Lipid–Protein and Protein–Protein Interactions in the Pulmonary Surfactant System and Their Role in Lung Homeostasis


Abstract
:1. Introduction
2. The Essential Role of Pulmonary Surfactant at the Lung Interface
2.1. Formation, Stability and Dynamics of the Surfactant Film: Protein and Lipid Interaction
2.2. Alteration of the Interfacial Surfactant Film is Cause of Lung Injury
3. Surfactant Homeostasis in the Alveolar Spaces
3.1. Synthesis, Storage and Secretion of Pulmonary Surfactant
3.2. Surfactant Structures in the Alveolar Spaces
3.3. Surfactant Recycling and Catabolism
3.4. The Importance of Surfactant Anabolism/Catabolism Balance and Its Alteration
4. Anti-/Pro-Inflammatory Balance
4.1. Interaction with Bacterial Lipopolysaccharide
4.2. Interaction with Pattern-Recognition Receptors
5. Antimicrobial Effect
6. Alveolar Epithelial Homeostasis
6.1. Modulation of Apoptosis
6.2. Efferocytosis
6.3. Tissue Repair
7. Conclusions: Surfactant as a Hub in Alveolar Homeostasis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP binding cassette subfamily A member 1 |
| ABCA3 | ATP binding cassette subfamily A member 3 |
| ABCG3 | ATP binding cassette subfamily G member 3 |
| AE1C | alveolar epithelial type I cells |
| AE2C | alveolar epithelial type II cells |
| AM | alveolar macrophages |
| ARDS | acute respiratory distress syndrome |
| C1q | complement component 1q |
| CCT | cytidine triphosphate-phosphocoline cytidyltransferase |
| CD14 | cluster of differentiation 14 |
| CD91 | cluster of differentiation 91 |
| CRD | carbohydrate recognition domain |
| DPPC | dipalmitoylphosphatidylcholine |
| EMC3 | endoplasmic reticulum membrane protein complex subunit 3 |
| ER | endoplasmic reticulum |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| gp340 | glycoprotein 340 |
| GPR116 | G-protein coupled receptor 116 |
| IFN-γ | interferon gamma |
| LA | large aggregates |
| LB | lamellar bodies |
| LBP | lipopolysaccharide binding protein |
| LPCAT1 | acyl CoA:lysophosphatidylcholine acyltransferase |
| LPS | lipopolysaccharide |
| MD2 | myeloid differentiation factor 2 |
| NETs | neutrophil extracellular traps |
| P63 | tumor protein 63 |
| PAP | pulmonary alveolar proteinosis |
| PC | phosphatidylcholine |
| PG | phosphatidylglycerol |
| PI | phosphatidylinositol |
| PLA2 | phospholipase A2 |
| POPG | palmitoyloleoylphosphatidylglycerol |
| RCT | reverse cholesterol transport |
| RDS | respiratory disease syndrome |
| SA | small aggregates |
| SARS | severe acute respiratory syndrome |
| SIRPα | signal inhibitory regulatory protein alfa |
| SNARE | N-ethylmaleimide-sensitive factor attachment receptor |
| SP-A | surfactant protein A |
| SP-B | surfactant protein B |
| SP-BN | N-terminal propeptide of SP-B |
| SP-C | surfactant protein C |
| SP-D | surfactant protein D |
| SP-R210 | surfactant protein receptor 210 |
| SR-A | class A scavenger receptor |
| STAR10 | steroidogenic acute regulatory-related lipid transfer protein 10 |
| TGF-β | transforming growth factor β |
| TLR4 | Toll-like receptor 4 |
References
- Crapo, J.D.; Barry, B.E.; Gehr, P.; Bachofen, M.; Weibel, E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982, 125, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Goerke, J. Pulmonary surfactant: Functions and molecular composition. Biochim. Biophys. Acta 1998, 1408, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Avery, M.E.; Mead, J. Surface properties in relation to atelectasis and hyaline membrane disease. AMA J. Dis. Child. 1959, 97, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.A. Surface tension of lung extracts. Proc. Soc. Exp. Biol. Med. 1957, 95, 170–172. [Google Scholar] [CrossRef]
- Wright, J.R. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 2005, 5, 58–68. [Google Scholar] [CrossRef]
- Creuwels, L.A.; van Golde, L.M.; Haagsman, H.P. The pulmonary surfactant system: Biochemical and clinical aspects. Lung 1997, 175, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Curstedt, T.; Jornvall, H.; Robertson, B.; Bergman, T.; Berggren, P. Two hydrophobic low-molecular-mass protein fractions of pulmonary surfactant. Characterization and biophysical activity. Eur. J. Biochem. 1987, 168, 255–262. [Google Scholar] [CrossRef]
- Possmayer, F.; Nag, K.; Rodriguez, K.; Qanbar, R.; Schurch, S. Surface activity in vitro: Role of surfactant proteins. Comp. Biochem. Physiol. Mol. Integr. Physiol. 2001, 129, 209–220. [Google Scholar] [CrossRef]
- Yu, S.H.; Possmayer, F. Comparative studies on the biophysical activities of the low-molecular-weight hydrophobic proteins purified from bovine pulmonary surfactant. Biochim. Biophys. Acta 1988, 961, 337–350. [Google Scholar] [CrossRef]
- Perez-Gil, J. Structure of pulmonary surfactant membranes and films: The role of proteins and lipid-protein interactions. Biochim. Biophys. Acta 2008, 1778, 1676–1695. [Google Scholar] [CrossRef] [Green Version]
- Orgeig, S.; Hiemstra, P.S.; Veldhuizen, E.J.; Casals, C.; Clark, H.W.; Haczku, A.; Knudsen, L.; Possmayer, F. Recent advances in alveolar biology: Evolution and function of alveolar proteins. Respir. Physiol. Neurobiol. 2010, 173, S43–S54. [Google Scholar] [CrossRef] [Green Version]
- Perez-Gil, J.; Weaver, T.E. Pulmonary surfactant pathophysiology: Current models and open questions. Physiology (Bethesda) 2010, 25, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmeda, B.; Martinez-Calle, M.; Perez-Gil, J. Pulmonary surfactant metabolism in the alveolar airspace: Biogenesis, extracellular conversions, recycling. Ann. Anat. 2017, 209, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.R. Clearance and recycling of pulmonary surfactant. Am. J. Physiol. 1990, 259, L1–L12. [Google Scholar] [CrossRef] [PubMed]
- Casals, C.; Garcia-Fojeda, B.; Minutti, C.M. Soluble defense collagens: Sweeping up immune threats. Mol. Immunol. 2019, 112, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, B.C.; Nakata, K.; Bonella, F.; Campo, I.; Griese, M.; Hamilton, J.; Wang, T.; Morgan, C.; Cottin, V.; McCarthy, C. Pulmonary alveolar proteinosis. Nat. Rev. Dis. Primers 2019, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.; Perez-Gil, J. Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films. Chem. Phys. Lipids 2015, 185, 153–175. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.A. Functions of the alveolar lining. Am. Rev. Respir. Dis. 1977, 115, 67–71. [Google Scholar] [CrossRef]
- Keating, E.; Zuo, Y.Y.; Tadayyon, S.M.; Petersen, N.O.; Possmayer, F.; Veldhuizen, R.A. A modified squeeze-out mechanism for generating high surface pressures with pulmonary surfactant. Biochim. Biophys. Acta 2012, 1818, 1225–1234. [Google Scholar] [CrossRef] [Green Version]
- Stahlman, M.T.; Gray, M.P.; Falconieri, M.W.; Whitsett, J.A.; Weaver, T.E. Lamellar body formation in normal and surfactant protein B-deficient fetal mice. Lab. Investig. 2000, 80, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Ban, N.; Matsumura, Y.; Sakai, H.; Takanezawa, Y.; Sasaki, M.; Arai, H.; Inagaki, N. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J. Biol. Chem. 2007, 282, 9628–9634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobi, N.; Giolai, M.; Olmeda, B.; Miklavc, P.; Felder, E.; Walther, P.; Dietl, P.; Frick, M.; Perez-Gil, J.; Haller, T. A small key unlocks a heavy door: The essential function of the small hydrophobic proteins SP-B and SP-C to trigger adsorption of pulmonary surfactant lamellar bodies. Biochim. Biophys. Acta 2016, 1863, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Schurch, D.; Ospina, O.L.; Cruz, A.; Perez-Gil, J. Combined and independent action of proteins SP-B and SP-C in the surface behavior and mechanical stability of pulmonary surfactant films. Biophys. J. 2010, 99, 3290–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Fujita, Y.; Kogishi, K. Reconstitution of tubular myelin from synthetic lipids and proteins associated with pig pulmonary surfactant. Am. Rev. Respir. Dis. 1989, 140, 75–81. [Google Scholar] [CrossRef]
- Magoon, M.W.; Wright, J.R.; Baritussio, A.; Williams, M.C.; Goerke, J.; Benson, B.J.; Hamilton, R.L.; Clements, J.A. Subfractionation of lung surfactant. Implications for metabolism and surface activity. Biochim. Biophys. Acta 1983, 750, 18–31. [Google Scholar] [CrossRef]
- Gross, N.J.; Narine, K.R. Surfactant subtypes of mice: Metabolic relationships and conversion in vitro. J. Appl. Physiol. 1989, 67, 414–421. [Google Scholar] [CrossRef]
- Ikegami, M.; Grant, S.; Korfhagen, T.; Scheule, R.K.; Whitsett, J.A. Surfactant protein-D regulates the postnatal maturation of pulmonary surfactant lipid pool sizes. J. Appl. Physiol. 2009, 106, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Bates, S.R. P63 (CKAP4) as an SP-A receptor: Implications for surfactant turnover. Cell Physiol. Biochem. 2010, 25, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Rice, W.R.; Ross, G.F.; Singleton, F.M.; Dingle, S.; Whitsett, J.A. Surfactant-associated protein inhibits phospholipid secretion from type II cells. J. Appl. Physiol. 1987, 63, 692–698. [Google Scholar] [CrossRef]
