesis via regulation of post-translational modulation of hypoxia-inducible factor 1α. FASEB J. 2013, 28, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.M.; Prosser, H.C.; Dunn, L.L.; Vanags, L.Z.; Ridiandries, A.; Tsatralis, T.; Leece, L.; Clayton, Z.; Yuen, S.C.G.; Robertson, S.; et al. High Density Lipoproteins Rescue Diabetes-Impaired Angiogenesis via Scavenger Receptor Class B Type I. Diabetes 2016, 65, 3091–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, W.S. HDL-C vs HDL-P: How Changing One Letter Could Make a Difference in Understanding the Role of High-Density Lipoprotein in Disease. Clin. Chem. 2014, 60, e1–e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terasaka, N.; Yu, S.; Yvan-Charvet, L.; Wang, N.; Mzhavia, N.; Langlois, R.; Pagler, T.; Li, R.; Welch, C.L.; Goldberg, I.J.; et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J. Clin. Investig. 2008, 118, 3701–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, H.C.; Ng, M.K.C.; Bursill, C.A. The role of cholesterol efflux in mechanisms of endothelial protection by HDL. Curr. Opin. Lipidol. 2012, 23, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H. High-Density Lipoproteins as Biomarkers and Therapeutic Tools. In Impacts of Lifestyle, Diseases, and Environmental Stressors on HDL; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1. [Google Scholar]
- Grundy, S.M. Pre-Diabetes, Metabolic Syndrome, and Cardiovascular Risk. J. Am. Coll. Cardiol. 2012, 59, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, H.; Miller, M.; Nasir, K.; McEvoy, J.W.; Herrington, D.; Blumenthal, R.S.; Blaha, M.J. Primary Low Level of High-Density Lipoprotein Cholesterol and Risks of Coronary Heart Disease, Cardiovascular Disease, and Death: Results From the Multi-Ethnic Study of Atherosclerosis. Am. J. Epidemiol. 2016, 183, 875–883. [Google Scholar] [CrossRef] [Green Version]
- Kartono, T.; Mallapasi, M.N.; Mulawardi, M.; Laidding, S.R.; Aminyoto, M.; Prihantono, P. Correlation of HDL cholesterol serum and Wagner’s severity level of diabetic foot ulcers. Int. J. Res. Med. Sci. 2017, 5, 5129. [Google Scholar] [CrossRef] [Green Version]
- Ikura, K.; Hanai, K.; Shinjyo, T.; Uchigata, Y. HDL cholesterol as a predictor for the incidence of lower extremity amputation and wound-related death in patients with diabetic foot ulcers. Atheroscler 2015, 239, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Kadi, H.; Ozyurt, H.; Ceyhan, K.; Koc, F.; Celik, A.; Burucu, T. The Relationship between High-Density Lipoprotein Cholesterol and Coronary Collateral Circulation in Patients with Coronary Artery Disease. J. Investig. Med. 2012, 60, 808–812. [Google Scholar] [CrossRef]
- Prashanth, V.; Rayman, G.; Ketan, D.; Driver, V.; Hartemann, A.; Londahl, M.; Piaggesi, A.; Apelqvist, J.; Attinger, C.; Game, F. Effectiveness of interventions to enhance healing of chronic foot ulcers in diabetes: A systematic review. Diabetes Metab. Res. Rev. 2019, 36, 3284. [Google Scholar]
