Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections


1
Department of Avian Infectious Diseases, Shanghai Veterinary Research Institute. Chinese Academy of Agricultural Science, Shanghai 200241, China
2
Jiangsu Co-Innovation Center for Prevention and Control of Important Animal Infectious Disease and Zoonoses, Yangzhou University, Yangzhou 225009, China
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(10), 3689; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103689
Submission received: 13 March 2020
/
Revised: 14 May 2020
/
Accepted: 21 May 2020
/
Published: 23 May 2020
(This article belongs to the Special Issue Intracellular Organelle Rearrangement Induced by Pathogenic Infections)
Abstract
:Viruses have evolved different strategies to hijack subcellular organelles during their life cycle to produce robust infectious progeny. Successful viral reproduction requires the precise assembly of progeny virions from viral genomes, structural proteins, and membrane components. Such spatial and temporal separation of assembly reactions depends on accurate coordination among intracellular compartmentalization in multiple organelles. Here, we overview the rearrangement and morphology remodeling of virus-triggered intracellular organelles. Focus is given to the quality control of intracellular organelles, the hijacking of the modified organelle membranes by viruses, morphology remodeling for viral replication, and degradation of intracellular organelles by virus-triggered selective autophagy. Understanding the functional reprogram and morphological remodeling in the virus-organelle interplay can provide new insights into the development of broad-spectrum antiviral strategies.
1. Introduction
Viruses are obligate intracellular parasites that must rely on the cellular function for each stage of their life cycle [1,2]. To successfully enter a cell, enveloped viruses bind to surface-specific receptors through their transmembrane glycoproteins and subsequently activate intracellular signaling transduction to initiate entry; non-enveloped viruses bind through the capsid surface or projections from the capsid [3]. Viral penetration into the host cell is followed by genome uncoating, genome expression and replication, assembly of new virions, and their egress [4]. To maintain homeostasis, a fundamental function of the membrane-bound organelles is used as a scaffold to compartmentalize cellular trafficking and secretory signaling. Upon viral infection, the membranes of the intracellular organelles are remodeled and utilized by viruses as platforms to coordinate the accumulation of viral and cellular components required for efficient replication [4,5].
In addition to the rearrangement of intracellular organelles, massive viral infection also leads to the accumulation of damaged organelles, misfolded proteins, and other macromolecules. Autophagy is a conserved catabolic multistep process that non-selectively or selectively delivers large cytoplasmic proteins, including damaged organelles, into specific double-membrane autophagosome vesicles, and shuttles to the vacuole/lysosomes for degradation and recycling [6]. The process of autophagic regulation is divided into several steps: initiation, elongation, fusion, and degradation [7]. The specific targeting of cytoplasmic substrates for degradation through autophagosome depends mainly on specific cargo receptors, which contain an LC3-interacting region (LIR) motif and ubiquitin binding domain [8]. To date, several adaptor proteins, including p62/SQSTM1 [9,10,11,12], AMBRA1 [13], NBR1 [14], optineurin/OTPN [15,16], TAXIBP1 [17], CALCOCO2/NDP52 [18], BNIP3L/NIX [19,20], and BNIP3 [21], PHB2 [22], FUNDC1 [23], Cardiolipin [24], and FAM134B [25], have been identified as being involved in the recognition of cargo substrates for degradation. Most viruses activate and utilize the autophagic machinery for infectious progeny with notable exceptions, such as sindbis virus (SIV) [26,27], herpesviruses (α-, β-, and γ-) [28,29,30], human parainfluenza virus typ3 (HPIV3) [31], and human immunodeficiency virus type 1 (HIV-1) [32]. During viral infection, based on degradation substrates such as the mitochondria, peroxisome, endoplasmic reticulum (ER), lysosome, and nucleus, the selective autophagy of organelles is called mitophagy, pexophagy, ER-phagy, lysophagy, and nucleophagy, respectively [33,34].
Given the importance of membrane biogenesis in the interplay between the virus and the organelle, in this review, we briefly summarize our current knowledge about viruses’ modification of membranes morphology and biogenesis of intercellular organelles to support viral infection progeny. Moreover, we describe the potential roles of selective autophagy in the regulation of intracellular organelles upon viral infection.
2. Rearrangement of Intracellular Organelles during Viral Infections
To maximize their viral replication and evade host antiviral responses, viruses have evolved a plethora of strategies to hijack cellular organelles [1,35,36]. Each step of viral replication is closely accompanied by the rearrangement of intracellular organelles.
2.1. Remodeling of the Mitochondria for Viral Replication
The mitochondria are highly dynamic organelles and form interconnected tubular networks, undergoing a balance between fusion and fission in response to intracellular and/or extracellular stresses [37] (Figure 1A,B). Mitochondrial fusion involves two sets of key GTPase proteins in mammals: the mitofusin GTPases (Mfns) (Mfn1 and Mfn2) of the outer mitochondrial membrane (OMM) and optic atrophy 1 (OPA1) of the inner mitochondrial membrane (IMM) [38,39,40,41,42]. The Mfns mediate OMM fusion and cristae integrity [43]. However, the OPA1 mediates IMM fusion and cristae integrity by regulating of the mRNA splicing forms, membrane potential, and the adenosine triphosphate (ATP)-dependent diverse cellular proteases [39,40,41]. Subsequently, OMM fusions are followed by IMM fusion processes, resulting in the concomitant mixing of the mitochondrial contents and merging of two individual mitochondria. In a previous study, Cipolat et al. identified that OPA1 specific functional cross-talk with Mfn1 rather than Mfn2 is involved in the mitochondrial fusion of OMM [44]. Mitochondrial fission is a complex process that includes two distinct steps: an initial constriction of mitochondrial membranes and membrane scission. The initial constriction step narrows the mitochondrial tube diameter at the ER-mitochondria intersection zones where ER tubules wrap around the OMM. Manor et al. suggested that actin-nucleating protein spire 1C localizes to the mitochondria, directly links the mitochondria to the actin cytoskeleton and the ER, and finally promotes actin polymerization at the ER-mitochondria intersections [45]. The membrane scission of the mitochondria is primarily regulated by dynamic relative GTPase protein (DRP-1) [46]. The mitochondrial localization of DRP-1 is a cytosolic factor promoting mitochondrial fission, which powers the constriction and division of the mitochondria primarily through post-translational modification (e.g., phosphorylation) (reviewed by Lee et al. [47]). Recent studies have reported that the recruitment of DRP-1 in mammalian cells requires several accessory proteins, such as the mitochondrial fission protein 1 (Fis-1) and mitochondrial fission factor (Mff) [48]. Although such proteins are proposed to constitute the fission complex of the mitochondria, mediating mitochondrial fission using this complex has remained unclear.
Viruses have evolved several strategies to remodel the mitochondria for viral replication and assembly, including spatial distribution, morphology remodeling, and metabolism reprogramming. To maximize the effectiveness of DNA replication, African swine fever virus (ASFV) infection recruits the mitochondria around the viral factories, associated with the morphology change and accumulation of the mitochondria. It was speculated that the translation and ATP synthesis are coupled and compartmentalized around viral factories to promote virus replication [49] (Figure 1C). Normal mitochondria are dynamic organelles, and form interconnected tubular networks [37,50] (Figure 1A). The cristae remodeling of the IMM determines the assembly and stability of respiratory chain supercomplexes and respiratory efficiency [51]. In general, the mitochondrial elongation process is associated with the dimerization and activation of the ATPase function to produce additional energy [50,52]. NDV induces the hyper-fusion of the mitochondria in infected A549 cells (unpublished data), which is similar to the characteristic of Dengue virus (DENV) [53] and severe acute respiratory syndrome-coronavirus (SARS-CoVs) [54]. Notably, except for vaccinia virus (VV) [55], most viruses exploit aerobic glycolysis of the mitochondria for the production of viral progeny [36].
Moreover, viral infections may increase the inter-organellar interactions of the mitochondria with other organelles for replication. Rubella virus (RUBV) [56] and Bunyamwera virus (BUNV) [57] infections increase the membrane interactions among mitochondria, ER, and Golgi (Figure 1C), which is consistent with that of the ER-mitochondria contract that serves as a platform for inter-organellar communication [58].
To date, several reports have argued the role of Mfns in innate immunity [60,61,62]. The interaction of Mfns with the adaptor mitochondrial antiviral signaling protein (MAVS) (also called IPS-1, Cardif, or VISA) at the mitochondrial associated membrane (MAM) leads the initiation of the IFN signaling pathway [63,64]. Meanwhile, MAVS was also reported to interact with MFN2, which leads to the inhibition of inflammatory cytokine production, suggesting the MAM plays a complex role in the regulation of innate immunity [61] (detailed in review [64]). Castanier et al. also identified the cross-modulation relationship between mitochondrial dynamic and retinoic acid-inducible gene I protein (RIG-I) like receptor (RLR) signaling activation [60]. Certain viruses, such as influenza A virus (IAV) [65], measles virus (MV) [66], hepatitis B virus (HBV) [67,68,69,70], and hepatitis C virus (HCV) [67], induce selective autophagy to degrade fragmented mitochondria and evade innate immunity. Meanwhile, the non-structural (NS) protein 4B of DENV induces mitochondrial elongation via inactivation of DRP-1 and dampens the activation of RLR signal pathway to promote replication [53]. Similarly, the open reading frame-9b (ORF-9b) encoded by SARS-CoVs causes mitochondrial elongation via triggering DRP-1 degradation, and inhibits RLR signaling [54].
Collectively, viruses have evolved several strategies to hijack the mitochondria for viral genome replication and assembly, including the remodeling of mitochondrial morphology and distribution, the regulation of the fusion–fission machinery complex, and the synthesis of ATP production.
2.2. Rearrangement of ER and Unfolded Protein Response (UPR) during Viral Infection
The ER, a single continuous membrane, consists of two primary structural subdomains: the nuclear envelope and the peripheral ER (a polygonal network) [71]. The nuclear envelope of ER consists of two flat membrane bilayers; the peripheral ER is composed of membrane cisternae and dynamic interconnected tubules [71,72]. The ER is the largest intracellular endomembrane system and has multiple complex functions, including Ca2+ storage, fatty acid synthesis, ion homeostasis, and, in particular, the quality control of newly synthesized proteins [73]. The accumulation of misfolded or unfolded proteins in the ER lumen is known as ER stress [74]. UPR and ER-associated degradation (ERAD) signaling are central to maintain the quality control of the ER [74,75]. The UPR is a signaling cascade aimed at eliminating misfolding proteins and increasing folding capacity in lumen [74]. The protein-folding conditions in the ER lumen is primarily sensed by three integrated signaling transducers: activating transcription factor 6 (ATF6) [76], double-stranded RNA-activated protein kinase-like kinase (PERK), and inositol requiring enzyme 1α (IRE1α) [58,77] (Figure 2). Each branch uses a distinct mechanism to drive the transcription of UPR signal transduction, such as ATF6 by regulated proteolysis, PERK by translational control, and IRE1 by non-conventional mRNA splicing [77]. By contrast, ERAD recognizes misfolded proteins and retro-translocates such proteins into the cytoplasm for degradation by the ubiquitin-proteasome-dependent ERAD and the autophagy-lysosome dependent ERAD [75,78].