- Martinez-Calle, M.; Olmeda, B.; Dietl, P.; Frick, M.; Perez-Gil, J. Pulmonary surfactant protein SP-B promotes exocytosis of lamellar bodies in alveolar type II cells. FASEB J. 2018, 32, 4600–4611. [Google Scholar] [CrossRef] [Green Version]
- Rider, E.D.; Ikegami, M.; Jobe, A.H. Localization of alveolar surfactant clearance in rabbit lung cells. Am. J. Physiol. 1992, 263, L201–L209. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.D.; Malur, A.; Barna, B.P.; Ghosh, S.; Kavuru, M.S.; Malur, A.G.; Thomassen, M.J. Targeted PPAR{gamma} deficiency in alveolar macrophages disrupts surfactant catabolism. J. Lipid Res. 2010, 51, 1325–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, E.M.; Ramani, R.; Szasz, T.P.; Morley, S.C. Inhaled GM-CSF in neonatal mice provides durable protection against bacterial pneumonia. Sci. Adv. 2019, 5, eaax3387. [Google Scholar] [CrossRef] [Green Version]
- Sallese, A.; Suzuki, T.; McCarthy, C.; Bridges, J.; Filuta, A.; Arumugam, P.; Shima, K.; Ma, Y.; Wessendarp, M.; Black, D.; et al. Targeting cholesterol homeostasis in lung diseases. Sci. Rep. 2017, 7, 10211. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.B.; Ammit, A.J.; Gelissen, I.C. Examining the role of ABC lipid transporters in pulmonary lipid homeostasis and inflammation. Respir. Res. 2017, 18, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, C.; Waring, A.; Zasadzinski, J.A. Keeping lung surfactant where it belongs: Protein regulation of two-dimensional viscosity. Biophys. J. 2005, 89, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Stachowiak, J.C.; Hayden, C.C.; Sanchez, M.A.; Wang, J.; Bunker, B.C.; Voigt, J.A.; Sasaki, D.Y. Targeting proteins to liquid-ordered domains in lipid membranes. Langmuir 2011, 27, 1457–1462. [Google Scholar] [CrossRef]
- Chavarha, M.; Khoojinian, H.; Schulwitz, L.E., Jr.; Biswas, S.C.; Rananavare, S.B.; Hall, S.B. Hydrophobic surfactant proteins induce a phosphatidylethanolamine to form cubic phases. Biophys. J. 2010, 98, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Chavarha, M.; Loney, R.W.; Rananavare, S.B.; Hall, S.B. An anionic phospholipid enables the hydrophobic surfactant proteins to alter spontaneous curvature. Biophys. J. 2013, 104, 594–603. [Google Scholar] [CrossRef] [Green Version]
- Chavarha, M.; Loney, R.W.; Rananavare, S.B.; Hall, S.B. Hydrophobic surfactant proteins strongly induce negative curvature. Biophys. J. 2015, 109, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Chavarha, M.; Loney, R.W.; Kumar, K.; Rananavare, S.B.; Hall, S.B. Differential effects of the hydrophobic surfactant proteins on the formation of inverse bicontinuous cubic phases. Langmuir 2012, 28, 16596–16604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baoukina, S.; Tieleman, D.P. Lung surfactant protein SP-B promotes formation of bilayer reservoirs from monolayer and lipid transfer between the interface and subphase. Biophys. J. 2011, 100, 1678–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Rodriguez, E.; Pascual, A.; Arroyo, R.; Floros, J.; Perez-Gil, J. Human Pulmonary Surfactant Protein SP-A1 Provides Maximal Efficiency of Lung Interfacial Films. Biophys. J. 2016, 111, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Schurch, S.; Possmayer, F.; Cheng, S.; Cockshutt, A.M. Pulmonary SP-A enhances adsorption and appears to induce surface sorting of lipid extract surfactant. Am. J. Physiol. 1992, 263, L210–L218. [Google Scholar] [CrossRef] [PubMed]
- Liekkinen, J.; Enkavi, G.; Javanainen, M.; Olmeda, B.; Perez-Gil, J.; Vattulainen, I. Pulmonary Surfactant Lipid Reorganization Induced by the Adsorption of the Oligomeric Surfactant Protein B Complex. J. Mol. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Calle, M.; Prieto, M.; Olmeda, B.; Fedorov, A.; Loura, L.M.S.; Perez-Gil, J. Pulmonary surfactant protein SP-B nanorings induce the multilamellar organization of surfactant complexes. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183216. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, F.; Ospina, O.L.; Mingarro, I.; Rodriguez-Crespo, I.; Perez-Gil, J. Palmitoylation of pulmonary surfactant protein SP-C is critical for its functional cooperation with SP-B to sustain compression/expansion dynamics in cholesterol-containing surfactant films. Biophys. J. 2010, 99, 3234–3243. [Google Scholar] [CrossRef] [Green Version]
- Roldan, N.; Goormaghtigh, E.; Perez-Gil, J.; Garcia-Alvarez, B. Palmitoylation as a key factor to modulate SP-C-lipid interactions in lung surfactant membrane multilayers. Biochim. Biophys. Acta 2015, 1848, 184–191. [Google Scholar] [CrossRef]
- Serrano, A.G.; Cruz, A.; Rodriguez-Capote, K.; Possmayer, F.; Perez-Gil, J. Intrinsic structural and functional determinants within the amino acid sequence of mature pulmonary surfactant protein SP-B. Biochemistry 2005, 44, 417–430. [Google Scholar] [CrossRef]
- Serrano, A.G.; Ryan, M.; Weaver, T.E.; Perez-Gil, J. Critical structure-function determinants within the N-terminal region of pulmonary surfactant protein SP-B. Biophys. J. 2006, 90, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.A.; Qi, X.; Serrano, A.G.; Ikegami, M.; Perez-Gil, J.; Johansson, J.; Weaver, T.E. Mapping and analysis of the lytic and fusogenic domains of surfactant protein B. Biochemistry 2005, 44, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Cabre, E.J.; Malmstrom, J.; Sutherland, D.; Perez-Gil, J.; Otzen, D.E. Surfactant protein SP-B strongly modifies surface collapse of phospholipid vesicles: Insights from a quartz crystal microbalance with dissipation. Biophys. J. 2009, 97, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmeda, B.; Garcia-Alvarez, B.; Gomez, M.J.; Martinez-Calle, M.; Cruz, A.; Perez-Gil, J. A model for the structure and mechanism of action of pulmonary surfactant protein B. FASEB J. 2015, 29, 4236–4247. [Google Scholar] [CrossRef]
- Bernardino de la Serna, J.; Vargas, R.; Picardi, V.; Cruz, A.; Arranz, R.; Valpuesta, J.M.; Mateu, L.; Perez-Gil, J. Segregated ordered lipid phases and protein-promoted membrane cohesivity are required for pulmonary surfactant films to stabilize and protect the respiratory surface. Faraday Discuss. 2013, 161, 535–548; discussion 563–589. [Google Scholar] [CrossRef] [PubMed]
- Cabre, E.J.; Loura, L.M.; Fedorov, A.; Perez-Gil, J.; Prieto, M. Topology and lipid selectivity of pulmonary surfactant protein SP-B in membranes: Answers from fluorescence. Biochim. Biophys. Acta 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Gil, J.; Casals, C.; Marsh, D. Interactions of hydrophobic lung surfactant proteins SP-B and SP-C with dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylglycerol bilayers studied by electron spin resonance spectroscopy. Biochemistry 1995, 34, 3964–3971. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.; Marsh, D.; Perez-Gil, J. Rotational dynamics of spin-labelled surfactant-associated proteins SP-B and SP-C in dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylglycerol bilayers. Biochim. Biophys. Acta 1998, 1415, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Ingenito, E.P.; Mora, R.; Mark, L. Pivotal role of anionic phospholipids in determining dynamic behavior of lung surfactant. Am. J. Respir. Crit. Care Med. 2000, 161, 831–838. [Google Scholar] [CrossRef]
- Parra, E.; Alcaraz, A.; Cruz, A.; Aguilella, V.M.; Perez-Gil, J. Hydrophobic pulmonary surfactant proteins SP-B and SP-C induce pore formation in planar lipid membranes: Evidence for proteolipid pores. Biophys. J. 2013, 104, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Walters, R.W.; Jenq, R.R.; Hall, S.B. Distinct steps in the adsorption of pulmonary surfactant to an air-liquid interface. Biophys. J. 2000, 78, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Taneva, S.G.; Keough, K.M. Dynamic surface properties of pulmonary surfactant proteins SP-B and SP-C and their mixtures with dipalmitoylphosphatidylcholine. Biochemistry 1994, 33, 14660–14670. [Google Scholar] [CrossRef] [PubMed]
- Klenz, U.; Saleem, M.; Meyer, M.C.; Galla, H.J. Influence of lipid saturation grade and headgroup charge: A refined lung surfactant adsorption model. Biophys. J. 2008, 95, 699–709. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Wintergalen, A.; Sieber, M.; Galla, H.J.; Amrein, M.; Guckenberger, R. Distribution of the surfactant-associated protein C within a lung surfactant model film investigated by near-field optical microscopy. Biophys. J. 2000, 78, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Almlen, A.; Stichtenoth, G.; Linderholm, B.; Haegerstrand-Bjorkman, M.; Robertson, B.; Johansson, J.; Curstedt, T. Surfactant proteins B and C are both necessary for alveolar stability at end expiration in premature rabbits with respiratory distress syndrome. J. Appl. Physiol. 2008, 104, 1101–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Saiedy, M.; Tarokh, A.; Nelson, S.; Hossini, K.; Green, F.; Ling, C.C.; Prenner, E.J.; Amrein, M. The role of multilayers in preventing the premature buckling of the pulmonary surfactant. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.; Moleiro, L.H.; Lopez-Montero, I.; Cruz, A.; Monroy, F.; Perez-Gil, J. A combined action of pulmonary surfactant proteins SP-B and SP-C modulates permeability and dynamics of phospholipid membranes. Biochem. J. 2011, 438, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almlen, A.; Walther, F.J.; Waring, A.J.; Robertson, B.; Johansson, J.; Curstedt, T. Synthetic surfactant based on analogues of SP-B and SP-C is superior to single-peptide surfactants in ventilated premature rabbits. Neonatology 2010, 98, 91–99. [Google Scholar] [CrossRef]