- Rayman, G.; Vas, P.; Dhatariya, K.; Driver, V.; Hartemann, A.; Londahl, M.; Piaggesi, A.; Apelqvist, J.; Attinger, C.; Game, F.; et al. Guidelines on use of interventions to enhance healing of chronic foot ulcers in diabetes (IWGDF 2019 update). Diabetes/Metab. Res. Rev. 2020, 36, e3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonkwo, U.A.; DiPietro, L.A. Diabetes and Wound Angiogenesis. Int. J. Mol. Sci. 2017, 18, 1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-J.; Lan, Y.-M.; Ou, M.-Q.; Ji, L.-Q.; Lin, S.-D. Expression of miR-217 and HIF-1α/VEGF pathway in patients with diabetic foot ulcer and its effect on angiogenesis of diabetic foot ulcer rats. J. Endocrinol. Investig. 2019, 42, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Brem, H.; Tomic-Canic, M. Cellular and molecular basis of wound healing in diabetes. J. Clin. Investig. 2007, 117, 1219–1222. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.A.; Bond, S.J.; Gupta, S.; Yacoub, O.A.; Anderson, G.L. Angiogenesis Inhibitor TNP-470 Inhibits Murine Cutaneous Wound Healing. J. Surg. Res. 1999, 82, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Roman, C.D.; Choy, H.; Nanney, L.; Riordan, C.; Parman, K.; Johnson, D.; Beauchamp, R.D. Vascular Endothelial Growth Factor-Mediated Angiogenesis Inhibition and Postoperative Wound Healing in Rats. J. Surg. Res. 2002, 105, 43–47. [Google Scholar] [CrossRef]
- Galiano, R.D.; Tepper, O.M.; Pelo, C.R.; Bhatt, K.A.; Callaghan, M.; Bastidas, N.; Bunting, S.; Steinmetz, H.G.; Gurtner, G.C. Topical Vascular Endothelial Growth Factor Accelerates Diabetic Wound Healing through Increased Angiogenesis and by Mobilizing and Recruiting Bone Marrow-Derived Cells. Am. J. Pathol. 2004, 164, 1935–1947. [Google Scholar] [CrossRef] [Green Version]
- Sasmaz, H.; Yilmaz, M.B.; Madanmohan, T.; Nandeesha, H.; Pavithran, P. Coronary Collaterals in Obese Patients: Impact of Metabolic Syndrome. Angiology 2008, 60, 164–168. [Google Scholar] [CrossRef]
- Brackbill, M.L.; Sytsma, C.S.; Sykes, K. Perioperative Outcomes of Coronary Artery Bypass Grafting: Effects of Metabolic Syndrome and Patient’s Sex. Am. J. Crit. Care 2009, 18, 468–473. [Google Scholar] [CrossRef]
- Hoffmann, R.; Stellbrink, E.; Schröder, J.; Gräwe, A.; Vogel, G.; Blindt, R.; Kelm, M.; Radke, P.W. Impact of the Metabolic Syndrome on Angiographic and Clinical Events after Coronary Intervention Using Bare-Metal or Sirolimus-Eluting Stents. Am. J. Cardiol. 2007, 100, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Berge, K.G.; Canner, P.L.; Hainline, A. High-density lipoprotein cholesterol and prognosis after myocardial infarction. Circulation 1982, 66, 1176–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharjee, S.; Roe, M.; Amsterdam, E.A.; Holmes, D.N.; Boden, W.E. Relation of Admission High-Density Lipoprotein Cholesterol Level and in-Hospital Mortality in Patients with Acute Non-ST Segment Elevation Myocardial Infarction (from the National Cardiovascular Data Registry). Am. J. Cardiol. 2013, 112, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Rha, S.-W.; Elnagar, A.; Choi, B.G.; Im, S.I.; Kim, S.; Na, J.O.; Han, S.; Choi, C.U.; Lim, H.E.; et al. The impact of low level of high-density lipoprotein cholesterol on 6-month angiographic and 2-year clinical outcomes in acute myocardial infarction patients undergoing primary percutaneous coronary intervention. J. Am. Coll. Cardiol. 2012, 59, E18. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Kim, W.; Woo, J.S.; Lee, T.W.; Ihm, C.G.; Kim, Y.G.; Moon, J.Y.; Lee, S.H.; Jeong, M.H.; Jeong, K.H.; et al. The Predictive Role of Serum Triglyceride to High-Density Lipoprotein Cholesterol Ratio According to Renal Function in Patients with Acute Myocardial Infarction. PLoS ONE 2016, 11, e0165484. [Google Scholar] [CrossRef]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Cooper, M.E. Pathogenesis of diabetic nephropathy. J. Diabetes Investig. 