A series of studies has reported that viral infections reshape the morphology and membrane remodeling of ER [1,71], and exploit various strategies to hijack the three branches of UPR for viral replication (Figure 2 and Table 1). The possible explanations were summarized as follows: first, the large malleable surface area of ER is used as a physical scaffold to protect viral RNA from degradation by cellular mRNA decay machinery [73,79]. RNA viruses have evolved several strategies to avoid the cellular mRNA decay machinery [79]. Second, viruses, particularly most RNA viruses, remodel the ER membrane to form a variety of structures for infectious progeny [5], including single-membrane spherule vesicles, double-membrane vesicles, convoluted membranes, and single-membrane sheets in the ER lumen [71]. Tenorio et al. identified that δNS and μNS of reovirus caused tubulation and fragmentation of the ER, respectively, to re-build replication sites [80], indicating that viral proteins play different roles in the rearrangement of ER membranes. Similarly, the NS4A of DENV induces the membrane arrangement of ER lumen in a 2K-regulated manner [81]. Third, viruses recruit the ER membranes into the replication and assembly compartments. The viral cytoplasmic replication site of VV [82,83], equine arteritis virus (EAV) [84], and polivirus (PV) [85] is derived directly from the ER membrane. Moreover, ASFV structural protein p54 plays an important role in the recruitment and transformation of the ER membranes into the envelope precursors [86]. Fourth, viruses increase the capacity and spatial rearrangement to increase ER biogenesis, including membrane protein synthesis, fatty acid change, and Ca2+ storage [73]. For enveloped viruses, the key molecular chaperone of ER, including Bip/GRP78 and calnexin/calreticulin, assists the folding of the extracellular domains of viral membrane glycoproteins, such as GP2a of PRRSV [87], and hemagglutinin-neuraminidase (HN) and fusion (F) proteins of NDV [88], when they translocate into the lumen of the ER. Meanwhile, the reprograming of ER biogenesis, such as Ca2+ storage, is required for viral replication, including HCV [89] and ASFV [90]. Fifth, viruses co-opt or subvert the ERAD processes to re-establish ER homeostasis, which actively exports the malformed proteins from the ER for degradation. Human cytomegalovirus (HCMV) [91] and IAV [92] exploit the ERAD pathway to benefit viral replication. Finally, the membrane remodeling of ER may suppress the activation of host immunity. Upon viral infections, particularly DNA viruses, stimulator of interferon genes (STING), an activated ER adaptor of the cyclic GMP-AMP synthase (cGAS)-STING signaling pathway, translocates from the ER to the ER-Golgi-intermediate compartment (ERGIC) and the Golgi apparatus, and then activates downstream molecules [93,94,95]. Therefore, we speculate that the morphology remodeling and membrane modification of ER induced by viruses may be involved in the regulation of STING trafficking, EARD degradation, and post-translational modification, and eventually evade the activation of cGAS-STING pathway (Figure 3).
2.3. Rearrangement of Peroxisome for Infectious Progeny
The peroxisomes are single membrane-bounded organelles that function in numerous metabolic pathways, including β-oxidation of long-chain fatty acids, detoxification of hydrogen peroxide, and synthesis of ether phospholipids and bile acids [113,114]. Notably, the mitochondria and peroxisomes share common functions in the β-oxidation of fatty acids and the reduction of damaging peroxides. Proliferation of peroxisome is largely mediated by growth and division. Peroxisomal division in mammalian cells comprises multiple processes, including membrane deformation, elongation, constriction, and fission [115]. With the exception of peroxin (PEX)-11, the peroxisomes and mitochondria share common fission machinery, including DRP-1, Mff, and Fis1 [116,117]. The fission machinery of peroxisome is orchestrated by PEX-11β and mitochondrial fission factors [115]. Mitochondrial-derived vesicles (MDVs) are involved in the transportation of mitochondrial-anchored protein ligase (MAPL), a mitochondrial outer membrane, to peroxisomes [118]. The retromer complex containing vacuolar protein sorting (Vps) 5, Vps 26, and Vps 29, a known component of vesicle transport from the endosome to the Golgi apparatus, also regulates the transport of MAPL as a binding partner from the mitochondria to peroxisomes [119].
Viruses regulate the morphology and biogenesis of peroxisomes to promote progeny replication [35]. For instance, the C-terminal of the rotavirus VP4 protein is directly located in peroxisomes via its conserved peroxisomal targeting signal [120]. Meanwhile, viruses have exploited the myristoyl-CoA produced by peroxisome biogenesis for the N-myristoylation of viral proteins [35], such as ASFV [121], indicating that peroxisomal lipid metabolism contributes to viral replication. Another typical example is the tomato bushy stunt virus (TBSV), a member of the Tombusviridae family, which infects a variety of plant species. McCartney et al. reported that TBSV induced the rearrangement of peroxisomes and caused vesiculation of the peroxisomal membrane, where it provided a scaffold for virus anchoring and server as the sites of viral RNA synthesis [122]. In the absence of peroxisomes, TBSV also exploits the surface of the ER membrane as a viral factory for viral replication and assembly [123]. It is suggestive of the remarkable flexibility of the virus to use host membranes for replication.
2.4. Hijacking of Golgi Apparatus for Infectious Progeny
The Golgi apparatus is a highly dynamic organelle whose function primarily includes saccule formation and utilization of such saccules in vesicle formation at the opposite face for delivery [124]. The normal cellular secretory pathway, bidirectional transport between the ER and Golgi apparatus, is mediated by tubulovesicular transport containers that depend on two coat protein complexes, COP-I and COP-II. COP-II establishes a membrane flow from the ER to the Golgi complex [125]. However, COP-I coat acts in retrograde transport from the Golgi back to the ER [126].
In general, certain viruses utilize the cellular trafficking and secretory pathway of the ER-Golgi transport system to replicate/release their progeny [1]. PV utilizes the components of the ADP-ribosylation factor (Arf) family of small GTPases [127] and cellular COP-II proteins [128] to form membrane-bound replication complex for viral replication, indicating that PV hijacks the components of the cellular secretory pathway for replication. As shown in Table 2, metonaviridae [56] and peribunyaviridae [57] hijack the Golgi complex to re-construct it as a viral factory for viral replication. For example, RUBV [56] and BUNV [57] infections modify cell structural morphology and remodel the Golgi complex as a viral factory during the entire life cycle. Furthermore, host secretory signaling is also crucial for innate and acquired immune responses, such as the exportation of proinflammatory and antiviral cytokines. Nearly 25 years ago, Doedens et al. reported that the 2B and 3A proteins of PV inhibited cellular protein secretion by directly blocking the transportation from the ER to the Golgi apparatus [129], indicating that the functional secretory protein is not indispensable for viral RNA replication. Mechanistically, Dodd et al. identified that the inhibition of 3A protein of PV on the ER to Golgi limited the antiviral cytokine secretion of native immune response and inflammation, including interleukin-6, interleukin-8, and β-interferon [130]. Deitz et al. also identified that PV 3A protein reduced the presentation of new antigens on the cell surface [131]. Considering that the ER adaptor STING was also located on the Golgi and ERGIC [93], we hypothesized that the membrane remodeling and modification of Golgi induced by viruses might also be involved in the regulation of cGAS-STING pathways (Figure 3). Collectively, all these data suggest that enteroviruses, such as PV and CVB, have evolved a series of strategies to hijack cellular trafficking and secretion for viral replication.
2.5. Role of the Lysosome and Endosome in Viral Infections
The lysosome, an acidic and membrane-bound organelle, acts as a cellular recycling center and is filled with a number of hydrolases [152]. The lysosome-based degradation processes are subject to reciprocal regulation [153]. Lysosomes degrade unwanted materials that are delivered either from outside via the endocytic pathway or from inside via the autophagic pathway [153,154]. For viral replication and assembly, certain viruses, including Alphaviruses [146], such as semliki forest virus (SFV) [145], exploit the membrane surface of the endosome and lysosome as a viral factory. Similarly, RUBV also can modify the membrane of lysosomes for a viral factory [142]. Meanwhile, the Toll-like receptors (TLR), such as TLR 3/7/9, are located on the endosome, indicating that the endosome also plays an important role in innate immunity [94]. Therefore, we speculate that another possible strategy is that viruses, particularly DNA viruses, evade the TLR-mediated activation of the NF-κB and transcription of proinflammatory cytokines. HBV infection suppresses TLR-9 expression and prevents TLR9 promoter activity in human primary B cells [155]. Interestingly, DENV, a positive-stand RNA virus, activates the TLR9 by inducing mtDNA release in human dendritic cells [156]. Additionally, the endosomal-lysosomal sorting system is a complex and dynamic vesicular sorting signaling, which is fundamental to maintain homeostasis [157]. Viruses, particularly enveloped viruses such as HIV [151], have evolved several strategies to hijack the endosomal sorting complex required for the transport (ESCRT) complex to facilitate viral budding. Collectively, all these data indicate that different viruses utilize different strategies to hijack the endosome/lysosome for viral progeny.
3. Degradation of Intracellular Organelles by Virus-Triggered Selective Autophagy
Macroautophagy was initially described as a non-selective degradation process [33]. However, selective autophagy is characterized as a highly regulated and specific degradation pathway targeting damaged organelles [158]. The initiation of autophagy includes the formation of the phagophore from membrane precursors. The phagophore elongates by the ubiquitin-like conjugation systems and LC3-II-phosphatidylethanolamine to form the autophagosome [33]. The autophagosome sequesters within damaged intracellular organelles, such as the mitochondria, ER, peroxisome, nucleus, and lysosome, and undergoes fusion with a lysosome to form an autolysosome, where degradation occurs [33]. Depending on the targeted organelles, selective autophagy can be divided into mitophagy, pexophagy, ER-phage, lysophagy, nucleophagy, etc. [33] (Figure 3). In most cases, viral infection leads to the severe damage of intracellular organelles, which subsequently initiates selective autophagy to degrade these damaged organelles. Therefore, selective autophagy is the protective mechanism for cells to maintain cell homeostasis. In contrast, in some cases, selective autophagy could be utilized by a virus to promote their replication. Here, we address the principal mechanism of selective autophagy triggered by viral infection, with an emphasis on mitophagy and pexophagy, which has been best described to date.
3.1. Overview of Mitophagy and Pexophagy Signaling
The selective elimination of damaged mitochondria is termed mitophagy and is a type of macroautophagy [6] (Figure 3). The fragmented mitochondria are easier to recognize by autophagosome than the elongated mitochondria because of imbalanced fission–fusion of the mitochondria [160,161,162,163]. The canonical ubiquitin-dependent PTEN-induced putative kinase protein 1(PINK1)-Parkin mitophagy signal has been validated in multiple model systems by different approaches [164,165,166]. Mitochondrial PINK1 is rapidly turned over on the bioenergetically well-coupled mitochondria by proteolysis by presenilin-associated rhomboid like protein (PARL) [167], but PINK1 is selectively stabilized on the mitochondria with loss of membrane potential (Δψm) [168,169]. Selective accumulation of PINK1 recruits downstream Parkin, a cytosolic RING-between-RING E3 ligase, to the impaired mitochondria [169]. In turn, Parkin-induced mitophagy is strictly dependent on depolarization-induced accumulation [167,169]. Chen et al. previously reported that PINK1-phosphorylated Mfn2 as a receptor-mediated Parkin recruitment to the damaged mitochondria [166]. Meanwhile, the phosphorylated-ubiquitin on ser 65 (p-Ser65-Ub) chains is also identified as a potent Parkin activator and receptor, which leads to the onset of mitophagy [170,171,172]. Notably, Lazarou et al. recently reported that PINK1 recruits NDP52 and OPTN cargo receptors, but not p62/SQSTM1, to directly activate mitophagy, independently of Parkin [173]. Similarly, to protect against ischemic brain injury, BNIP3L/NIX-mediated mitophagy is independent of Parkin [19,20]. Furthermore, upon mitophagy induction, AMBRA1 binds to the autophagosome adapter LC3 to initiate a powerful mitophagy, promoting canonical Parkin PINK1-dependent and Parkin-independent mitochondrial clearance [13]. All the aforementioned data show that Parkin may act as an amplifier, which is not indispensable for mitophagy. Intriguingly, the protein kinase PINK1 and Parkin are also involved in the generation of MDVs [174].