- Cabre, E.J.; Martinez-Calle, M.; Prieto, M.; Fedorov, A.; Olmeda, B.; Loura, L.M.S.; Perez-Gil, J. Homo- and hetero-oligomerization of hydrophobic pulmonary surfactant proteins SP-B and SP-C in surfactant phospholipid membranes. J. Biol. Chem. 2018, 293, 9399–9411. [Google Scholar] [CrossRef] [Green Version]
- Roldan, N.; Nyholm, T.K.M.; Slotte, J.P.; Perez-Gil, J.; Garcia-Alvarez, B. Effect of Lung Surfactant Protein SP-C and SP-C-Promoted Membrane Fragmentation on Cholesterol Dynamics. Biophys. J. 2016, 111, 1703–1713. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Gil, L.; Perez-Gil, J.; Goormaghtigh, E. Cholesterol modulates the exposure and orientation of pulmonary surfactant protein SP-C in model surfactant membranes. Biochim. Biophys. Acta 2009, 1788, 1907–1915. [Google Scholar] [CrossRef]
- Orgeig, S.; Daniels, C.B. The roles of cholesterol in pulmonary surfactant: Insights from comparative and evolutionary studies. Comp. Biochem. Physiol. Mol. Integr. Physiol. 2001, 129, 75–89. [Google Scholar] [CrossRef]
- Bernardino de la Serna, J.; Perez-Gil, J.; Simonsen, A.C.; Bagatolli, L.A. Cholesterol rules: Direct observation of the coexistence of two fluid phases in native pulmonary surfactant membranes at physiological temperatures. J. Biol. Chem. 2004, 279, 40715–40722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roldan, N.; Perez-Gil, J.; Morrow, M.R.; Garcia-Alvarez, B. Divide & Conquer: Surfactant Protein SP-C and Cholesterol Modulate Phase Segregation in Lung Surfactant. Biophys. J. 2017, 113, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Gunasekara, L.; Schurch, S.; Schoel, W.M.; Nag, K.; Leonenko, Z.; Haufs, M.; Amrein, M. Pulmonary surfactant function is abolished by an elevated proportion of cholesterol. Biochim. Biophys. Acta 2005, 1737, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Leonenko, Z.; Gill, S.; Baoukina, S.; Monticelli, L.; Doehner, J.; Gunasekara, L.; Felderer, F.; Rodenstein, M.; Eng, L.M.; Amrein, M. An elevated level of cholesterol impairs self-assembly of pulmonary surfactant into a functional film. Biophys. J. 2007, 93, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vockeroth, D.; Gunasekara, L.; Amrein, M.; Possmayer, F.; Lewis, J.F.; Veldhuizen, R.A. Role of cholesterol in the biophysical dysfunction of surfactant in ventilator-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L117–L125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Gil, L.; Schurch, D.; Goormaghtigh, E.; Perez-Gil, J. Pulmonary surfactant protein SP-C counteracts the deleterious effects of cholesterol on the activity of surfactant films under physiologically relevant compression-expansion dynamics. Biophys. J. 2009, 97, 2736–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruwisch, J.; Sehlmeyer, K.; Roldan, N.; Garcia-Alvarez, B.; Perez-Gil, J.; Weaver, T.E.; Ochs, M.; Knudsen, L.; Lopez-Rodriguez, E. Air Space Distension Precedes Spontaneous Fibrotic Remodeling and Impaired Cholesterol Metabolism in the Absence of Surfactant Protein C. Am. J. Respir. Cell Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Palaniyar, N.; Ikegami, M.; Korfhagen, T.; Whitsett, J.; McCormack, F.X. Domains of surfactant protein A that affect protein oligomerization, lipid structure and surface tension. Comp. Biochem. Physiol. Mol. Integr. Physiol. 2001, 129, 109–127. [Google Scholar] [CrossRef]
- Casals, C. Role of surfactant protein A (SP-A)/lipid interactions for SP-A functions in the lung. Pediatr. Pathol. Mol. Med. 2001, 20, 249–268. [Google Scholar] [CrossRef]
- Saenz, A.; Canadas, O.; Bagatolli, L.A.; Sanchez-Barbero, F.; Johnson, M.E.; Casals, C. Effect of surfactant protein A on the physical properties and surface activity of KL4-surfactant. Biophys. J. 2007, 92, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Barbero, F.; Strassner, J.; Garcia-Canero, R.; Steinhilber, W.; Casals, C. Role of the degree of oligomerization in the structure and function of human surfactant protein A. J. Biol. Chem. 2005, 280, 7659–7670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiansen, J.Q.; Keating, E.; Aspros, A.; Yao, L.J.; Bosma, K.J.; Yamashita, C.M.; Lewis, J.F.; Veldhuizen, R.A. Cholesterol-mediated surfactant dysfunction is mitigated by surfactant protein A. Biochim. Biophys. Acta 2015, 1848, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Barbero, F.; Rivas, G.; Steinhilber, W.; Casals, C. Structural and functional differences among human surfactant proteins SP-A1, SP-A2 and co-expressed SP-A1/SP-A2: Role of supratrimeric oligomerization. Biochem. J. 2007, 406, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Verdugo, I.; Wang, G.; Floros, J.; Casals, C. Structural analysis and lipid-binding properties of recombinant human surfactant protein a derived from one or both genes. Biochemistry 2002, 41, 14041–14053. [Google Scholar] [CrossRef]
- Martinez-Calle, M.; Alonso, A.; Perez-Gil, J.; Olmeda, B. Native supramolecular protein complexes in pulmonary surfactant: Evidences for SP-A/SP-B interactions. J. Proteom. 2019, 207, 103466. [Google Scholar] [CrossRef]
- Wang, G.; Taneva, S.; Keough, K.M.; Floros, J. Differential effects of human SP-A1 and SP-A2 variants on phospholipid monolayers containing surfactant protein B. Biochim. Biophys. Acta 2007, 1768, 2060–2069. [Google Scholar] [CrossRef] [Green Version]
- Persson, A.V.; Gibbons, B.J.; Shoemaker, J.D.; Moxley, M.A.; Longmore, W.J. The major glycolipid recognized by SP-D in surfactant is phosphatidylinositol. Biochemistry 1992, 31, 12183–12189. [Google Scholar] [CrossRef]
- Taneva, S.G.; Keough, K.M. Adsorption of pulmonary surfactant protein SP-A to monolayers of phospholipids containing hydrophobic surfactant protein SP-B or SP-C: Potential differential role for tertiary interaction of lipids, hydrophobic proteins, and SP-A. Biochemistry 2000, 39, 6083–6093. [Google Scholar] [CrossRef]
- Bates, S.R.; Dodia, C.; Tao, J.Q.; Fisher, A.B. Surfactant protein-A plays an important role in lung surfactant clearance: Evidence using the surfactant protein-A gene-targeted mouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L325–L333. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C.; Chander, A. Inhibition of lung calcium-independent phospholipase A2 by surfactant protein A. Am. J. Physiol. 1994, 267, L335–L341. [Google Scholar] [CrossRef] [PubMed]
- Nag, K.; Munro, J.G.; Inchley, K.; Schurch, S.; Petersen, N.O.; Possmayer, F. SP-B refining of pulmonary surfactant phospholipid films. Am. J. Physiol. 1999, 277, L1179–L1189. [Google Scholar] [CrossRef]
- Sano, H.; Kuroki, Y.; Honma, T.; Ogasawara, Y.; Sohma, H.; Voelker, D.R.; Akino, T. Analysis of chimeric proteins identifies the regions in the carbohydrate recognition domains of rat lung collectins that are essential for interactions with phospholipids, glycolipids, and alveolar type II cells. J. Biol. Chem. 1998, 273, 4783–4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikegami, M.; Na, C.L.; Korfhagen, T.R.; Whitsett, J.A. Surfactant protein D influences surfactant ultrastructure and uptake by alveolar type II cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L552–L561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuzawa, T.; Ishida, J.; Kato, A.; Ichinose, T.; Ariestanti, D.M.; Takahashi, T.; Ito, K.; Abe, J.; Suzuki, T.; Wakana, S.; et al. Lung surfactant levels are regulated by Ig-Hepta/GPR116 by monitoring surfactant protein D. PLoS ONE 2013, 8, e69451. [Google Scholar] [CrossRef] [Green Version]
- Hobi, N.; Ravasio, A.; Haller, T. Interfacial stress affects rat alveolar type II cell signaling and gene expression. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L117–L129. [Google Scholar] [CrossRef] [Green Version]
- Ravasio, A.; Hobi, N.; Bertocchi, C.; Jesacher, A.; Dietl, P.; Haller, T. Interfacial sensing by alveolar type II cells: A new concept in lung physiology? Am. J. Physiol. Cell Physiol. 2011, 300, C1456–C1465. [Google Scholar] [CrossRef]
- Knudsen, L.; Ochs, M. The micromechanics of lung alveoli: Structure and function of surfactant and tissue components. Histochem. Cell Biol. 2018, 150, 661–676. [Google Scholar] [CrossRef] [Green Version]
- Ruhl, N.; Lopez-Rodriguez, E.; Albert, K.; Smith, B.J.; Weaver, T.E.; Ochs, M.; Knudsen, L. Surfactant Protein B Deficiency Induced High Surface Tension: Relationship between Alveolar Micromechanics, Alveolar Fluid Properties and Alveolar Epithelial Cell Injury. Int. J. Mol. Sci. 2019, 20, 4243. [Google Scholar] [CrossRef] [Green Version]
- Echaide, M.; Autilio, C.; Arroyo, R.; Perez-Gil, J. Restoring pulmonary surfactant membranes and films at the respiratory surface. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1725–1739. [Google Scholar] [CrossRef]