2011, 2, 243–247. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Appelhoff, R.J.; Tian, Y.-M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J. Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-inducible Factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.-H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and Inhibitory Domains of Hypoxia-inducible Factor 1α. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, J.A.; Jiang, B.-H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-Inducible Factor-1 Mediates Activation of Cultured Vascular Endothelial Cells by Inducing Multiple Angiogenic Factors. Circ. Res. 2003, 93, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Pengchun, Y.; Kerstin, W.; Dubrac, A.; Tung, J.K.; Alves, T.C.; Fang, J.S.; Xie, Y.; Zhu, J.; Chen, Z.; De Smet, F. FGF-dependent metabolic control of vascular development. Nature 2017, 545, 224–228. [Google Scholar]
- Hristov, M. Endothelial Progenitor Cells Isolation and Characterization. Trends Cardiovasc. Med. 2003, 13, 201–206. [Google Scholar] [CrossRef]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, Z.; Foo, S.-Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [Green Version]
- Sumi, M.; Sata, M.; Miura, S.-I.; Rye, K.-A.; Toya, N.; Kanaoka, Y.; Yanaga, K.; Ohki, T.; Saku, K.; Nagai, R. Reconstituted High-Density Lipoprotein Stimulates Differentiation of Endothelial Progenitor Cells and Enhances Ischemia-Induced Angiogenesis. Arter. Thromb. Vasc. Biol. 2007, 27, 813–818. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Vergeer, M.; Bisoendial, R.J.; op‘T Roodt, J.; Levels, H.; Birjmohun, R.S.; Kuivenhoven, J.A.; Basser, R.; Rabelink, T.J.; Kastelein, J.J.P.; et al. Reconstituted HDL infusion restores endothelial function in patients with type 2 diabetes mellitus. Diabetologia 2008, 51, 1081–1084. [Google Scholar] [CrossRef] [Green Version]
- Van Oostrom, O.; Nieuwdorp, M.; Westerweel, P.; Hoefer, I.; Basser, R.; Stroes, E.; Verhaar, M.C. Reconstituted HDL Increases Circulating Endothelial Progenitor Cells in Patients with Type 2 Diabetes. Arter. Thromb. Vasc. Biol. 2007, 27, 1864–1865. [Google Scholar] [CrossRef] [Green Version]
- Prosser, H.; Ng, M.; Bursill, C. Multifunctional Regulation of Angiogenesis by High Density Lipoproteins. Hear. Lung Circ. 2012, 21, S45. [Google Scholar] [CrossRef] [Green Version]
- Theofilatos, D.; Fotakis, P.; Valanti, E.; Sanoudou, D.; Zannis, V.; Kardassis, D. HDL-apoA-I induces the expression of angiopoietin like 4 (ANGPTL4) in endothelial cells via a PI3K/AKT/FOXO1 signaling pathway. Metabolism 2018, 87, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.C.; Chan, J.S.K.; Goh, C.Q.; Gounko, N.V.; Luo, B.; Wang, X.; Foo, S.; Wong, M.T.C.; Choong, C.; Kersten, S.; et al. Angiopoietin-like 4 Stimulates STAT3-mediated iNOS Expression and Enhances Angiogenesis to Accelerate Wound Healing in Diabetic Mice. Mol. Ther. 2014, 22, 1593–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Jang, C.; Dharaneeswaran, H.; Li, J.; Bhide, M.; Yang, S.; Li, K.; Arany, Z. Endothelial pyruvate kinase M2 maintains vascular integrity. J. Clin. Investig. 2018, 128, 4543–4556. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, N.H.; Harris, R.A. Role of Pyruvate Dehydrogenase Kinase 4 in Regulation of Blood Glucose Levels. Korean Diabetes J. 2010, 34, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ma, K.; Sadana, P.; Chowdhury, F.; Gaillard, S.; Wang, F.; McDonnell, N.P.; Unterman, T.G.; Elam, M.B.; Park, E.A. Estrogen-related Receptors Stimulate Pyruvate Dehydrogenase Kinase Isoform 4 Gene Expression. J. Biol. Chem. 2006, 281, 39897–39906. [Google Scholar] [CrossRef] [Green Version]