Peroxisome homeostasis is regulated by division and pexophagic degradation. The degradation of the aberrant peroxisomes by selective autophagy is known as pexophagy [33,114,175] (Figure 3). Four different types of pexophagy are identified in mammalian cells [115], including ubiquitin-mediated pexophagy [176], NBR1-induced pexophagy [177], PEX3-induced pexophagy [178], and PEX14-LC3 interaction-mediated pexophagy [179]. Compared with pexophagy in yeast [114], the underlying mechanisms of pexophagy in mammalian cells are less elucidated. The ubiquitination of mammalian PEX5 [180,181] and PMP70 [181] has been identified in pexophagy. In response to reaction oxygen species, ataxia-telangiectasia-mutated kinase phosphorylates PEX5 and eventually links the peroxisome with the adaptor p62/SQSTM1 for pexophagy [180]. During amino acid starvation, the peroxisomal E3 ubiquitin ligase PEX-2 ubiquitinates downstream PEX5 and PMP70 and subsequently degrades peroxisome in an NBR1-dependent manner [181].
Under various stresses, complex signaling pathways are involved in the activation and regulation of mitophagy and pexophagy. The detailed mechanism needs to be further elucidated in future research.
3.2. Virus Triggers Mitophagy and Pexophagy to Suppress MAVS-Dependent RLR Signaling
The RLR-dependent type I interferon responses are regulated by MAVSs, which are initially thought to be only located in the OMM of the mitochondria [182]. Upon viral infection, MAVSs bind to RLRs and promote the activation of downstream signal transduction [93,94]. Khan et al. reported that HCV attenuated the innate immunity via Parkin-dependent recruitment linear ubiquitin assembly complex to the mitochondria [183]. Similarly, Edmonston strain (MV-Edm), an attenuated MV, triggered MAVS degradation via p62/SQSTM1-mediated mitophagy to weaken the RLR signal [66]. The matrix protein (M) of HPIV3 [184] and PB1-F2 of IAV [185] induce the mitophagy Parkin-PINK1-independent pathway to suppress innate immunity. More importantly, Dixit et al. recently identified that partial MAVSs are also located on the peroxisome for antiviral signal transduction [159]. Upon viral infection, the mitochondria and peroxisomes are not just simple metabolic organelles, but rather serve as a critical subcellular platform for antiviral immunity, which expands our understanding about the integration of antiviral networks of the intracellular organelles. Mitochondrial MAVSs may mediate the proapoptotic signaling of innate immune activity against viral infections [186,187]. Previous reports suggested that HCV proteolytically cleaved the MAVS from the mitochondria by NS3/4A [188,189,190] at cysteine 508 amino residue rather than degrading the MAVS to cripple innate immunity [191]. Horner et al. further identified that NS3/4A of HCV targeted the mitochondrial-associated membrane (MAM) and cleaved MAVSs from the MAM, but not from the mitochondria to ablate the immune actions of the MAVS signalosome during HCV infection [192]. Taken together, various viruses have evolved a plethora of strategies to exploit mitophagy and pexophagy to suppress MAVS-dependent RLR signaling to maximize their own replication.
3.3. ER-Phagy (reticulophagy) Trigged by Viral Infection
Different viruses have exploited different strategies to utilize the UPR of the ER for viral replication (Table 1). In our laboratory, we found that NDV activated all three branches of the UPR of the ER to facilitate NDV replication [105]. Synergistic expression of HN and F of virulent NDV is necessary for the UPR activation of the ER [88]. However, the exact mechanism of how a virus leads to the accumulation of unfolded proteins in the lumen and utilizes the UPR of the ER needs to be further investigated. The selective degradation of ER is termed ER-phagy [193,194]. Upon the stimulation of an ER stress inducer, the signaling networks of ATF6, PERK, and IRE1α, and cellular Ca2+, are necessary for the activation of ER-phagy at different stages, including induction, vesicle nucleation, and elongation [194] (Figure 3). PERK and IRE1α branches of ER stress are indispensable for DENV-induced autophagy [98]. To date, FAM134 [25], BNIP3/Nix [21], and p62/SQSTM1 [12] have been identified as cargo receptors of ER-phagy. Additionally, Tomar et al. [195] reported that TRIM13, an ER-resident ubiquitin E3 ligase, regulates the ER-phagy process and ER stress. Considering the important role of the ER-localized STING in anti-viral innate immunity [93,95], we speculate that ER-phagy may be involved in the inhibition of the cGAS-STING pathway during virus infections, particularly DNA virus infection (Figure 3). The precise underlying mechanisms of ER-phagy upon viral infection need to be further investigated.
3.4. Lysophagy and Nucleophagy Trigged by Viral Infection
The selective autophagy is initiated by isolation membranes. Subsequently, the isolation membranes are close to one another to form double membrane bound autophagosomes, and eventually fuse with lysosomes for degradation [6,34]. Notably, the autophagosome does not have the ability to degrade its contents. Only after fusion with the lysosome, which provides an acidic environment and hydrolases, can the autophagosome degrade the autophagosomal contents. Numerous inducers can trigger lysosomal membrane permeabilization and the consequent leakage of the lysosomal content into the cytosol, which eventually leads to so-called “lysosomal cell death” [152]. The removal of damaged lysosomes by selected autophagy is termed lysophagy [33,34] (Figure 3). Moreover, nucleophagy is a selective autophagy, which selectively removes damaged or non-essential material by the autophagy pathway [113,196] (Figure 3). Recently, Unterholznoer et al. have identified IFI16, a PYHIN protein, as a new DNA sensor [197]. Considering the nuclear distribution of IFI16, we speculate that nucleophagy may involve the IFI16-dependent innate immunity (Figure 3). Compared with mitophagy and pexophagy, many questions regarding the molecular details of lysophagy and nucleophagy pathways should be further elucidated. One of the interesting questions is how the nucleus and lysosome are sequestered by the phagophore and recognized by the cargo adaptor in response to viral infections.
4. Concluding Remarks and Future Perspectives
In the current review, we present a brief overview of the quality control strategies of intracellular organelles in mammalian cells in response to viral infection. Although distinct steps of the viral life cycle have long been known to associate with the abundant membrane rearrangement of intracellular organelles [4], a detailed understanding of the interplay of virus and host, particularly the interaction between individual viral protein and organelle component, has remained unclear. Several important scientific questions remain unelucidated. First, what is the mechanism to coerce the host translational machinery into synthesizing viral proteins in the face of ongoing infectious progeny? Second, how do viral and cellular proteins contribute to the re-construction of viral replication factories in different subcellular membranes sites? Third, what are the viral proteins and cellular factors that are required for membrane remodeling and that metabolize reprogramming in virus-infected cells?
Moreover, although we have made great progress in the understanding of selective autophagy, the assembly site of a double membrane has remained unclear. The assembly of the phagophore may require various membranes, including the ER [198], ER-Golgi intermediate compartments (ERGIC) [199], ER-mitochondria junctions [200], mitochondria [201], mitochondrial-derived vesicles (MDVs) [202], Golgi-endosomal membranes [203], and the plasma membrane [204]. Upon DNA or RNA viral infection, further work is needed to decipher the exact phagophore assembly site of selection autophagy during viral infections. More importantly it remains unclear how the host cell initiates the “eat-me” signal for the elimination and the elongation of the phagophore membrane around targeted organelles. Every selective autophagy pathway requires a specific cargo receptor, which bridges ubiquitinated organelles to LC3/Atg8 family membranes to link with the autophagy machinery. In mammalian cells, several cargo proteins, including p62/SQSTM1, CALCOCO2/NDP52, NBR1, optineurin/OPTN, AMBRA1, BNIP3L/NIX, BNIP3, FUNDC1, TAXIBP1, cardiolipin, prohibitin-2/PHB-2, and FAM134B (Table 3), have been identified; however, the exact processes of recognition and specific selection of damaged organelles for degradation by selective autophagosomes during viral infection remain poorly understood. Notably, Lazarou et al. identified that NDP52 and OPTN are the primary receptors for PINK1-dependent mitophagy, independent of Parkin. PINK1-generated phospho-ubiquitin directly serves as the “eat-me” signal on the mitochondria [173], which extends our understanding of classical PINK1-Parkin mitophagy signaling. Furthermore, Tank-binding kinase 1 is involved in the phosphorylation of cargo receptors, including OPTN and NDP52, to create an “eat-me” signal at the autophagy-relevant site [15]. The difference between the autophagy receptor NDP52 and p62 determines the species-specific impact of the autophagy machinery during CHIKV infection, indicating that a receptor may regulate viral infection in a species-dependent manner [205]. Recently, cardiolipin of the inner mitochondrial membrane phospholipid was found to serve as an “eat-me” signal for mitochondrial clearance from neuronal cells [24]. Meanwhile, Wei et al. [22] recently identified the prohibitin-2/PHB-2 as a specific mitophagy receptor of IMM for autophagic degradation. Interestingly, the matrix protein of HPIV [184] and the PB1-F2 protein of IAV [185] can be a receptor in the induction of mitophagy. Specific selection of cargo proteins during damaged organelle degradation may be primarily dependent on the targeted organelle and viral characteristic.
Extensive improvement of our understanding of the cross-talk between viruses and organelles must depend on the innovative applications of new techniques and materials [206], such as the single-particle tracking method, the ribopuromycylation method, single cell RNA-seq, three-dimensional (3D)-reconstruction of electron microscopy, image-based genome-wide RNA interference screens, haploid genetic screens, yeast two-hybrid screens, a modern ultra-structural technique, and, in particular, a high-throughput and genome-scale CRISPR-Cas screening technique. Electron tomography and 3D imaging technology are being successfully applied to the study of virus-cell interactions, such as for EAV [84], RUBV [56], and BUNV [57]. Similarly, Ertel et al. recently revealed new interior and exterior features of RNA replication compartments of the non-human flock house nodavirus via a cryo-electron tomography technique [144]. Based on image-based genome-wide siRNA screening, Orvedahi et al. identified that SMAD specific E3 ubiquitin protein ligase 1 (SMURF1), as a newly recognized mediator, functions in mitophagy triggered by viral infection but not in starvation-induced autophagy [207]. Researchers should keep a closer eye on advanced technological breakthroughs and combine these advanced technological breakthroughs into a comprehensive understanding of virus–organelle interaction. The discovery of the underlying virus–host molecular mechanism already has an overlapping function to multiple viruses, which advance the discovery of host druggable targets and development of broad-spectrum antiviral approaches.
Author Contributions
S.R. wrote the manuscript draft. S.R. and Y.S. prepared and drew the schematic diagram. Y.S. and C.D. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Key Research and Development Program of China (No. 2018YFD0500100) and the National Natural Science Foundation of China (No. 31872453, 31530074).
Conflicts of Interest
All listed authors declare no competing financial interest.