- Pierrakos, C.; Karanikolas, M.; Scolletta, S.; Karamouzos, V.; Velissaris, D. Acute respiratory distress syndrome: Pathophysiology and therapeutic options. J. Clin. Med. Res. 2012, 4, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogee, L.M. Genetic causes of surfactant protein abnormalities. Curr. Opin. Pediatr. 2019, 31, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Veldhuizen, R.A.; McCaig, L.A.; Akino, T.; Lewis, J.F. Pulmonary surfactant subfractions in patients with the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 152, 1867–1871. [Google Scholar] [CrossRef] [PubMed]
- Autilio, C.; Echaide, M.; Shankar-Aguilera, S.; Bragado, R.; Amidani, D.; Salomone, F.; Perez-Gil, J.; De Luca, D. Surfactant Injury in the Early Phase of Severe Meconium Aspiration Syndrome. Am. J. Respir. Cell Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Amin, R.S.; Wert, S.E.; Baughman, R.P.; Tomashefski, J.F., Jr.; Nogee, L.M.; Brody, A.S.; Hull, W.M.; Whitsett, J.A. Surfactant protein deficiency in familial interstitial lung disease. J. Pediatr. 2001, 139, 85–92. [Google Scholar] [CrossRef]
- Han, S.; Mallampalli, R.K. The Role of Surfactant in Lung Disease and Host Defense against Pulmonary Infections. Ann. Am. Thorac. Soc. 2015, 12, 765–774. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chen, S.; Xu, Y.; Liu, X.; Xiong, P.; Fu, Y. Surfactant protein A expression and distribution in human lung samples from smokers with or without chronic obstructive pulmonary disease in China. Medicine 2020, 99, e19118. [Google Scholar] [CrossRef]
- Pilecki, B.; Wulf-Johansson, H.; Stottrup, C.; Jorgensen, P.T.; Djiadeu, P.; Nexoe, A.B.; Schlosser, A.; Hansen, S.W.K.; Madsen, J.; Clark, H.W.; et al. Surfactant Protein D Deficiency Aggravates Cigarette Smoke-Induced Lung Inflammation by Upregulation of Ceramide Synthesis. Front. Immunol. 2018, 9, 3013. [Google Scholar] [CrossRef]
- Betsuyaku, T.; Kuroki, Y.; Nagai, K.; Nasuhara, Y.; Nishimura, M. Effects of ageing and smoking on SP-A and SP-D levels in bronchoalveolar lavage fluid. Eur. Respir. J. 2004, 24, 964–970. [Google Scholar] [CrossRef]
- Ilumets, H.; Mazur, W.; Toljamo, T.; Louhelainen, N.; Nieminen, P.; Kobayashi, H.; Ishikawa, N.; Kinnula, V.L. Ageing and smoking contribute to plasma surfactant proteins and protease imbalance with correlations to airway obstruction. BMC Pulm. Med. 2011, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Pulmonary Diseases and Ageing. Subcell. Biochem. 2019, 91, 45–74. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.L.; Tan, Q.; Holst, R.; Christiansen, L.; Hansen, N.C.; Hojland, A.T.; Wulf-Johansson, H.; Schlosser, A.; Titlestad, I.L.; Vestbo, J.; et al. Surfactant protein D is a candidate biomarker for subclinical tobacco smoke-induced lung damage. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L887–L895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaioannou, A.I.; Kostikas, K.; Manali, E.D.; Papadaki, G.; Roussou, A.; Spathis, A.; Mazioti, A.; Tomos, I.; Papanikolaou, I.; Loukides, S.; et al. Serum Levels of Surfactant Proteins in Patients with Combined Pulmonary Fibrosis and Emphysema (CPFE). PLoS ONE 2016, 11, e0157789. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.P.; Mao, Q.H.; Yang, L. Effect of pulmonary surfactant combined with mechanical ventilation on oxygenation functions and expressions of serum transforming growth factor-beta1 (TGF-beta1) and bone morphogenetic protein 7 (BMP-7) of neonatal respiratory distress syndrome. Eur. Rev. Med Pharmacol. Sci. 2017, 21, 4357–4361. [Google Scholar]
- Ley, B.; Brown, K.K.; Collard, H.R. Molecular biomarkers in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L681–L691. [Google Scholar] [CrossRef]
- Beers, M.F.; Hamvas, A.; Moxley, M.A.; Gonzales, L.W.; Guttentag, S.H.; Solarin, K.O.; Longmore, W.J.; Nogee, L.M.; Ballard, P.L. Pulmonary surfactant metabolism in infants lacking surfactant protein B. Am. J. Respir. Cell Mol. Biol. 2000, 22, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.C.; Wert, S.E.; Bachurski, C.J.; Stahlman, M.T.; Stripp, B.R.; Weaver, T.E.; Whitsett, J.A. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc. Natl. Acad. Sci. USA 1995, 92, 7794–7798. [Google Scholar] [CrossRef] [Green Version]
- Nogee, L.M.; Wert, S.E.; Proffit, S.A.; Hull, W.M.; Whitsett, J.A. Allelic heterogeneity in hereditary surfactant protein B (SP-B) deficiency. Am. J. Respir. Crit. Care Med. 2000, 161, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, A.E., 3rd; Wert, S.E.; Ikegami, M.; Whitsett, J.A.; Hamvas, A.; White, F.V.; Piedboeuf, B.; Jobin, C.; Guttentag, S.; Nogee, L.M. Prolonged survival in hereditary surfactant protein B (SP-B) deficiency associated with a novel splicing mutation. Pediatr. Res. 2000, 48, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Tredano, M.; Griese, M.; de Blic, J.; Lorant, T.; Houdayer, C.; Schumacher, S.; Cartault, F.; Capron, F.; Boccon-Gibod, L.; Lacaze-Masmonteil, T.; et al. Analysis of 40 sporadic or familial neonatal and pediatric cases with severe unexplained respiratory distress: Relationship to SFTPB. Am. J. Med Genet. Part A 2003, 119A, 324–339. [Google Scholar] [CrossRef]
- Hamvas, A.; Nogee, L.M.; Mallory, G.B., Jr.; Spray, T.L.; Huddleston, C.B.; August, A.; Dehner, L.P.; deMello, D.E.; Moxley, M.; Nelson, R.; et al. Lung transplantation for treatment of infants with surfactant protein B deficiency. J. Pediatr. 1997, 130, 231–239. [Google Scholar] [CrossRef]
- Leibel, S.L.; Winquist, A.; Tseu, I.; Wang, J.; Luo, D.; Shojaie, S.; Nathan, N.; Snyder, E.; Post, M. Reversal of Surfactant Protein B Deficiency in Patient Specific Human Induced Pluripotent Stem Cell Derived Lung Organoids by Gene Therapy. Sci. Rep. 2019, 9, 13450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baekvad-Hansen, M.; Dahl, M.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Surfactant protein-B 121ins2 heterozygosity, reduced pulmonary function, and chronic obstructive pulmonary disease in smokers. Am. J. Respir. Crit. Care Med. 2010, 181, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Glasser, S.W.; Detmer, E.A.; Ikegami, M.; Na, C.L.; Stahlman, M.T.; Whitsett, J.A. Pneumonitis and emphysema in sp-C gene targeted mice. J. Biol. Chem. 2003, 278, 14291–14298. [Google Scholar] [CrossRef] [Green Version]
- Griese, M.; Lorenz, E.; Hengst, M.; Schams, A.; Wesselak, T.; Rauch, D.; Wittmann, T.; Kirchberger, V.; Escribano, A.; Schaible, T.; et al. Surfactant proteins in pediatric interstitial lung disease. Pediatr. Res. 2016, 79, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Mulugeta, S.; Nureki, S.; Beers, M.F. Lost after translation: Insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L507–L525. [Google Scholar] [CrossRef] [Green Version]
- Nathan, N.; Taytard, J.; Duquesnoy, P.; Thouvenin, G.; Corvol, H.; Amselem, S.; Clement, A. Surfactant protein A: A key player in lung homeostasis. Int. J. Biochem. Cell Biol. 2016, 81, 151–155. [Google Scholar] [CrossRef]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Maitra, M.; Wang, Y.; Gerard, R.D.; Mendelson, C.R.; Garcia, C.K. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J. Biol. Chem. 2010, 285, 22103–22113. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, L.; Wang, L.; Qiao, L.; Liang, J.; Yan, H.; Zhao, K.; Liu, X.; Wang, L. Identification and examination of a novel 9-bp insert/deletion polymorphism on porcine SFTPA1 exon 2 associated with acute lung injury using an oleic acid-acute lung injury model. Anim. Sci. J. Nihon chikusan Gakkaiho 2015, 86, 573–578. [Google Scholar] [CrossRef]
- Hallman, M.; Haataja, R. Surfactant protein polymorphisms and neonatal lung disease. Semin. Perinatol. 2006, 30, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Greenwood, C.M.; Eguale, T.; Kifle, A.; Beyene, J.; Habte, A.; Tadesse, A.; Gebrexabher, H.; Britton, S.; Schurr, E. Variants of the SFTPA1 and SFTPA2 genes and susceptibility to tuberculosis in Ethiopia. Hum. Genet. 2006, 118, 752–759. [Google Scholar] [CrossRef]
- Yang, H.Y.; Li, H.; Wang, Y.G.; Xu, C.Y.; Zhao, Y.L.; Ma, X.G.; Li, X.W.; Chen, H. Correlation analysis between single nucleotide polymorphisms of pulmonary surfactant protein A gene and pulmonary tuberculosis in the Han population in China. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2014, 26, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Thorenoor, N.; Wu, R.; DiAngelo, S.L.; Ye, M.; Thomas, N.J.; Liao, X.; Lin, T.R.; Warren, S.; Floros, J. Genetic Association of Pulmonary Surfactant Protein Genes, SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD With Cystic Fibrosis. Front. Immunol. 2018, 9, 2256. [Google Scholar] [CrossRef] [PubMed]
- Hallman, M.; Haataja, R.; Marttila, R. Surfactant proteins and genetic predisposition to respiratory distress syndrome. Semin. Perinatol. 2002, 26, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Ikegami, M.; Moon, C.; Naren, A.P.; Shannon, J.M. Lysophosphatidylcholine Acyltransferase 1 (LPCAT1) Specifically Interacts with Phospholipid Transfer Protein StarD10 to Facilitate Surfactant Phospholipid Trafficking in Alveolar Type II Cells. J. Biol. Chem. 2015, 290, 18559–18574. [Google Scholar] [CrossRef] [Green Version]