- Aragonés, J.; Schneider, M.; Van Geyte, K.; Fraisl, P.; Dresselaers, T.; Mazzone, M.; Dirkx, R.; Zacchigna, S.; Lemieux, H.; Jeoung, N.H.; et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat. Genet. 2008, 40, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Primer, K.; Solly, E.; Psaltis, P.; Tan, J.; Bursill, C. High-density Lipoproteins Rescue Diabetes-impaired Angiogenesis by Restoring Cellular Metabolic Reprogramming Responses to Hypoxia. Heart Lung Circ. 2019, 28, 365. [Google Scholar] [CrossRef] [Green Version]
- Fisslthaler, B.; Fleming, I. Activation and Signaling by the AMP-Activated Protein Kinase in Endothelial Cells. Circ. Res. 2009, 105, 114–127. [Google Scholar] [CrossRef]
- Dagher, Z.; Ruderman, N.B.; Tornheim, K.; Ido, Y. Acute Regulation of Fatty Acid Oxidation and AMP-Activated Protein Kinase in Human Umbilical Vein Endothelial Cells. Circ. Res. 2001, 88, 1276–1282. [Google Scholar] [CrossRef]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.S.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boros, L.G.; Lee, P.; Brandes, J.; Cascante, M.; Muscarella, P.; Schirmer, W.; Melvin, W.S.; Ellison, E. Nonoxidative pentose phosphate pathways and their direct role in ribose synthesis in tumors: Is cancer a disease of cellular glucose metabolism? Med. Hypotheses 1998, 50, 55–59. [Google Scholar] [CrossRef]
- Prasai, P.K.; Shrestha, B.; Orr, A.W.; Pattillo, C.B. Decreases in GSH:GSSG activate vascular endothelial growth factor receptor 2 (VEGFR2) in human aortic endothelial cells. Redox Biol. 2018, 19, 22–27. [Google Scholar] [CrossRef]
- Rudnicki, M.; Abdifarkosh, G.; Nwadozi, E.; Ramos, S.V.; Makki, A.; Sepa-Kishi, D.M.; Ceddia, R.B.; Perry, C.G.; Roudier, E.; Haas, T.L. Endothelial-specific FoxO1 depletion prevents obesity-related disorders by increasing vascular metabolism and growth. eLife 2018, 7, 7. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, E.-J.; Kim, D.-K.; Lee, J.-M.; Park, S.B.; Lee, I.-K.; Harris, R.A.; Lee, M.-O.; Choi, H.-S. Hypoxia induces PDK4 gene expression through induction of the orphan nuclear receptor ERRgamma. PLoS ONE 2012, 7, 115–125. [Google Scholar]
- Sawada, N.; Jiang, A.; Takizawa, F.; Safdar, A.; Manika, A.; Tesmenitsky, Y.; Kang, K.-T.; Bischoff, J.; Kalwa, H.; Sartoretto, J.L.; et al. Endothelial PGC-1α mediates vascular dysfunction in diabetes. Cell Metab. 2014, 19, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Apse, K.; Pang, J.; Stanton, R.C. High Glucose Inhibits Glucose-6-phosphate Dehydrogenase via cAMP in Aortic Endothelial Cells. J. Biol. Chem. 2000, 275, 40042–40047. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.A.; Umanah, G.K.E.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagné, J.-P.; Poirier, G.G.; Dawson, V.L.; Dawson, V.L. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [Green Version]