Abbreviations
| ATF6 | Activating transcription factor 6 |
| ATP | Adenosine triphosphate |
| AMBRA1 | Autophagy and beclin-1 regulator 1 |
| ASFV | African swine fever virus |
| BUNV | Bunyamwera virus |
| BNIP3L/NIX | BCL2 interacting protein 3 like |
| BNIP3 | BCL2 interacting protein 3 |
| cGAS | Cyclic GMP-AMP synthase |
| CVB | Coxsackievirus B |
| CHIKV | Chikungunya virus |
| CALCOCO2/NDP52 | Calcium binding and coiled-coil domain 2/Antigen nuclear dot 52 kDa protein |
| COP | Coat protein complex |
| CV | Coxsackieviruses |
| DENV | Dengue virus |
| DRP-1 | Dynamic relative GTPase protein |
| ERGIC | ER-Golgi-intermediate compartment |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| EAV | Equine arteritis virus |
| FHV | Flock house virus |
| FUNDC1 | FUN14 domain containing 1 |
| FAM134B | Family with sequence similarity 134, member B |
| HIV | Human immune deficiency |
| HPIV3 | Human parainfluenza virus typ3 |
| HCV | Hepatitis C virus |
| HSV-1 | Human herpes simplex virus-1 |
| HBV | Hepatitis B virus |
| IAV | Influenza A virus |
| IMM | Inner mitochondrial membranes |
| IBV | Avian coronavirus infectious bronchitis virus |
| IAV | Influenza A virus |
| IRE1α | Inositol requiring enzyme 1α |
| JEV | Japanese encephalitis virus |
| LC3 | Light chain 3 |
| LIR | LC3-interacting region |
| MHV | Murine hepatitis virus |
| Mfns | Mitofusin GTPases |
| Mfn1 | Mitofusin 1 |
| Mfn2 | Mitofusin 2 |
| Mff | Mitochondrial fission factor |
| MAM | Mitochondrial-associated-membrane |
| MCMV | Murine cytomegalovirus |
| MV | Measles virus |
| MAPL | Mitochondrial-anchored protein ligase |
| MDVs | Mitochondrial-derived vesicles |
| MAVS | Mitochondrial antiviral signaling protein |
| NDV | Newcastle disease virus |
| NBR1 | Next to BRCA1 gene 1 protein |
| OMM | Out mitochondrial membranes |
| OPA1 | Optic atrophy 1 |
| OPTN | Optineurin |
| PERK | Double-stranded RNA-activated protein kinase-like kinase |
| PRRSV | Porcine reproductive and respiratory syndrome virus |
| PHB2 | Prohibitin 2 |
| PEX | Peroxin |
| PV | Polioviruses |
| PINK1 | PTEN-induced putative kinase protein 1 |
| RUBV | Rubella virus |
| ROS | Reactive oxygen species |
| Ref | Reference |
| RIG-I | Retinoic acid-inducible gene I protein |
| STING | Stimulator of interferon genes |
| SIV | Sindbis virus |
| SFV | Semliki forest virus |
| SARS | Severe acute respiratory syndrome |
| TBSV | Tomato bushy stunt virus |
| TBEV | Tick-borne encephalitis virus |
| TAXIBP1 | Tax1 binding protein 1 |
| UPR | Unfolded proteins response |
| UPRmt | Unfolded protein response (UPR) of mitochondria |
References
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011, 9, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Helenius, A. Virus entry at a glance. J. Cell Sci. 2013, 126, 1289–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Paul, P.; Munz, C. Autophagy and Mammalian Viruses: Roles in Immune Response, Viral Replication, and Beyond. Adv. Virus Res. 2016, 95, 149–195. [Google Scholar]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. BioMed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ni, H.M.; Guo, F.; Ding, Y.; Shi, Y.H.; Lahiri, P.; Frohlich, L.F.; Rulicke, T.; Smole, C.; Schmidt, V.C.; et al. Sequestosome 1/p62 Protein Is Associated with Autophagic Removal of Excess Hepatic Endoplasmic Reticulum in Mice. J. Biol. Chem. 2016, 291, 18663–18674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Tumbarello, D.A.; Manna, P.T.; Allen, M.; Bycroft, M.; Arden, S.D.; Kendrick-Jones, J.; Buss, F. The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PLoS Pathog. 2015, 11, e1005174. [Google Scholar] [CrossRef] [Green Version]
- Till, A.; Lipinski, S.; Ellinghaus, D.; Mayr, G.; Subramani, S.; Rosenstiel, P.; Franke, A. Autophagy receptor CALCOCO2/NDP52 takes center stage in Crohn disease. Autophagy 2013, 9, 1256–1257. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e10. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Chang, B.; Brulois, K.F.; Castro, K.; Min, C.K.; Rodgers, M.A.; Shi, M.; Ge, J.; Feng, P.; Oh, B.H.; et al. Kaposi’s sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 2013, 87, 12499–12503. [Google Scholar] [CrossRef] [Green Version]
- Mouna, L.; Hernandez, E.; Bonte, D.; Brost, R.; Amazit, L.; Delgui, L.R.; Brune, W.; Geballe, A.P.; Beau, I.; Esclatine, A. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 2016, 12, 327–342. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Lazarow, P.B. Viruses exploiting peroxisomes. Curr. Opin. Microbiol. 2011, 14, 458–469. [Google Scholar] [CrossRef]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 2015, 4, e08828. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Yoon, Y. Mitochondrial fission: Regulation and ER connection. Mol. Cells 2014, 37, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Castello, A.; Quintas, A.; Sanchez, E.G.; Sabina, P.; Nogal, M.; Carrasco, L.; Revilla, Y. Regulation of host translational machinery by African swine fever virus. PLoS Pathog. 2009, 5, e1000562. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, K.A.; Camarda, R.; Lagunoff, M. Vaccinia virus requires glutamine but not glucose for efficient replication. J. Virol. 2014, 88, 4366–4374. [Google Scholar] [CrossRef] [Green Version]
- Fontana, J.; Lopez-Iglesias, C.; Tzeng, W.P.; Frey, T.K.; Fernandez, J.J.; Risco, C. Three-dimensional structure of Rubella virus factories. Virology 2010, 405, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Fontana, J.; Lopez-Montero, N.; Elliott, R.M.; Fernandez, J.J.; Risco, C. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cell Microbiol. 2008, 10, 2012–2028. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014, 25, 528–537. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, K.; Oshiumi, H.; Takeda, M.; Ishihara, N.; Yanagi, Y.; Seya, T.; Kawabata, S.; Koshiba, T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009, 2, ra47. [Google Scholar] [CrossRef] [PubMed]
- Onoguchi, K.; Onomoto, K.; Takamatsu, S.; Jogi, M.; Takemura, A.; Morimoto, S.; Julkunen, I.; Namiki, H.; Yoneyama, M.; Fujita, T. Virus-infection or 5’ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. 2010, 6, e1001012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Martinvalet, D. The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 2018, 9, 336. [Google Scholar] [CrossRef]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.M.; Beltinger, C.; Wei, J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [Green Version]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, R.E.; Wang, S.; Liu, T.Y.; Rapoport, T.A. Reconstitution of the tubular endoplasmic reticulum network with purified components. Nature 2017, 543, 257–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verchot, J. How does the stressed out ER find relief during virus infection? Curr. Opin. Virol. 2016, 17, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.J.; Xiong, Z.; Lu, X.; Ye, M.; Han, X.; Jiang, R. ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cell. Signal. 2014, 26, 332–342. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629. [Google Scholar] [CrossRef]
- Moon, S.L.; Barnhart, M.D.; Wilusz, J. Inhibition and avoidance of mRNA degradation by RNA viruses. Curr. Opin. Microbiol. 2012, 15, 500–505. [Google Scholar] [CrossRef] [Green Version]
- Tenorio, R.; Fernandez de Castro, I.; Knowlton, J.J.; Zamora, P.F.; Lee, C.H.; Mainou, B.A.; Dermody, T.S.; Risco, C. Reovirus sigmaNS and muNS Proteins Remodel the Endoplasmic Reticulum to Build Replication Neo-Organelles. mBio 2018, 9, e01253-18. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risco, C.; Rodriguez, J.R.; Lopez-Iglesias, C.; Carrascosa, J.L.; Esteban, M.; Rodriguez, D. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 2002, 76, 1839–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husain, M.; Moss, B. Evidence against an essential role of COPII-mediated cargo transport to the endoplasmic reticulum-Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J. Virol. 2003, 77, 11754–11766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoops, K.; Barcena, M.; Limpens, R.W.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Ultrastructural characterization of arterivirus replication structures: Reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 2012, 86, 2474–2487. [Google Scholar] [CrossRef] [Green Version]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.M.; Garcia-Escudero, R.; Salas, M.L.; Andres, G. African swine fever virus structural protein p54 is essential for the recruitment of envelope precursors to assembly sites. J. Virol. 2004, 78, 4299–4313. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Chai, Y.; Song, J.; Liu, T.; Chen, P.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. Reprogramming the unfolded protein response for replication by porcine reproductive and respiratory syndrome virus. PLoS Pathog. 2019, 15, e1008169. [Google Scholar] [CrossRef]
- Ren, S.; Rehman, Z.U.; Shi, M.; Yang, B.; Liu, P.; Yin, Y.; Qu, Y.; Meng, C.; Yang, Z.; Gao, X.; et al. Hemagglutinin-neuraminidase and fusion proteins of virulent Newcastle disease virus cooperatively disturb fusion–fission homeostasis to enhance mitochondrial function by activating the unfolded protein response of endoplasmic reticulum and mitochondrial stress. Vet. Res. 2019, 50, 37. [Google Scholar]
- Targett-Adams, P.; Boulant, S.; Douglas, M.W.; McLauchlan, J. Lipid metabolism and HCV infection. Viruses 2010, 2, 1195–1217. [Google Scholar] [CrossRef] [Green Version]
- Cobbold, C.; Brookes, S.M.; Wileman, T. Biochemical requirements of virus wrapping by the endoplasmic reticulum: Involvement of ATP and endoplasmic reticulum calcium store during envelopment of African swine fever virus. J. Virol. 2000, 74, 2151–2160. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Palaniyandi, S.; Zhu, I.; Tang, J.; Li, W.; Wu, X.; Ochsner, S.P.; Pauza, C.D.; Cohen, J.I.; Zhu, X. Human cytomegalovirus evades antibody-mediated immunity through endoplasmic reticulum-associated degradation of the FcRn receptor. Nat. Commun. 2019, 10, 3020. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.I.; Ko, D.H.; Shin, N.; Pyo, C.W.; Choi, S.Y. Endoplasmic reticulum-associated degradation potentiates the infectivity of influenza A virus by regulating the host redox state. Free Radic. Biol. Med. 2019, 135, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Fung, T.S.; Liao, Y.; Liu, D.X. The endoplasmic reticulum stress sensor IRE1alpha protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Kuo, S.H.; Lin, C.Y.; Fu, P.J.; Lin, Y.S.; Yeh, T.M.; Liu, H.S. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 2018, 8, 489. [Google Scholar] [CrossRef]
- Wang, Q.; Xin, X.; Wang, T.; Wan, J.; Ou, Y.; Yang, Z.; Yu, Q.; Zhu, L.; Guo, Y.; Wu, Y.; et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J. Virol. 2019, 93, e00887-19. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Xu, A.; Wu, X.; Zhang, Y.; Guo, Y.; Guo, F.; Pan, Z.; Kong, L. Japanese encephalitis virus induces apoptosis by the IRE1/JNK pathway of ER stress response in BHK-21 cells. Arch. Virol. 2016, 161, 699–703. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Achazi, K.; Niedrig, M. Tick-borne encephalitis virus triggers inositol-requiring enzyme 1 (IRE1) and transcription factor 6 (ATF6) pathways of unfolded protein response. Virus Res. 2013, 178, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, I.H.; Zhang, M.S.; Powers, L.S.; Shao, J.Q.; Baltrusaitis, J.; Rutkowski, D.T.; Legge, K.; Monick, M.M. Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 2012, 287, 4679–4689. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, W.; Niu, Q.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; Ding, C. eIF2alpha-CHOP-BCl-2/JNK and IRE1alpha-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Disease 2019, 10, 891. [Google Scholar] [CrossRef] [Green Version]
- Stahl, S.; Burkhart, J.M.; Hinte, F.; Tirosh, B.; Mohr, H.; Zahedi, R.P.; Sickmann, A.; Ruzsics, Z.; Budt, M.; Brune, W. Cytomegalovirus downregulates IRE1 to repress the unfolded protein response. PLoS Pathog. 2013, 9, e1003544. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Chapa, T.J.; Gualberto, N.; Yu, D. Murine cytomegalovirus targets transcription factor ATF4 to exploit the unfolded-protein response. J. Virol. 2012, 86, 6712–6723. [Google Scholar] [CrossRef] [Green Version]