- Agassandian, M.; Mallampalli, R.K. Surfactant phospholipid metabolism. Biochim. Biophys. Acta 2013, 1831, 612–625. [Google Scholar] [CrossRef] [Green Version]
- Maniscalco, W.M.; Wilson, C.M.; Gross, I.; Gobran, L.; Rooney, S.A.; Warshaw, J.B. Development of glycogen and phospholipid metabolism in fetal and newborn rat lung. Biochim. Biophys. Acta 1978, 530, 333–346. [Google Scholar] [CrossRef]
- Ridsdale, R.; Post, M. Surfactant lipid synthesis and lamellar body formation in glycogen-laden type II cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L743–L751. [Google Scholar] [CrossRef]
- Torday, J.; Hua, J.; Slavin, R. Metabolism and fate of neutral lipids of fetal lung fibroblast origin. Biochim. Biophys. Acta 1995, 1254, 198–206. [Google Scholar] [CrossRef]
- Torday, J.S.; Rehan, V.K. On the evolution of the pulmonary alveolar lipofibroblast. Exp. Cell Res. 2016, 340, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Zhou, R.; Rehg, J.E.; Jackowski, S. Role of phosphocholine cytidylyltransferase alpha in lung development. Mol. Cell. Biol. 2007, 27, 975–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Bridges, J.P.; Ikegami, M.; Brilli, L.L.; Chen, X.; Mason, R.J.; Shannon, J.M. LPCAT1 regulates surfactant phospholipid synthesis and is required for transitioning to air breathing in mice. J. Clin. Investig. 2010, 120, 1736–1748. [Google Scholar] [CrossRef] [Green Version]
- Butler, P.L.; Mallampalli, R.K. Cross-talk between remodeling and de novo pathways maintains phospholipid balance through ubiquitination. J. Biol. Chem. 2010, 285, 6246–6258. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Sorokina, E.M.; Li, H.; Zhou, S.; Raabe, T.; Feinstein, S.I. A novel lysophosphatidylcholine acyl transferase activity is expressed by peroxiredoxin 6. J. Lipid Res. 2016, 57, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.Z.; Manevich, Y.; Baldwin, J.L.; Dodia, C.; Yu, K.; Feinstein, S.I.; Fisher, A.B. Interaction of surfactant protein A with peroxiredoxin 6 regulates phospholipase A2 activity. J. Biol. Chem. 2006, 281, 7515–7525. [Google Scholar] [CrossRef] [Green Version]
- Batenburg, J.J.; Haagsman, H.P. The lipids of pulmonary surfactant: Dynamics and interactions with proteins. Prog. Lipid Res. 1998, 37, 235–276. [Google Scholar] [CrossRef]
- Roszell, B.R.; Tao, J.Q.; Yu, K.J.; Huang, S.; Bates, S.R. Characterization of the Niemann-Pick C pathway in alveolar type II cells and lamellar bodies of the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L919–L932. [Google Scholar] [CrossRef] [Green Version]
- Foster, C.D.; Zhang, P.X.; Gonzales, L.W.; Guttentag, S.H. In vitro surfactant protein B deficiency inhibits lamellar body formation. Am. J. Respir. Cell Mol. Biol. 2003, 29, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochs, M. The closer we look the more we see? Quantitative microscopic analysis of the pulmonary surfactant system. Cell. Physiol. Biochem. 2010, 25, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Gray, J.M.; Notarfrancesco, K.L.; Gonzales, L.W.; Koval, M.; Feinstein, S.I.; Ballard, P.L.; Fisher, A.B.; Shuman, H. Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J. Biol. Chem. 2002, 277, 22147–22155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarbock, R.; Kaltenborn, E.; Frixel, S.; Wittmann, T.; Liebisch, G.; Schmitz, G.; Griese, M. ABCA3 protects alveolar epithelial cells against free cholesterol induced cell death. Biochim. Biophys. Acta 2015, 1851, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Cheong, N.; Zhang, H.; Madesh, M.; Zhao, M.; Yu, K.; Dodia, C.; Fisher, A.B.; Savani, R.C.; Shuman, H. ABCA3 is critical for lamellar body biogenesis in vivo. J. Biol. Chem. 2007, 282, 23811–23817. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Snowball, J.M.; Xu, Y.; Na, C.L.; Weaver, T.E.; Clair, G.; Kyle, J.E.; Zink, E.M.; Ansong, C.; Wei, W.; et al. EMC3 coordinates surfactant protein and lipid homeostasis required for respiration. J. Clin. Investig. 2017, 127, 4314–4325. [Google Scholar] [CrossRef] [Green Version]
- LeVine, A.M.; Whitsett, J.A.; Gwozdz, J.A.; Richardson, T.R.; Fisher, J.H.; Burhans, M.S.; Korfhagen, T.R. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J. Immunol. 2000, 165, 3934–3940. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, M.; Whitsett, J.A.; Jobe, A.; Ross, G.; Fisher, J.; Korfhagen, T. Surfactant metabolism in SP-D gene-targeted mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L468–L476. [Google Scholar] [CrossRef] [Green Version]
- Ashino, Y.; Ying, X.; Dobbs, L.G.; Bhattacharya, J. [Ca(2+)](i) oscillations regulate type II cell exocytosis in the pulmonary alveolus. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L5–L13. [Google Scholar] [CrossRef] [Green Version]
- Bridges, J.P.; Ludwig, M.G.; Mueller, M.; Kinzel, B.; Sato, A.; Xu, Y.; Whitsett, J.A.; Ikegami, M. Orphan G protein-coupled receptor GPR116 regulates pulmonary surfactant pool size. Am. J. Respir. Cell Mol. Biol. 2013, 49, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.Y.; Hilton, M.B.; Seaman, S.; Haines, D.C.; Nagashima, K.; Burks, C.M.; Tessarollo, L.; Ivanova, P.T.; Brown, H.A.; Umstead, T.M.; et al. Essential regulation of lung surfactant homeostasis by the orphan G protein-coupled receptor GPR116. Cell Rep. 2013, 3, 1457–1464. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Filuta, A.; Ludwig, M.G.; Seuwen, K.; Jaros, J.; Vidal, S.; Arora, K.; Naren, A.P.; Kandasamy, K.; Parthasarathi, K.; et al. Epithelial Gpr116 regulates pulmonary alveolar homeostasis via Gq/11 signaling. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R.; Kistler, G.S.; Tondury, G. A stereologic electron microscope study of "tubular myelin figures" in alveolar fluids of rat lungs. Z. Zellforsch. Mikrosk. Anat. 1966, 69, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Guo, X.; Diangelo, S.; Thomas, N.J.; Floros, J. Humanized SFTPA1 and SFTPA2 transgenic mice reveal functional divergence of SP-A1 and SP-A2: Formation of tubular myelin in vivo requires both gene products. J. Biol. Chem. 2010, 285, 11998–12010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikegami, M.; Korfhagen, T.R.; Whitsett, J.A.; Bruno, M.D.; Wert, S.E.; Wada, K.; Jobe, A.H. Characteristics of surfactant from SP-A-deficient mice. Am. J. Physiol. 1998, 275, L247–L254. [Google Scholar] [CrossRef]
- Kramer, B.W.; Ikegami, M.; Jobe, A.H. Surfactant phospholipid catabolic rate is pool size dependent in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L842–L848. [Google Scholar] [CrossRef]
- Zhang, L.; Ikegami, M.; Crouch, E.C.; Korfhagen, T.R.; Whitsett, J.A. Activity of pulmonary surfactant protein-D (SP-D) in vivo is dependent on oligomeric structure. J. Biol. Chem. 2001, 276, 19214–19219. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, A.D.; Kurak, K.; Moussavian, B.; Whitsett, J.A.; Wert, S.E.; Hull, W.M.; McNanie, J.; Ikegami, M. Preferential uptake of small-aggregate fraction of pulmonary surfactant in vitro. Am. J. Physiol. 1997, 273, L468–L477. [Google Scholar] [CrossRef]
- Miles, P.R.; Ma, J.Y.; Bowman, L. Degradation of pulmonary surfactant disaturated phosphatidylcholines by alveolar macrophages. J. Appl. Physiol. 1988, 64, 2474–2481. [Google Scholar] [CrossRef]
- de Aguiar Vallim, T.Q.; Lee, E.; Merriott, D.J.; Goulbourne, C.N.; Cheng, J.; Cheng, A.; Gonen, A.; Allen, R.M.; Palladino, E.N.D.; Ford, D.A.; et al. ABCG1 regulates pulmonary surfactant metabolism in mice and men. J. Lipid Res. 2017, 58, 941–954. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Arif, A.A.; Guo, J.; Ha, Z.; Lee-Sayer, S.S.M.; Poon, G.F.T.; Dosanjh, M.; Roskelley, C.D.; Huan, T.; Johnson, P. CD44 Loss Disrupts Lung Lipid Surfactant Homeostasis and Exacerbates Oxidized Lipid-Induced Lung Inflammation. Front. Immunol. 2020, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, B.; Trapnell, B.C. The molecular basis of pulmonary alveolar proteinosis. Clin. Immunol. 2010, 135, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Wallace, A.M.; Schneider, D.A.; Burg, E.; Kim, J.; Alekseeva, E.; Ubags, N.D.; Cool, C.D.; Fang, L.; Suratt, B.T.; et al. AIBP augments cholesterol efflux from alveolar macrophages to surfactant and reduces acute lung inflammation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griese, M.; Bonella, F.; Costabel, U.; de Blic, J.; Tran, N.B.; Liebisch, G. Quantitative Lipidomics in Pulmonary Alveolar Proteinosis. Am. J. Respir. Crit. Care Med. 2019, 200, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Madison, M.C.; Landers, C.T.; Gu, B.H.; Chang, C.Y.; Tung, H.Y.; You, R.; Hong, M.J.; Baghaei, N.; Song, L.Z.; Porter, P.; et al. Electronic cigarettes disrupt lung lipid homeostasis and innate immunity independent of nicotine. J. Clin. Investig. 2019, 129, 4290–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slovinsky, W.S.; Shaghaghi, H.; Para, R.; Romero, F.; Summer, R. Alcohol-induced lipid dysregulation impairs glycolytic responses to LPS in alveolar macrophages. Alcohol 2020, 83, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Fessler, M.B.; Summer, R.S. Surfactant Lipids at the Host-Environment Interface. Metabolic Sensors, Suppressors, and Effectors of Inflammatory Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 624–635. [Google Scholar] [CrossRef] [Green Version]