- Devalaraja-Narashimha, K.; Padanilam, B.J. PARP-1 inhibits glycolysis in ischemic kidneys. J. Am. Soc. Nephrol. 2008, 20, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmetz, P.R.; Balko, C.; Gabbay, K.H. The Sorbitol Pathway and the Complications of Diabetes. N. Engl. J. Med. 1973, 288, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Randall, W.C.; Taylor, A.H.; Kappler, F.; Walker, M.; Brown, T.; Szwergold, B.S. Fructose-3-phosphate production and polyol pathway metabolism in diabetic rat hearts. Metabolism 1997, 46, 1333–1338. [Google Scholar] [CrossRef]
- Szwergold, B.; Kappler, F.; Brown, T. Identification of fructose 3-phosphate in the lens of diabetic rats. Science 1990, 247, 451–454. [Google Scholar] [CrossRef]
- Hamada, Y.; Araki, N.; Horiuchi, S.; Hotta, N. Role of polyol pathway in nonenzymatic glycation. Nephrol. Dial. Transplant. 1996, 11, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Tamarat, R.; Silvestre, J.-S.; Huijberts, M.; Benessiano, J.; Ebrahimian, T.G.; Duriez, M.; Wautier, M.-P.; Wautier, J.L.; Levy, B. Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 8555–8560. [Google Scholar] [CrossRef] [Green Version]
- Matafome, P.; Sena, C.; Seiça, R. Methylglyoxal, obesity, and diabetes. Endocrine 2013, 43, 472–484. [Google Scholar] [CrossRef]
- Federici, M. Insulin-Dependent Activation of Endothelial Nitric Oxide Synthase Is Impaired by O-Linked Glycosylation Modification of Signaling Proteins in Human Coronary Endothelial Cells. Circulation 2002, 106, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Namba, T.; Koike, H.; Murakami, K.; Aoki, M.; Makino, H.; Hashiya, N.; Ogihara, T.; Kaneda, Y.; Kohno, M.; Morishita, R. Angiogenesis Induced by Endothelial Nitric Oxide Synthase Gene Through Vascular Endothelial Growth Factor Expression in a Rat Hindlimb Ischemia Model. Circulation 2003, 108, 2250–2257. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Soesanto, Y.; McClain, D. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting Akt activity in endothelial cells. Arter. Thromb. Vasc. Biol. 2008, 28, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, H.O.; Tarshoby, M.; Monestel, R.; Hook, G.; Cronin, J.; Johnson, A.; Bayazeed, B.; Baron, A.D. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J. Clin. Investig. 1997, 100, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, H.O.; Paradisi, G.; Hook, G.; Crowder, K.; Cronin, J.; Baron, A.D. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes 2000, 49, 1231–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kreutzenberg, S.V.; Crepaldi, C.; Marchetto, S.; Calo, L.; Tiengo, A.; Del Prato, S.; Avogaro, A. Plasma Free Fatty Acids and Endothelium-Dependent Vasodilation: Effect of Chain-Length and Cyclooxygenase Inhibition. J. Clin. Metab. 2000, 85, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primer, K.R.; Psaltis, P.J.; Tan, J.T.M.; Bursill, C.A. The Role of High-Density Lipoproteins in Endothelial Cell Metabolism and Diabetes-Impaired Angiogenesis. Int. J. Mol. Sci. 2020, 21, 3633. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103633
Primer KR, Psaltis PJ, Tan JTM, Bursill CA. The Role of High-Density Lipoproteins in Endothelial Cell Metabolism and Diabetes-Impaired Angiogenesis. International Journal of Molecular Sciences. 2020; 21(10):3633. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103633
Chicago/Turabian StylePrimer, Khalia R., Peter J. Psaltis, Joanne T.M. Tan, and Christina A. Bursill. 2020. "The Role of High-Density Lipoproteins in Endothelial Cell Metabolism and Diabetes-Impaired Angiogenesis" International Journal of Molecular Sciences 21, no. 10: 3633. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103633