- Burnett, H.F.; Audas, T.E.; Liang, G.; Lu, R.R. Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones 2012, 17, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Su, C.; Jiang, Z.; Zheng, C. Herpes Simplex Virus 1 UL41 Protein Suppresses the IRE1/XBP1 Signal Pathway of the Unfolded Protein Response via Its RNase Activity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma (1) 34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, M.; Arias, C.; Mohr, I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 2007, 81, 3377–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Gao, B.; Ye, L.; Han, X.; Wang, W.; Kong, L.; Fang, X.; Zeng, Y.; Zheng, H.; Li, S.; et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007, 124, 44–49. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Till, A.; Lakhani, R.; Burnett, S.F.; Subramani, S. Pexophagy: The selective degradation of peroxisomes. Int. J. Cell Biol. 2012, 2012, 512721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honsho, M.; Yamashita, S.-I.; Fujiki, Y. Peroxisome homeostasis: Mechanisms of division and selective degradation of peroxisomes in mammals. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 984–991. [Google Scholar] [CrossRef]
- Delille, H.K.; Schrader, M. Targeting of hFis1 to peroxisomes is mediated by Pex19p. J. Biol. Chem. 2008, 283, 31107–31115. [Google Scholar] [CrossRef] [Green Version]
- Schrader, M. Shared components of mitochondrial and peroxisomal division. Biochim. Biophys. Acta 2006, 1763, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010, 20, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Mohan, K.V.; Som, I.; Atreya, C.D. Identification of a type 1 peroxisomal targeting signal in a viral protein and demonstration of its targeting to the organelle. J. Virol. 2002, 76, 2543–2547. [Google Scholar] [CrossRef] [Green Version]
- Aguado, B.; Vinuela, E.; Alcami, A. African swine fever virus fatty acid acylated proteins. Virology 1991, 185, 942–945. [Google Scholar] [CrossRef]
- McCartney, A.W.; Greenwood, J.S.; Fabian, M.R.; White, K.A.; Mullen, R.T. Localization of the tomato bushy stunt virus replication protein p33 reveals a peroxisome-to-endoplasmic reticulum sorting pathway. Plant Cell 2005, 17, 3513–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonczyk, M.; Pathak, K.B.; Sharma, M.; Nagy, P.D. Exploiting alternative subcellular location for replication: Tombusvirus replication switches to the endoplasmic reticulum in the absence of peroxisomes. Virology 2007, 362, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James Morre, D.; Mollenhauer, H.H. Microscopic morphology and the origins of the membrane maturation model of Golgi apparatus function. Int. Rev. Cytol. 2007, 262, 191–218. [Google Scholar]
- Barlowe, C.; Orci, L.; Yeung, T.; Hosobuchi, M.; Hamamoto, S.; Salama, N.; Rexach, M.F.; Ravazzola, M.; Amherdt, M.; Schekman, R. COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994, 77, 895–907. [Google Scholar] [CrossRef]
- Rabouille, C.; Klumperman, J. Opinion: The maturing role of COPI vesicles in intra-Golgi transport. Nat. Rev. Mol. Cell Biol. 2005, 6, 812–817. [Google Scholar] [CrossRef]
- Belov, G.A.; Altan-Bonnet, N.; Kovtunovych, G.; Jackson, C.L.; Lippincott-Schwartz, J.; Ehrenfeld, E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 2007, 81, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Rust, R.C.; Landmann, L.; Gosert, R.; Tang, B.L.; Hong, W.; Hauri, H.P.; Egger, D.; Bienz, K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 2001, 75, 9808–9818. [Google Scholar] [CrossRef] [Green Version]
- Doedens, J.R.; Kirkegaard, K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995, 14, 894–907. [Google Scholar] [CrossRef]
- Dodd, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J. Virol. 2001, 75, 8158–8165. [Google Scholar] [CrossRef] [Green Version]
- Deitz, S.B.; Dodd, D.A.; Cooper, S.; Parham, P.; Kirkegaard, K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 2000, 97, 13790–13795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Beato, R.; Salas, M.L.; Vinuela, E.; Salas, J. Role of the host cell nucleus in the replication of African swine fever virus DNA. Virology 1992, 188, 637–649. [Google Scholar] [CrossRef]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Katsafanas, G.C.; Moss, B. Colocalization of Transcription and Translation within Cytoplasmic Poxvirus Factories Coordinates Viral Expression and Subjugates Host Functions. Cell Host Microbe 2007, 2, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, K.W.; van der Meer, Y.; Roos, N.; Snijder, E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar] [CrossRef] [Green Version]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J.; Moscona, A. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Proteins 3, 4, and 6 Induce Double-Membrane Vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [Green Version]
- Bost, A.G.; Prentice, E.; Denison, M.R. Mouse hepatitis virus replicase protein complexes are translocated to sites of M protein accumulation in the ERGIC at late times of infection. Virology 2001, 285, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weber, F.; Kochs, G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef] [Green Version]
- Mohl, B.P.; Bartlett, C.; Mankouri, J.; Harris, M. Early events in the generation of autophagosomes are required for the formation of membrane structures involved in hepatitis C virus genome replication. J. Gen. Virol. 2016, 97, 680–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magliano, D.; Marshall, J.A.; Bowden, D.S.; Vardaxis, N.; Meanger, J.; Lee, J.Y. Rubella virus replication complexes are virus-modified lysosomes. Virology 1998, 240, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.J.; Schwartz, M.D.; Ahlquist, P. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J. Virol. 2001, 75, 11664–11676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertel, K.J.; Benefield, D.; Castano-Diez, D.; Pennington, J.G.; Horswill, M.; den Boon, J.A.; Otegui, M.S.; Ahlquist, P. Cryo-electron tomography reveals novel features of a viral RNA replication compartment. eLife 2017, 6, e25940. [Google Scholar] [CrossRef]
- Kujala, P.; Ikaheimonen, A.; Ehsani, N.; Vihinen, H.; Auvinen, P.; Kaariainen, L. Biogenesis of the Semliki Forest Virus RNA Replication Complex. J. Virol. 2001, 75, 3873–3884. [Google Scholar] [CrossRef] [Green Version]
- Froshauer, S.; Kartenbeck, J.; Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol. 1988, 107, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Spuul, P.; Balistreri, G.; Kaariainen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest Virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef] [Green Version]
- Baggen, J.; Thibaut, H.J.; Strating, J.; van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16, 368–381. [Google Scholar] [CrossRef]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 2019, 17, 67–81. [Google Scholar] [CrossRef]
- Shi, X.; Kohl, A.; Leonard, V.H.; Li, P.; McLees, A.; Elliott, R.M. Requirement of the N-terminal region of orthobunyavirus nonstructural protein NSm for virus assembly and morphogenesis. J. Virol. 2006, 80, 8089–8099. [Google Scholar] [CrossRef] [Green Version]
- Gomez, C.; Hope, T.J. The ins and outs of HIV replication. Cell. Microbiol. 2005, 7, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Tout, I.; Gomes, M.; Ainouze, M.; Marotel, M.; Pecoul, T.; Durantel, D.; Vaccarella, S.; Dubois, B.; Loustaud-Ratti, V.; Walzer, T.; et al. Hepatitis B Virus Blocks the CRE/CREB Complex and Prevents TLR9 Transcription and Function in Human B Cells. J. Immunol. 2018, 201, 2331–2344. [Google Scholar] [CrossRef]
- Lai, J.H.; Wang, M.Y.; Huang, C.Y.; Wu, C.H.; Hung, L.F.; Yang, C.Y.; Ke, P.Y.; Luo, S.F.; Liu, S.J.; Ho, L.J. Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep. 2018, 19, e46182. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Kwon, S.K.; Lee, S.H.; Kitada, T.; Kim, J.M.; Chung, J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 2008, 377, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Koyano, F.; Kimura, M.; Kosako, H.; Saeki, Y.; Tanaka, K.; Matsuda, N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 2015, 209, 111–128. [Google Scholar] [CrossRef]
- Ordureau, A.; Heo, J.M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Zientara-Rytter, K.; Subramani, S. Autophagic degradation of peroxisomes in mammals. Biochem. Soc. Trans. 2016, 44, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [Green Version]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Abe, K.; Tatemichi, Y.; Fujiki, Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 2014, 10, 1549–1564. [Google Scholar] [CrossRef] [Green Version]
- Hara-Kuge, S.; Fujiki, Y. The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp. Cell Res. 2008, 314, 3531–3541. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. PLoS Pathog. 2016, 12, e1005693. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, L.; Li, Z.; Zhong, Y.; Tang, Q.; Qin, Y.; Chen, M. The Matrix Protein of Human Parainfluenza Virus Type 3 Induces Mitophagy that Suppresses Interferon Responses. Cell Host Microbe 2017, 21, 538–547.e4. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Holm, G.H.; Zurney, J.; Tumilasci, V.; Leveille, S.; Danthi, P.; Hiscott, J.; Sherry, B.; Dermody, T.S. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J. Biol. Chem. 2007, 282, 21953–21961. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.Y.; Chiang, R.L.; Chang, T.H.; Liao, C.L.; Lin, Y.L. The interferon stimulator mitochondrial antiviral signaling protein facilitates cell death by disrupting the mitochondrial membrane potential and by activating caspases. J. Virol. 2010, 84, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Foy, E.; Li, K.; Sumpter, R., Jr.; Loo, Y.M.; Johnson, C.L.; Wang, C.; Fish, P.M.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 2986–2991. [Google Scholar] [CrossRef] [Green Version]
- Loo, Y.M.; Owen, D.M.; Li, K.; Erickson, A.K.; Johnson, C.L.; Fish, P.M.; Carney, D.S.; Wang, T.; Ishida, H.; Yoneyama, M.; et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 6001–6006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, S.; Gallagher, C.M.; Walter, P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J. Cell Sci. 2014, 127, 4078–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Tomar, D.; Singh, R.; Singh, A.K.; Pandya, C.D.; Singh, R. TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Biochim. Biophys. Acta 2012, 1823, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2013, 2, e00947. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, Y.; Munro, S. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol. Biol. Cell 2010, 21, 3998–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Judith, D.; Mostowy, S.; Bourai, M.; Gangneux, N.; Lelek, M.; Lucas-Hourani, M.; Cayet, N.; Jacob, Y.; Prevost, M.C.; Pierre, P.; et al. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013, 14, 534–544. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Bartenschlager, R. Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells. Viruses 2015, 7, 6316–6345. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [Green Version]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Morphology remodeling and spatial redistribution of mitochondria triggered by virus infections. (A) The morphological diagram of the mitochondria. Mitochondria form a dynamic network pool, which constantly undergoes rearrangement and turnover. The equilibrium regulation of mitochondrial fusion–fission is essential to maintain the integrity of mitochondria [59]. The morphology of mitochondria was divided into hyper-fused (elongated), tubular (normal), short tubes, and fragmented [50]. (B) Regulation of mitochondrial fusion and fission. Mitochondrial fusion is mediated by mitofusin GTPases MFN1 and MFN2 at the outer mitochondrial membrane (OMM), and OPA1 at the inner mitochondrial membrane (IMM). Mitochondrial fission is driven by the fission machinery complex, which consists of DRP-1, Fis1, and MFF. Mitochondrial hyper-fusion is a pro-survival type, which can increase the ATP production and membrane potential (Δψm), and decrease reactive oxygen species (ROS) and mitophagy [50,59]. (C) Proposed model for the nuclear aggregation of mitochondria and the possible interplay among intracellular organelles in response to virus infections. * symbol indicates the possible interaction site. Representative virus that increases the interactions among intracellular organelles is shown with purple rectangle. African swine fever virus, ASFV; Rubella virus, RUBV; Bunyamwera virus, BUNV.