- Zemans, R.L.; Henson, P.M.; Henson, J.E.; Janssen, W.J. Conceptual approaches to lung injury and repair. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 1), S9–S15. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Meunier, E.; Broz, P. Evolutionary Convergence and Divergence in NLR Function and Structure. Trends Immunol. 2017, 38, 744–757. [Google Scholar] [CrossRef]
- Casals, C.; Campanero-Rhodes, M.A.; Garcia-Fojeda, B.; Solis, D. The Role of Collectins and Galectins in Lung Innate Immune Defense. Front. Immunol. 2018, 9, 1998. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ciechanowicz, A.K.; Kaplan, A.R.; Wang, L.; Zhang, P.X.; Lu, Y.C.; Tobin, R.E.; Tobin, B.A.; Cohn, L.; Zeiss, C.J.; et al. Surfactant protein C dampens inflammation by decreasing JAK/STAT activation during lung repair. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L882–L892. [Google Scholar] [CrossRef] [PubMed]
- Voelker, D.R.; Numata, M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019, 294, 4282–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Seydel, U.; Oikawa, M.; Fukase, K.; Kusumoto, S.; Brandenburg, K. Intrinsic conformation of lipid A is responsible for agonistic and antagonistic activity. Eur. J. Biochem. 2000, 267, 3032–3039. [Google Scholar] [CrossRef]
- Hancock, R.E.; Mutharia, L.M.; Chan, L.; Darveau, R.P.; Speert, D.P.; Pier, G.B. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: A class of serum-sensitive, nontypable strains deficient in lipopolysaccharide O side chains. Infect. Immun. 1983, 42, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Fomsgaard, A.; Conrad, R.S.; Galanos, C.; Shand, G.H.; Hoiby, N. Comparative immunochemistry of lipopolysaccharides from typable and polyagglutinable Pseudomonas aeruginosa strains isolated from patients with cystic fibrosis. J. Clin. Microbiol. 1988, 26, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, A.D.; Welte, W.; Hofmann, E.; Lindner, B.; Holst, O.; Coulton, J.W.; Diederichs, K. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure 2000, 8, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Schromm, A.B.; Brandenburg, K.; Loppnow, H.; Moran, A.P.; Koch, M.H.; Rietschel, E.T.; Seydel, U. Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. Eur. J. Biochem. 2000, 267, 2008–2013. [Google Scholar] [CrossRef]
- Mueller, M.; Lindner, B.; Dedrick, R.; Schromm, A.B.; Seydel, U. Endotoxin: Physical requirements for cell activation. J. Endotoxin Res. 2005, 11, 299–303. [Google Scholar] [CrossRef]
- Rosadini, C.V.; Kagan, J.C. Early innate immune responses to bacterial LPS. Curr. Opin. Immunol. 2017, 44, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol. Med. 2011, 3, 513–527. [Google Scholar] [CrossRef]
- Triantafilou, M.; Brandenburg, K.; Kusumoto, S.; Fukase, K.; Mackie, A.; Seydel, U.; Triantafilou, K. Combinational clustering of receptors following stimulation by bacterial products determines lipopolysaccharide responses. Biochem. J. 2004, 381, 527–536. [Google Scholar] [CrossRef]
- van Rozendaal, B.A.; van de Lest, C.H.; van Eijk, M.; van Golde, L.M.; Voorhout, W.F.; van Helden, H.P.; Haagsman, H.P. Aerosolized endotoxin is immediately bound by pulmonary surfactant protein D in vivo. Biochim. Biophys. Acta 1999, 1454, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Canadas, O.; Keough, K.M.; Casals, C. Bacterial lipopolysaccharide promotes destabilization of lung surfactant-like films. Biophys. J. 2011, 100, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Glasser, S.W.; Maxfield, M.D.; Ruetschilling, T.L.; Akinbi, H.T.; Baatz, J.E.; Kitzmiller, J.A.; Page, K.; Xu, Y.; Bao, E.L.; Korfhagen, T.R. Persistence of LPS-induced lung inflammation in surfactant protein-C-deficient mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Augusto, L.; Le Blay, K.; Auger, G.; Blanot, D.; Chaby, R. Interaction of bacterial lipopolysaccharide with mouse surfactant protein C inserted into lipid vesicles. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L776–L785. [Google Scholar] [CrossRef]
- Augusto, L.A.; Li, J.; Synguelakis, M.; Johansson, J.; Chaby, R. Structural basis for interactions between lung surfactant protein C and bacterial lipopolysaccharide. J. Biol. Chem. 2002, 277, 23484–23492. [Google Scholar] [CrossRef] [Green Version]
- Clark, H.W.; Mackay, R.M.; Deadman, M.E.; Hood, D.W.; Madsen, J.; Moxon, E.R.; Townsend, J.P.; Reid, K.B.M.; Ahmed, A.; Shaw, A.J.; et al. Crystal Structure of a Complex of Surfactant Protein D (SP-D) and Haemophilus influenzae Lipopolysaccharide Reveals Shielding of Core Structures in SP-D-Resistant Strains. Infect. Immun. 2016, 84, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Kalina, M.; Blau, H.; Riklis, S.; Kravtsov, V. Interaction of surfactant protein A with bacterial lipopolysaccharide may affect some biological functions. Am. J. Physiol. 1995, 268, L144–L151. [Google Scholar] [CrossRef] [PubMed]
- Kuan, S.F.; Rust, K.; Crouch, E. Interactions of surfactant protein D with bacterial lipopolysaccharides. Surfactant protein D is an Escherichia coli-binding protein in bronchoalveolar lavage. J. Clin. Investig. 1992, 90, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Verdugo, I.; Sanchez-Barbero, F.; Soldau, K.; Tobias, P.S.; Casals, C. Interaction of SP-A (surfactant protein A) with bacterial rough lipopolysaccharide (Re-LPS), and effects of SP-A on the binding of Re-LPS to CD14 and LPS-binding protein. Biochem. J. 2005, 391, 115–124. [Google Scholar] [CrossRef] [Green Version]
- DeSilva, N.S.; Ofek, I.; Crouch, E.C. Interactions of surfactant protein D with fatty acids. Am. J. Respir. Cell Mol. Biol. 2003, 29, 757–770. [Google Scholar] [CrossRef]
- Reinhardt, A.; Wehle, M.; Geissner, A.; Crouch, E.C.; Kang, Y.; Yang, Y.; Anish, C.; Santer, M.; Seeberger, P.H. Structure binding relationship of human surfactant protein D and various lipopolysaccharide inner core structures. J. Struct. Biol. 2016, 195, 387–395. [Google Scholar] [CrossRef]
- Littlejohn, J.R.; da Silva, R.F.; Neale, W.A.; Smallcombe, C.C.; Clark, H.W.; Mackay, R.A.; Watson, A.S.; Madsen, J.; Hood, D.W.; Burns, I.; et al. Structural definition of hSP-D recognition of Salmonella enterica LPS inner core oligosaccharides reveals alternative binding modes for the same LPS. PLoS ONE 2018, 13, e0199175. [Google Scholar] [CrossRef]
- Sahly, H.; Ofek, I.; Podschun, R.; Brade, H.; He, Y.; Ullmann, U.; Crouch, E. Surfactant protein D binds selectively to Klebsiella pneumoniae lipopolysaccharides containing mannose-rich O-antigens. J. Immunol. 2002, 169, 3267–3274. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Chiba, H.; Iwaki, D.; Sohma, H.; Voelker, D.R.; Kuroki, Y. Surfactant proteins A and D bind CD14 by different mechanisms. J. Biol. Chem. 2000, 275, 22442–22451. [Google Scholar] [CrossRef] [Green Version]
- Van Iwaarden, J.F.; Pikaar, J.C.; Storm, J.; Brouwer, E.; Verhoef, J.; Oosting, R.S.; van Golde, L.M.; van Strijp, J.A. Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem. J. 1994, 303 Pt 2, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Sohma, H.; Muta, T.; Nomura, S.; Voelker, D.R.; Kuroki, Y. Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J. Immunol. 1999, 163, 387–395. [Google Scholar]
- Garcia-Verdugo, I.; Canadas, O.; Taneva, S.G.; Keough, K.M.; Casals, C. Surfactant protein A forms extensive lattice-like structures on 1,2-dipalmitoylphosphatidylcholine/rough-lipopolysaccharide-mixed monolayers. Biophys. J. 2007, 93, 3529–3540. [Google Scholar] [CrossRef] [Green Version]
- Keese, S.P.; Brandenburg, K.; Roessle, M.; Schromm, A.B. Pulmonary surfactant protein A-induced changes in the molecular conformation of bacterial deep-rough LPS lead to reduced activity on human macrophages. Innate Immun. 2014, 20, 787–798. [Google Scholar] [CrossRef]
- Goh, B.C.; Wu, H.; Rynkiewicz, M.J.; Schulten, K.; Seaton, B.A.; McCormack, F.X. Elucidation of Lipid Binding Sites on Lung Surfactant Protein A Using X-ray Crystallography, Mutagenesis, and Molecular Dynamics Simulations. Biochemistry 2016, 55, 3692–3701. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.K.; Kim, S.J.; Rah, S.H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.O.; Park, B.S.; Yoon, T.Y.; et al. Reconstruction of LPS Transfer Cascade Reveals Structural Determinants within LBP, CD14, and TLR4-MD2 for Efficient LPS Recognition and Transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Stamme, C.; Muller, M.; Hamann, L.; Gutsmann, T.; Seydel, U. Surfactant protein a inhibits lipopolysaccharide-induced immune cell activation by preventing the interaction of lipopolysaccharide with lipopolysaccharide-binding protein. Am. J. Respir. Cell Mol. Biol. 2002, 27, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Kolomaznik, M.; Liskayova, G.; Kanjakova, N.; Hubcik, L.; Uhrikova, D.; Calkovska, A. The Perturbation of Pulmonary Surfactant by Bacterial Lipopolysaccharide and Its Reversal by Polymyxin B: Function and Structure. Int. J. Mol. Sci 2018, 19, 1964. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, J.; Mellors, J.W.; Ryan, J.L. Altered in vivo activity of liposome-incorporated lipopolysaccharide and lipid A. Infect. Immun. 1989, 57, 3357–3363. [Google Scholar] [CrossRef] [Green Version]