Figure 1.
Morphology remodeling and spatial redistribution of mitochondria triggered by virus infections. (A) The morphological diagram of the mitochondria. Mitochondria form a dynamic network pool, which constantly undergoes rearrangement and turnover. The equilibrium regulation of mitochondrial fusion–fission is essential to maintain the integrity of mitochondria [59]. The morphology of mitochondria was divided into hyper-fused (elongated), tubular (normal), short tubes, and fragmented [50]. (B) Regulation of mitochondrial fusion and fission. Mitochondrial fusion is mediated by mitofusin GTPases MFN1 and MFN2 at the outer mitochondrial membrane (OMM), and OPA1 at the inner mitochondrial membrane (IMM). Mitochondrial fission is driven by the fission machinery complex, which consists of DRP-1, Fis1, and MFF. Mitochondrial hyper-fusion is a pro-survival type, which can increase the ATP production and membrane potential (Δψm), and decrease reactive oxygen species (ROS) and mitophagy [50,59]. (C) Proposed model for the nuclear aggregation of mitochondria and the possible interplay among intracellular organelles in response to virus infections. * symbol indicates the possible interaction site. Representative virus that increases the interactions among intracellular organelles is shown with purple rectangle. African swine fever virus, ASFV; Rubella virus, RUBV; Bunyamwera virus, BUNV.
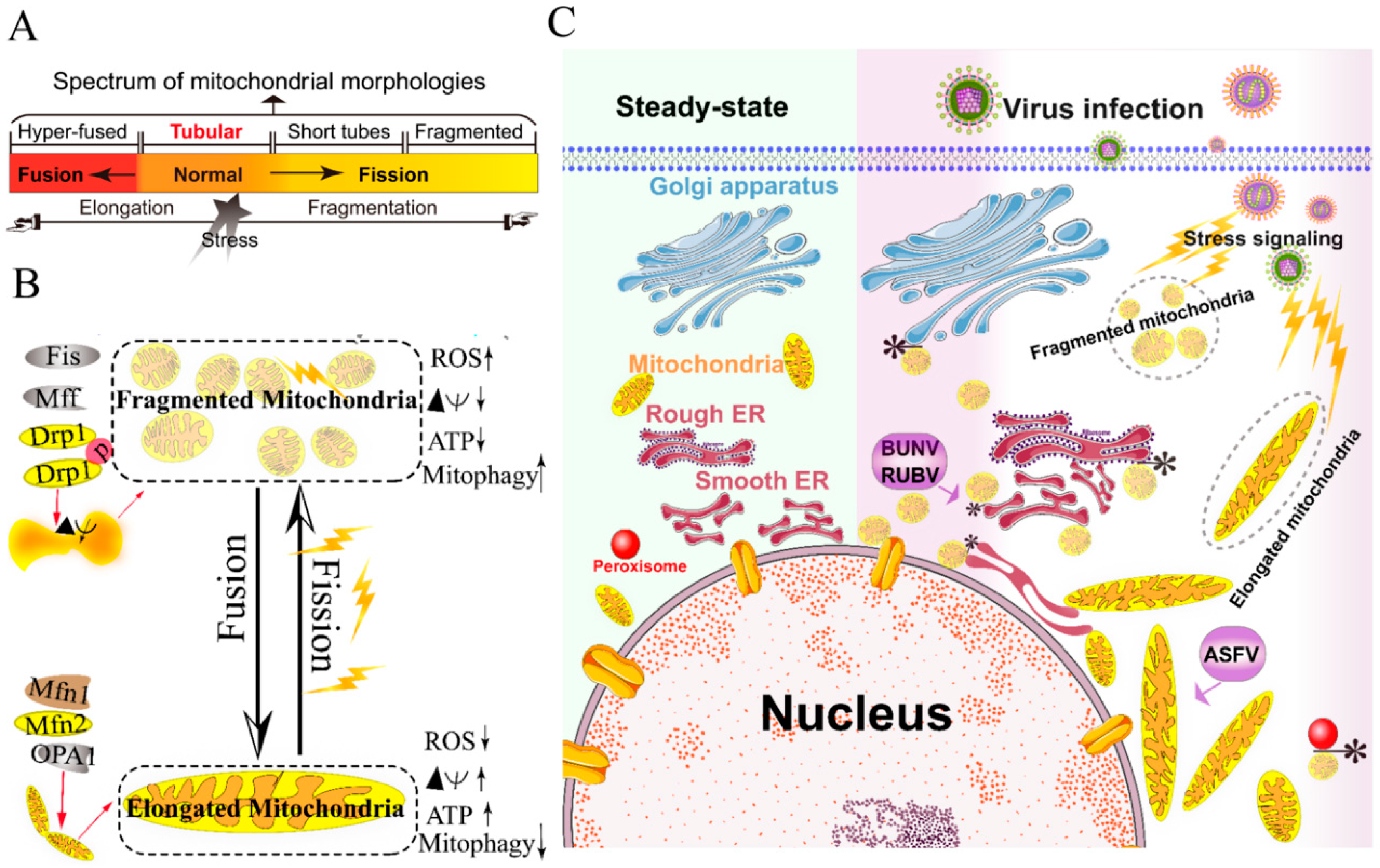
Figure 2.
Simplified diagram of the core element of the three unfolded protein response (UPR) signaling branches of the endoplasmic reticulum (ER). During different viral infections, the ER stress activates the three stress sensor proteins: IRE1α, ATF6, and PERK (detailed in reviews [77,96]). Each sensor uses a distinct mechanism of signal transduction to drive the transcription of UPR target genes and eventually work as feedback loops to mitigate the ER stress [77,96]. Upon ER stresses, ATF6, a transcriptional factor, translocate into the Golgi compartment, where it is cleaved by the site (1/2) protease. The N-terminal cytosolic domain of cleaved ATF6 is released into cytosol and then translocated into the nucleus where it binds to ER stress-response elements to activate target genes, including XBP-1 and C/EBP-homologous protein (CHOP) [76]. The activation of PERK inhibits general protein translation by the phosphorylation of eIF2α, enabling dedicated translation of transcripts, including ATF4, a key transducer. The IRE1 branch is regulated by non-conventional mRNA splicing [77,96]. Subsequently, the activated IRE1 processes XBP1 mRNA to generate the spliced form of XBP1 protein (XBP1s), which participates in the IRE1α-mediated UPR pathway in response to ER stresses [77,96]. Eventually, the activation of the cleaved ATF6 (N-ATF6), ATF4, and XBP1 transcription factors increases the protein-folding capacity in the ER lumen. Meanwhile, IRE1 and PERK sensors also decrease the load of proteins entering the ER [77,96].
Figure 2.
Simplified diagram of the core element of the three unfolded protein response (UPR) signaling branches of the endoplasmic reticulum (ER). During different viral infections, the ER stress activates the three stress sensor proteins: IRE1α, ATF6, and PERK (detailed in reviews [77,96]). Each sensor uses a distinct mechanism of signal transduction to drive the transcription of UPR target genes and eventually work as feedback loops to mitigate the ER stress [77,96]. Upon ER stresses, ATF6, a transcriptional factor, translocate into the Golgi compartment, where it is cleaved by the site (1/2) protease. The N-terminal cytosolic domain of cleaved ATF6 is released into cytosol and then translocated into the nucleus where it binds to ER stress-response elements to activate target genes, including XBP-1 and C/EBP-homologous protein (CHOP) [76]. The activation of PERK inhibits general protein translation by the phosphorylation of eIF2α, enabling dedicated translation of transcripts, including ATF4, a key transducer. The IRE1 branch is regulated by non-conventional mRNA splicing [77,96]. Subsequently, the activated IRE1 processes XBP1 mRNA to generate the spliced form of XBP1 protein (XBP1s), which participates in the IRE1α-mediated UPR pathway in response to ER stresses [77,96]. Eventually, the activation of the cleaved ATF6 (N-ATF6), ATF4, and XBP1 transcription factors increases the protein-folding capacity in the ER lumen. Meanwhile, IRE1 and PERK sensors also decrease the load of proteins entering the ER [77,96].

Figure 3.
Schematic diagram of the selective autophagy. Autophagy is a conserved catabolic process, which is artificially divided into several steps: initiation, elongation, fusion, and degradation [7]. The initiation of autophagy includes the formation of the phagophore, the initial sequestering compartment. The isolation membrane elongates and expands into a double-membrane structure called an autophagosome, which chooses its cargo (the damaged organelles indicated in this figure). Completion of the autophagosomes is followed by fusion with lysosomes to form autolysosomes, where the degradation of the cargo occurs [33]. The cargo adaptor interacts directly with the damaged intracellular organelles and an autophagy modifier of the ATG8/LC3 family, which functions as a bridge between polyubiquitinated cargo and autophagosome. The autophagy adaptors contain at least an LC3-interacting region (LIR) motif and a C-terminal ubiquitin-associated domain, which is responsible for binding to mono- and poly-ubiquitinated substrates. The selective autophagic organelles are often marked and dissipated for degradation by the addition of ubiquitin by E3 ubiquitin ligases and deubiquitinating enzymes. The adaptor mitochondrial antiviral signaling protein (MAVS) is located in both mitochondria and peroxisome [159], an important downstream adapter of RIG-I mediated antiviral signaling. The stimulator of interferon genes (STING) is located in the ER, an important downstream effector of the cGAS–STING pathway [94].
Figure 3.
Schematic diagram of the selective autophagy. Autophagy is a conserved catabolic process, which is artificially divided into several steps: initiation, elongation, fusion, and degradation [7]. The initiation of autophagy includes the formation of the phagophore, the initial sequestering compartment. The isolation membrane elongates and expands into a double-membrane structure called an autophagosome, which chooses its cargo (the damaged organelles indicated in this figure). Completion of the autophagosomes is followed by fusion with lysosomes to form autolysosomes, where the degradation of the cargo occurs [33]. The cargo adaptor interacts directly with the damaged intracellular organelles and an autophagy modifier of the ATG8/LC3 family, which functions as a bridge between polyubiquitinated cargo and autophagosome. The autophagy adaptors contain at least an LC3-interacting region (LIR) motif and a C-terminal ubiquitin-associated domain, which is responsible for binding to mono- and poly-ubiquitinated substrates. The selective autophagic organelles are often marked and dissipated for degradation by the addition of ubiquitin by E3 ubiquitin ligases and deubiquitinating enzymes. The adaptor mitochondrial antiviral signaling protein (MAVS) is located in both mitochondria and peroxisome [159], an important downstream adapter of RIG-I mediated antiviral signaling. The stimulator of interferon genes (STING) is located in the ER, an important downstream effector of the cGAS–STING pathway [94].
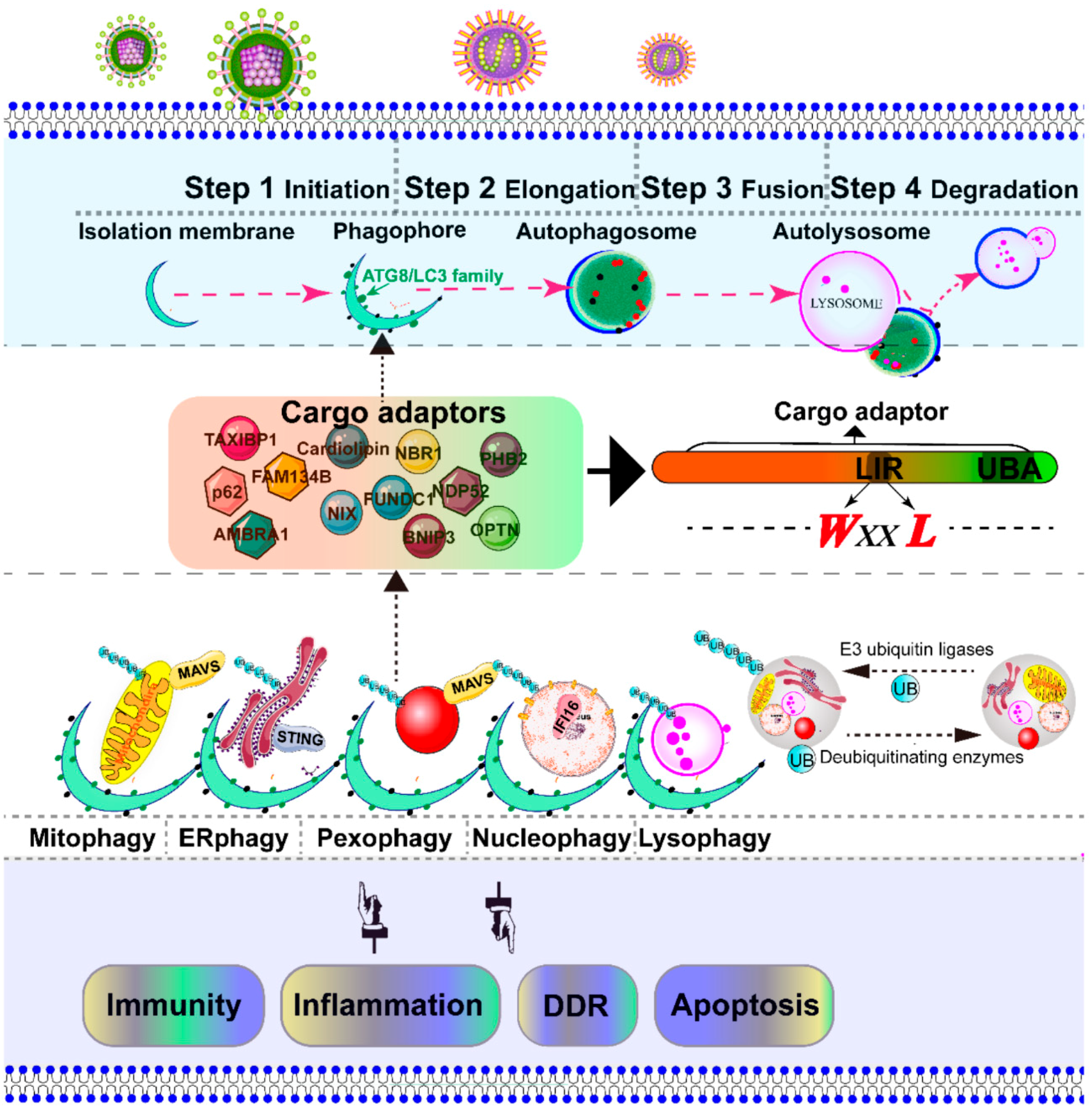

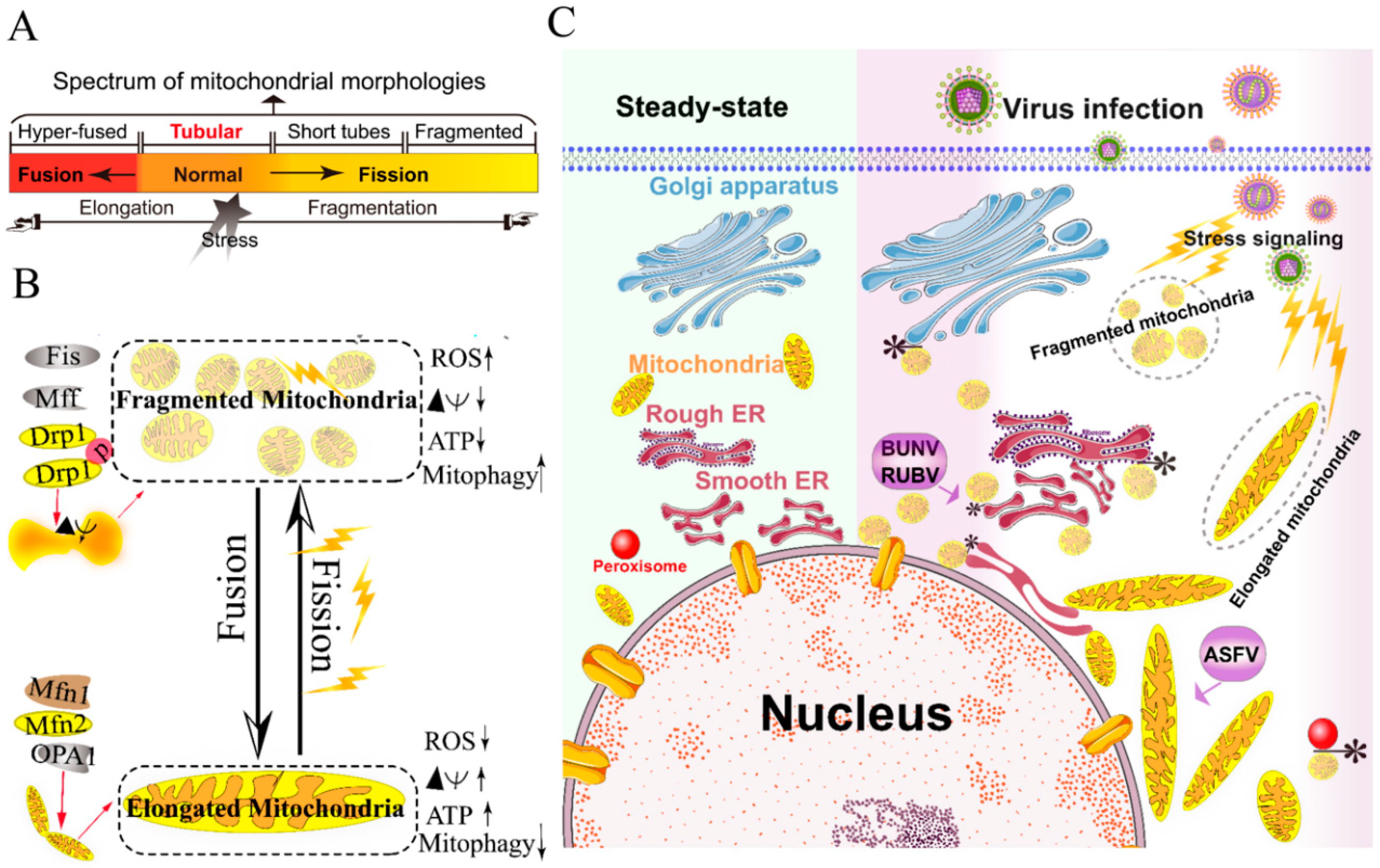{kind=link}
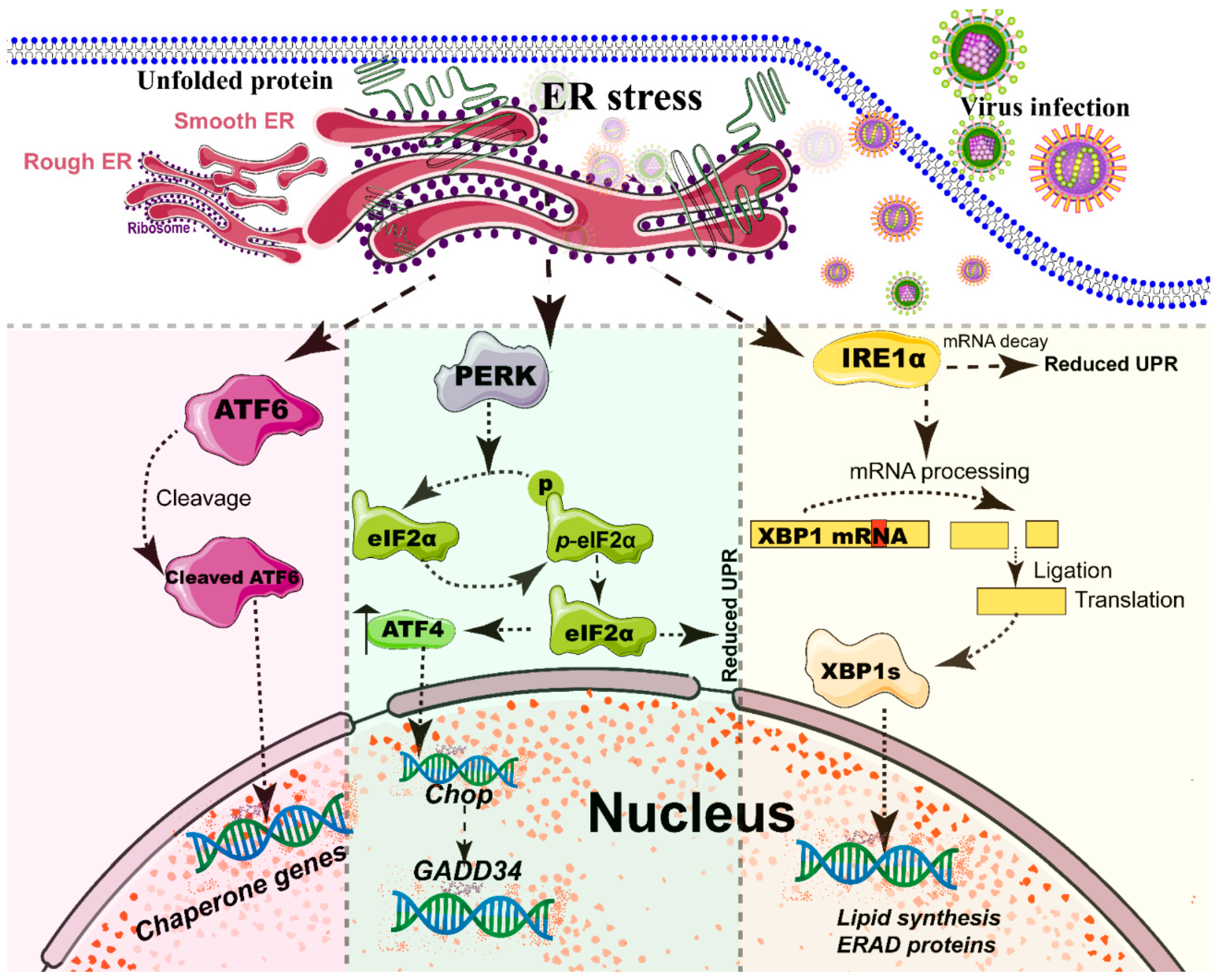{kind=link}
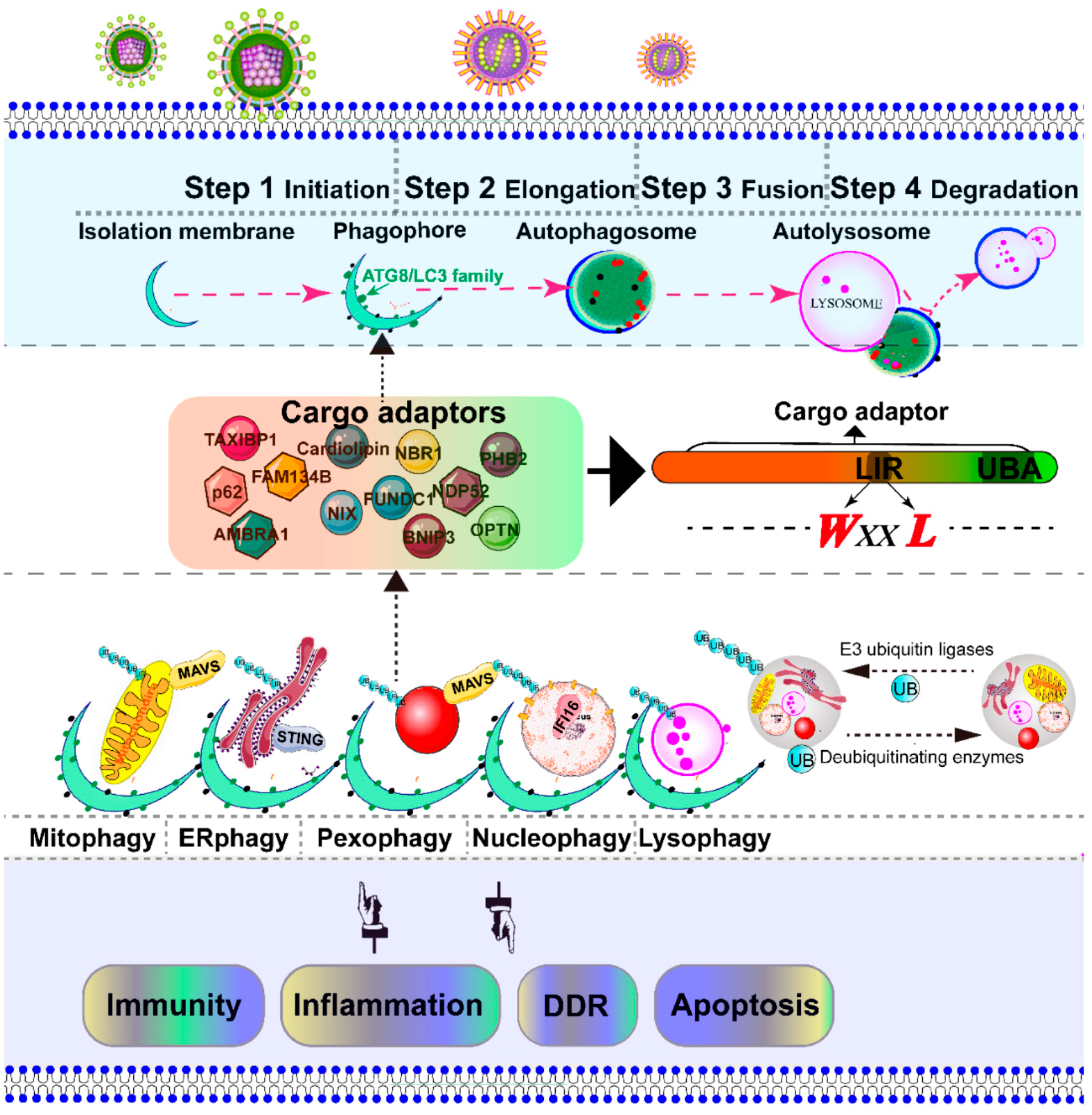{kind=link}
Table 1.
Viruses activate and exploit the UPR branch of ER for viral replication.
| Viruses | Family | Genome Structure | Virion Structure | Viral Protein | ATF6 | PERK | IRE1α | Ref |
|---|---|---|---|---|---|---|---|---|
| PRRSV | Arteriviridae | Linear, ssRNA(+) | Enveloped; Spherical | ? | × | √ | √ | [87] |
| IBV | Coronaviridae | Linear, ssRNA(+) | Enveloped; Spherical | ? | × | × | √ | [97] |
| DENV | Flaviridae | Linear, ssRNA(+) | Enveloped; Spherical | ? | × | √ | √ | [98] |
| JEV | Flaviridae | Linear, ssRNA(+) | Enveloped; Spherical | NS4B | √ | √ | √ | [99,100,101] |
| TBEV | Flaviridae | Linear, ssRNA(+) | Enveloped; Rounded | ? | √ | × | √ | [102] |
| HCV | Flaviridae | Linear, ssRNA(+) | Enveloped; Spherical | ? | √ | × | × | [103] |
| IAV | Orthomyxoviridae | Segmented, ssRNA (-) | Enveloped; Rounded | ? | × | √ | [104] | |
| NDV | Paramyxoviridae | Linear, ssRNA(-) | Enveloped; Spherical | F and HN | √ | √ | √ | [88,105] |
| MCMV | Herpesviridae | Linear, ds DNA | Enveloped; Spherical | ? | ? | √ | × | [106,107] |
| HSV-1 | Herpesviridae | Linear, dsDNA | Enveloped; Spherical | UL41/ICP0/γ134.5 | √ | √ | × | [108,109,110,111] |
| HBV | Hepadnaviridae | Circular dsDNA | Enveloped; Spherical | ? | √ | × | √ | [112] |
During different viral infections, the ER stress activates the three stress sensor proteins: IRE1α, ATF6, and PERK (review in the references [77,96]). The following abbreviations are used in this table: murine cytomegalovirus, MCMV; avian coronavirus infectious bronchitis virus, IBV; porcine reproductive and respiratory syndrome virus, PRRSV; hepatitis C virus, HCV; hepatitis B virus, HBV; influenza A virus, IAV; human herpes simplex virus-1, HSV-1; dengue virus, DENV; tick-borne encephalitis virus, TBEV; Japanese encephalitis virus, JEV; Newcastle disease virus, NDV. The symbols √, ×, and ? indicate activation, inhibition, and unknown, respectively.
Table 2.
Intracellular compartment sites for viral replication and assembly.
| Family | Viruses | Genome Structure | Virion Structure | Replication Site | Ref | Assembly Site | Ref |
|---|---|---|---|---|---|---|---|
| Asfarviridae | ASFV | Liner, dsDNA | Enveloped, spherical | Nuclear and cytoplasmic | [49,132,133] | ER | [86,90] |
| Poxviridae | VV | Liner, dsDNA | Enveloped, brick-shaped | cytoplasmic | [134] | ERGIC, ER | [82,83] |
| Arteriviridae/Arteriviruses | EAV, PRRSV | Liner, ssRNA (+) | Enveloped, spherical | Cytoplasmic double membranes | [84,135] | ER | [84] |
| Coronaviridae/Coronavirus | SARS/MHV | Liner, ssRNA (+) | Enveloped, spherical | Cytoplasmic double membranes | [136] | ERGIC, Golgi, ER | [137,138] |
| Flaviviridae/Hepacivirus | HCV | Linear, ssRNA(+) | Enveloped, spherical | Cytoplasmic | [139] | Autophagosome | [140,141] |
| Metonaviridae/Rubivirus | RUBV | Liner, ssRNA (+) | Enveloped, spherical isometric capsid? | Cytoplasmic | [56] | Golgi, Lysosome | [56,142] |
| Nodaviridae | FHV | Linear, ssRNA(+) | Non-envelop, icosahedral | Cytoplasmic | [143,144] | OMM | [143,144] |
| Togaviridae/Alphavirus | SFV | Liner, ssRNA (+) | Enveloped, spherical and icosahedral | Cytoplasmic | [145,146] | Endosome/Lysosome | [145,146,147] |