- Yamada, C.; Sano, H.; Shimizu, T.; Mitsuzawa, H.; Nishitani, C.; Himi, T.; Kuroki, Y. Surfactant protein A directly interacts with TLR4 and MD-2 and regulates inflammatory cellular response. Importance of supratrimeric oligomerization. J. Biol. Chem. 2006, 281, 21771–21780. [Google Scholar] [CrossRef] [Green Version]
- Augusto, L.A.; Synguelakis, M.; Johansson, J.; Pedron, T.; Girard, R.; Chaby, R. Interaction of pulmonary surfactant protein C with CD14 and lipopolysaccharide. Infect. Immun. 2003, 71, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Ohya, M.; Nishitani, C.; Sano, H.; Yamada, C.; Mitsuzawa, H.; Shimizu, T.; Saito, T.; Smith, K.; Crouch, E.; Kuroki, Y. Human pulmonary surfactant protein D binds the extracellular domains of Toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry 2006, 45, 8657–8664. [Google Scholar] [CrossRef]
- Nie, X.; Nishitani, C.; Yamazoe, M.; Ariki, S.; Takahashi, M.; Shimizu, T.; Mitsuzawa, H.; Sawada, K.; Smith, K.; Crouch, E.; et al. Pulmonary surfactant protein D binds MD-2 through the carbohydrate recognition domain. Biochemistry 2008, 47, 12878–12885. [Google Scholar] [CrossRef]
- Yamazoe, M.; Nishitani, C.; Takahashi, M.; Katoh, T.; Ariki, S.; Shimizu, T.; Mitsuzawa, H.; Sawada, K.; Voelker, D.R.; Takahashi, H.; et al. Pulmonary surfactant protein D inhibits lipopolysaccharide (LPS)-induced inflammatory cell responses by altering LPS binding to its receptors. J. Biol. Chem. 2008, 283, 35878–35888. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Asai, Y.; Ogawa, T. Treponemal phospholipids inhibit innate immune responses induced by pathogen-associated molecular patterns. J. Biol. Chem. 2003, 278, 44205–44213. [Google Scholar] [CrossRef] [Green Version]
- Kuronuma, K.; Mitsuzawa, H.; Takeda, K.; Nishitani, C.; Chan, E.D.; Kuroki, Y.; Nakamura, M.; Voelker, D.R. Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J. Biol. Chem. 2009, 284, 25488–25500. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y.; Shieh, C.C.; You, P.F.; Lei, H.Y.; Reid, K.B. Inhibitory effect of pulmonary surfactant proteins A and D on allergen-induced lymphocyte proliferation and histamine release in children with asthma. Am. J. Respir. Crit. Care Med. 1998, 158, 510–518. [Google Scholar] [CrossRef]
- Akashi, S.; Ogata, H.; Kirikae, F.; Kirikae, T.; Kawasaki, K.; Nishijima, M.; Shimazu, R.; Nagai, Y.; Fukudome, K.; Kimoto, M.; et al. Regulatory roles for CD14 and phosphatidylinositol in the signaling via toll-like receptor 4-MD-2. Biochem. Biophys. Res. Commun. 2000, 268, 172–177. [Google Scholar] [CrossRef]
- Kandasamy, P.; Numata, M.; Berry, K.Z.; Fickes, R.; Leslie, C.C.; Murphy, R.C.; Voelker, D.R. Structural analogs of pulmonary surfactant phosphatidylglycerol inhibit toll-like receptor 2 and 4 signaling. J. Lipid Res. 2016, 57, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Abate, W.; Alghaithy, A.A.; Parton, J.; Jones, K.P.; Jackson, S.K. Surfactant lipids regulate LPS-induced interleukin-8 production in A549 lung epithelial cells by inhibiting translocation of TLR4 into lipid raft domains. J. Lipid Res. 2010, 51, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Plociennikowska, A.; Zdioruk, M.I.; Traczyk, G.; Swiatkowska, A.; Kwiatkowska, K. LPS-induced clustering of CD14 triggers generation of PI(4,5)P2. J. Cell Sci. 2015, 128, 4096–4111. [Google Scholar] [CrossRef] [Green Version]
- Numata, M.; Kandasamy, P.; Voelker, D.R. Anionic pulmonary surfactant lipid regulation of innate immunity. Expert Rev. Respir. Med. 2012, 6, 243–246. [Google Scholar] [CrossRef]
- Pandit, H.; Madhukaran, S.P.; Nayak, A.; Madan, T. SP-A and SP-D in host defense against fungal infections and allergies. Front. Biosci. 2012, 4, 651–661. [Google Scholar] [CrossRef]
- Awasthi, S. Surfactant protein (SP)-A and SP-D as antimicrobial and immunotherapeutic agents. Recent Patents Anti-Infective Drug Discov. 2010, 5, 115–123. [Google Scholar] [CrossRef]
- Glasser, S.W.; Senft, A.P.; Whitsett, J.A.; Maxfield, M.D.; Ross, G.F.; Richardson, T.R.; Prows, D.R.; Xu, Y.; Korfhagen, T.R. Macrophage dysfunction and susceptibility to pulmonary Pseudomonas aeruginosa infection in surfactant protein C-deficient mice. J. Immunol. 2008, 181, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Glasser, S.W.; Witt, T.L.; Senft, A.P.; Baatz, J.E.; Folger, D.; Maxfield, M.D.; Akinbi, H.T.; Newton, D.A.; Prows, D.R.; Korfhagen, T.R. Surfactant protein C-deficient mice are susceptible to respiratory syncytial virus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L64–L72. [Google Scholar] [CrossRef] [Green Version]
- Ariki, S.; Nishitani, C.; Kuroki, Y. Diverse functions of pulmonary collectins in host defense of the lung. J. Biomed. Biotechnol. 2012, 2012, 532071. [Google Scholar] [CrossRef] [Green Version]
- LeVine, A.M.; Whitsett, J.A. Pulmonary collectins and innate host defense of the lung. Microbes Infect. 2001, 3, 161–166. [Google Scholar] [CrossRef]
- Ordonez, S.R.; Veldhuizen, E.J.A.; van Eijk, M.; Haagsman, H.P. Role of Soluble Innate Effector Molecules in Pulmonary Defense against Fungal Pathogens. Front. Microbiol. 2017, 8, 2098. [Google Scholar] [CrossRef]
- Nayak, A.; Dodagatta-Marri, E.; Tsolaki, A.G.; Kishore, U. An Insight into the Diverse Roles of Surfactant Proteins, SP-A and SP-D in Innate and Adaptive Immunity. Front. Immunol. 2012, 3, 131. [Google Scholar] [CrossRef] [Green Version]
- Piboonpocanun, S.; Chiba, H.; Mitsuzawa, H.; Martin, W.; Murphy, R.C.; Harbeck, R.J.; Voelker, D.R. Surfactant protein A binds Mycoplasma pneumoniae with high affinity and attenuates its growth by recognition of disaturated phosphatidylglycerols. J. Biol. Chem. 2005, 280, 9–17. [Google Scholar] [CrossRef] [Green Version]
- van de Wetering, J.K.; van Eijk, M.; van Golde, L.M.; Hartung, T.; van Strijp, J.A.; Batenburg, J.J. Characteristics of surfactant protein A and D binding to lipoteichoic acid and peptidoglycan, 2 major cell wall components of gram-positive bacteria. J. Infect. Dis 2001, 184, 1143–1151. [Google Scholar] [CrossRef]
- Carreto-Binaghi, L.E.; Aliouat el, M.; Taylor, M.L. Surfactant proteins, SP-A and SP-D, in respiratory fungal infections: Their role in the inflammatory response. Respir. Res. 2016, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.; Phipps, M.J.S.; Clark, H.W.; Skylaris, C.K.; Madsen, J. Surfactant Proteins A and D: Trimerized Innate Immunity Proteins with an Affinity for Viral Fusion Proteins. J. Innate Immun. 2019, 11, 13–28. [Google Scholar] [CrossRef]
- Brummer, E.; Stevens, D.A. Collectins and fungal pathogens: Roles of surfactant proteins and mannose binding lectin in host resistance. Med. Mycol. 2010, 48, 16–28. [Google Scholar] [CrossRef]
- van de Wetering, J.K.; van Remoortere, A.; Vaandrager, A.B.; Batenburg, J.J.; van Golde, L.M.; Hokke, C.H.; van Hellemond, J.J. Surfactant protein D binding to terminal alpha1-3-linked fucose residues and to Schistosoma mansoni. Am. J. Respir. Cell Mol. Biol. 2004, 31, 565–572. [Google Scholar] [CrossRef]
- Pikaar, J.C.; Voorhout, W.F.; van Golde, L.M.; Verhoef, J.; Van Strijp, J.A.; van Iwaarden, J.F. Opsonic activities of surfactant proteins A and D in phagocytosis of gram-negative bacteria by alveolar macrophages. J. Infect. Dis 1995, 172, 481–489. [Google Scholar] [CrossRef]
- Sever-Chroneos, Z.; Krupa, A.; Davis, J.; Hasan, M.; Yang, C.H.; Szeliga, J.; Herrmann, M.; Hussain, M.; Geisbrecht, B.V.; Kobzik, L.; et al. Surfactant protein A (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J. Biol. Chem. 2011, 286, 4854–4870. [Google Scholar] [CrossRef] [Green Version]
- Chroneos, Z.C.; Abdolrasulnia, R.; Whitsett, J.A.; Rice, W.R.; Shepherd, V.L. Purification of a cell-surface receptor for surfactant protein A. J. Biol. Chem. 1996, 271, 16375–16383. [Google Scholar] [CrossRef] [Green Version]
- Holmskov, U. Lung surfactant proteins (SP-A and SP-D) in non-adaptive host responses to infection. J. Leukoc. Biol 1999, 66, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Tino, M.J.; Wright, J.R. Glycoprotein-340 binds surfactant protein-A (SP-A) and stimulates alveolar macrophage migration in an SP-A-independent manner. Am. J. Respir. Cell Mol. Biol. 1999, 20, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Nadesalingam, J.; Reid, K.B.; Palaniyar, N. Collectin surfactant protein D binds antibodies and interlinks innate and adaptive immune systems. FEBS Lett. 2005, 579, 4449–4453. [Google Scholar] [CrossRef] [Green Version]
- Gardai, S.J.; Xiao, Y.Q.; Dickinson, M.; Nick, J.A.; Voelker, D.R.; Greene, K.E.; Henson, P.M. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003, 115, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Coya, J.M.; Akinbi, H.T.; Saenz, A.; Yang, L.; Weaver, T.E.; Casals, C. Natural Anti-Infective Pulmonary Proteins: In Vivo Cooperative Action of Surfactant Protein SP-A and the Lung Antimicrobial Peptide SP-BN. J. Immunol. 2015, 195, 1628–1636. [Google Scholar] [CrossRef] [Green Version]