| Tombusviridae | TBSV | Linear, ssRNA(+) | Non-envelop, icosahedral | Cytoplasmic | [123] | Peroxisome, ER | [122,123] |
| Picornaviridae/Enterovirus | CV/PV | Linear, ssRNA (+) | Non-envelop, spherical | Cytoplasmic | [148] | ER | [85,148] |
| Orthomyxoviridae | IAV | Segmented, ssRNA (−) | Enveloped; Rounded | Nuclear | [149] | ER, Golgi | [149] |
| Peribunyaviridae/Bunyavirus | BUNV | Segmented, ssRNA (−) | Enveloped, spherical | cytoplasmic | [57] | Golgi | [57,150] |
| Retroviridae/Lentivirus | HIV | Linear, ssRNA (+) | Enveloped, Spherical | Nuclear | [151] | ER or Golgi | [151] |
The following abbreviations are used in this table: African swine fever virus, ASFV; rubella virus, RUBV; Bunyamwera virus, BUNV; semliki forest virus, SFV; Flock house virus, FHV; tomato bushy stunt virus, TBSV; severe acute respiratory syndrome, SARS; murine hepatitis virus, MHV; porcine reproductive and respiratory syndrome virus, PRRSV; equine arteritis virus, EAV; influenza A virus, IAV; human immune deficiency, HIV; vaccinia virus, VV; poliovirus, PV; coxsackieviruses, CV; reticulum-Golgi intermediate compartment, ERGIC; outer mitochondrial membranes, OMM.
Table 3.
Cargo adaptors involved in the selective autophagy in mammals.
| Name | Mitophagy | ER-Phagy | Pexophagy | Nucleophagy | Lysophagy | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cargo Proteins | Mitochondria | Ref | ER | Ref | Peroxisome | Ref | Nucleus | Ref | Lysosome | Ref |
| p62/SQSTM1 | √ | [10] | √ | [12] | √ | [175,176,180] | ? | - | √ | [208,209] |
| NDP52 | √ | [173,210] | ? | - | ? | - | ? | - | ? | - |
| NBR1 | √ | [14] | ? | - | √ | [175,177,181] | ? | - | ? | - |
| OPTN | √ | [15,16,173,210] | ? | - | ? | - | ? | - | ? | - |
| AMBRA1 | √ | [13] | ? | - | ? | - | ? | - | ? | - |
| BNIP3L/NIX | √ | [19,20] | ? | - | ? | - | ? | - | ? | - |
| BNIP3 | √ | [21,211] | √ | [21] | ? | - | ? | - | ? | - |
| TAX1BP1 | √ | [15,17] | ? | - | ? | - | ? | - | ? | - |
| FUNDC1 | √ | [23] | ? | - | ? | - | ? | - | ? | - |
| Cardiolipin | √ | [24] | ? | - | ? | - | ? | - | ? | - |
| Prohibitin2 | √ | [22] | ? | ? | - | ? | - | - | ||
| FAM134B | ? | - | √ | [25] | ? | - | ? | - | ? | - |
The cargo adaptor functions as a bridge between the polyubiquitinated cargo and autophagosome, which is required for selective autophagy. The autophagy adaptors interact directly with the damaged intracellular organelles and an autophagy modifier of the ATG8/LC3 family. The symbols √, ×, and ? indicate activation, inhibition, and unknown, respectively.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ren, S.; Ding, C.; Sun, Y. Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections. Int. J. Mol. Sci. 2020, 21, 3689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103689
AMA Style
Ren S, Ding C, Sun Y. Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections. International Journal of Molecular Sciences. 2020; 21(10):3689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103689
Chicago/Turabian StyleRen, Shanhui, Chan Ding, and Yingjie Sun. 2020. "Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections" International Journal of Molecular Sciences 21, no. 10: 3689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103689
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.