- Beharka, A.A.; Gaynor, C.D.; Kang, B.K.; Voelker, D.R.; McCormack, F.X.; Schlesinger, L.S. Pulmonary surfactant protein A up-regulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J. Immunol. 2002, 169, 3565–3573. [Google Scholar] [CrossRef] [Green Version]
- Kudo, K.; Sano, H.; Takahashi, H.; Kuronuma, K.; Yokota, S.; Fujii, N.; Shimada, K.; Yano, I.; Kumazawa, Y.; Voelker, D.R.; et al. Pulmonary collectins enhance phagocytosis of Mycobacterium avium through increased activity of mannose receptor. J. Immunol. 2004, 172, 7592–7602. [Google Scholar] [CrossRef] [Green Version]
- Kuronuma, K.; Sano, H.; Kato, K.; Kudo, K.; Hyakushima, N.; Yokota, S.; Takahashi, H.; Fujii, N.; Suzuki, H.; Kodama, T.; et al. Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumoniae by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J. Biol. Chem. 2004, 279, 21421–21430. [Google Scholar] [CrossRef] [Green Version]
- Pondman, K.M.; Paudyal, B.; Sim, R.B.; Kaur, A.; Kouser, L.; Tsolaki, A.G.; Jones, L.A.; Salvador-Morales, C.; Khan, H.A.; Ten Haken, B.; et al. Pulmonary surfactant protein SP-D opsonises carbon nanotubes and augments their phagocytosis and subsequent pro-inflammatory immune response. Nanoscale 2017, 9, 1097–1109. [Google Scholar] [CrossRef] [Green Version]
- Noris, M.; Remuzzi, G. Overview of complement activation and regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Watford, W.T.; Wright, J.R.; Hester, C.G.; Jiang, H.; Frank, M.M. Surfactant protein A regulates complement activation. J. Immunol. 2001, 167, 6593–6600. [Google Scholar] [CrossRef] [Green Version]
- Douda, D.N.; Jackson, R.; Grasemann, H.; Palaniyar, N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J. Immunol. 2011, 187, 1856–1865. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zeng, M.; Stamler, J.S. Hemoglobin induction in mouse macrophages. Proc. Natl. Acad. Sci. USA 1999, 96, 6643–6647. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, R.; Khan, M.A.; Echaide, M.; Perez-Gil, J.; Palaniyar, N. SP-D attenuates LPS-induced formation of human neutrophil extracellular traps (NETs), protecting pulmonary surfactant inactivation by NETs. Commun. Biol. 2019, 2, 470. [Google Scholar] [CrossRef]
- Du, J.; Abdel-Razek, O.; Shi, Q.; Hu, F.; Ding, G.; Cooney, R.N.; Wang, G. Surfactant protein D attenuates acute lung and kidney injuries in pneumonia-induced sepsis through modulating apoptosis, inflammation and NF-kappaB signaling. Sci. Rep. 2018, 8, 15393. [Google Scholar] [CrossRef]
- Wu, H.; Kuzmenko, A.; Wan, S.; Schaffer, L.; Weiss, A.; Fisher, J.H.; Kim, K.S.; McCormack, F.X. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Investig. 2003, 111, 1589–1602. [Google Scholar] [CrossRef] [Green Version]
- Kuzmenko, A.I.; Wu, H.; McCormack, F.X. Pulmonary collectins selectively permeabilize model bacterial membranes containing rough lipopolysaccharide. Biochemistry 2006, 45, 2679–2685. [Google Scholar] [CrossRef] [Green Version]
- Canadas, O.; Garcia-Verdugo, I.; Keough, K.M.; Casals, C. SP-A permeabilizes lipopolysaccharide membranes by forming protein aggregates that extract lipids from the membrane. Biophys. J. 2008, 95, 3287–3294. [Google Scholar] [CrossRef] [Green Version]
- McCormack, F.X.; Gibbons, R.; Ward, S.R.; Kuzmenko, A.; Wu, H.; Deepe, G.S., Jr. Macrophage-independent fungicidal action of the pulmonary collectins. J. Biol. Chem. 2003, 278, 36250–36256. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Fojeda, B.; Gonzalez-Carnicero, Z.; de Lorenzo, A.; Minutti, C.M.; de Tapia, L.; Euba, B.; Iglesias-Ceacero, A.; Castillo-Lluva, S.; Garmendia, J.; Casals, C. Lung Surfactant Lipids Provide Immune Protection Against Haemophilus influenzae Respiratory Infection. Front. Immunol. 2019, 10, 458. [Google Scholar] [CrossRef] [Green Version]
- Dutta, K.; Nag, K.; Booth, V.; Smyth, E.; Dueck, H.; Fritzen-Garcia, M.; Ghosh, C.; Panda, A.K. Paradoxical Bactericidal Effects of Hydrophobic Lung Surfactant Proteins and Their Peptide Mimics Using Liposome Molecular Trojan. J. Oleo Sci. 2018, 67, 1043–1057. [Google Scholar] [CrossRef]
- Ryan, M.A.; Akinbi, H.T.; Serrano, A.G.; Perez-Gil, J.; Wu, H.; McCormack, F.X.; Weaver, T.E. Antimicrobial activity of native and synthetic surfactant protein B peptides. J. Immunol. 2006, 176, 416–425. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Johansson, J.; Ridsdale, R.; Willander, H.; Fitzen, M.; Akinbi, H.T.; Weaver, T.E. Surfactant protein B propeptide contains a saposin-like protein domain with antimicrobial activity at low pH. J. Immunol. 2010, 184, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, I.N.; De Luna, X.; White, M.R.; Hartshorn, K.L. The Role and Molecular Mechanism of Action of Surfactant Protein D in Innate Host Defense Against Influenza A Virus. Front. Immunol. 2018, 9, 1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Qahtani, A.A.; Murugaiah, V.; Bashir, H.A.; Pathan, A.A.; Abozaid, S.M.; Makarov, E.; Nal-Rogier, B.; Kishore, U.; Al-Ahdal, M.N. Full-length human surfactant protein A inhibits influenza A virus infection of A549 lung epithelial cells: A recombinant form containing neck and lectin domains promotes infectivity. Immunobiology 2019, 224, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Benne, C.A.; Kraaijeveld, C.A.; van Strijp, J.A.; Brouwer, E.; Harmsen, M.; Verhoef, J.; van Golde, L.M.; van Iwaarden, J.F. Interactions of surfactant protein A with influenza A viruses: Binding and neutralization. J. Infect. Dis 1995, 171, 335–341. [Google Scholar] [CrossRef]
- Mikerov, A.N.; Umstead, T.M.; Gan, X.; Huang, W.; Guo, X.; Wang, G.; Phelps, D.S.; Floros, J. Impact of ozone exposure on the phagocytic activity of human surfactant protein A (SP-A) and SP-A variants. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L121–L130. [Google Scholar] [CrossRef] [Green Version]
- Hartshorn, K.L.; Crouch, E.C.; White, M.R.; Eggleton, P.; Tauber, A.I.; Chang, D.; Sastry, K. Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J. Clin. Investig. 1994, 94, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Hartshorn, K.L.; White, M.R.; Shepherd, V.; Reid, K.; Jensenius, J.C.; Crouch, E.C. Mechanisms of anti-influenza activity of surfactant proteins A and D: Comparison with serum collectins. Am. J. Physiol. 1997, 273, L1156–L1166. [Google Scholar] [CrossRef]
- Reading, P.C.; Morey, L.S.; Crouch, E.C.; Anders, E.M. Collectin-mediated antiviral host defense of the lung: Evidence from influenza virus infection of mice. J. Virol. 1997, 71, 8204–8212. [Google Scholar] [CrossRef] [Green Version]
- Tecle, T.; White, M.R.; Crouch, E.C.; Hartshorn, K.L. Inhibition of influenza viral neuraminidase activity by collectins. Arch. Virol. 2007, 152, 1731–1742. [Google Scholar] [CrossRef]
- Gaiha, G.D.; Dong, T.; Palaniyar, N.; Mitchell, D.A.; Reid, K.B.; Clark, H.W. Surfactant protein A binds to HIV and inhibits direct infection of CD4+ cells, but enhances dendritic cell-mediated viral transfer. J. Immunol. 2008, 181, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Meschi, J.; Crouch, E.C.; Skolnik, P.; Yahya, K.; Holmskov, U.; Leth-Larsen, R.; Tornoe, I.; Tecle, T.; White, M.R.; Hartshorn, K.L. Surfactant protein D binds to human immunodeficiency virus (HIV) envelope protein gp120 and inhibits HIV replication. J. Gen. Virol. 2005, 86, 3097–3107. [Google Scholar] [CrossRef]
- Madsen, J.; Gaiha, G.D.; Palaniyar, N.; Dong, T.; Mitchell, D.A.; Clark, H.W. Surfactant Protein D modulates HIV infection of both T-cells and dendritic cells. PLoS ONE 2013, 8, e59047. [Google Scholar] [CrossRef] [Green Version]
- Ghildyal, R.; Hartley, C.; Varrasso, A.; Meanger, J.; Voelker, D.R.; Anders, E.M.; Mills, J. Surfactant protein A binds to the fusion glycoprotein of respiratory syncytial virus and neutralizes virion infectivity. J. Infect. Dis. 1999, 180, 2009–2013. [Google Scholar] [CrossRef]
- LeVine, A.M.; Elliott, J.; Whitsett, J.A.; Srikiatkhachorn, A.; Crouch, E.; DeSilva, N.; Korfhagen, T. Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2004, 31, 193–199. [Google Scholar] [CrossRef]
- Sano, H.; Nagai, K.; Tsutsumi, H.; Kuroki, Y. Lactoferrin and surfactant protein A exhibit distinct binding specificity to F protein and differently modulate respiratory syncytial virus infection. Eur. J. Immunol. 2003, 33, 2894–2902. [Google Scholar] [CrossRef]
- Hickling, T.P.; Malhotra, R.; Bright, H.; McDowell, W.; Blair, E.D.; Sim, R.B. Lung surfactant protein A provides a route of entry for respiratory syncytial virus into host cells. Viral Immunol. 2000, 13, 125–135. [Google Scholar] [CrossRef]
- van Iwaarden, J.F.; van Strijp, J.A.; Visser, H.; Haagsman, H.P.; Verhoef, J.; van Golde, L.M. Binding of surfactant protein A (SP-A) to herpes simplex virus type 1-infected cells is mediated by the carbohydrate moiety of SP-A. J. Biol. Chem. 1992, 267, 25039–25043. [Google Scholar]
- Leth-Larsen, R.; Zhong, F.; Chow, V.T.; Holmskov, U.; Lu, J. The SARS coronavirus spike glycoprotein is selectively recognized by lung surfactant protein D and activates macrophages. Immunobiology 2007, 212, 201–211. [Google Scholar] [CrossRef]
- Reading, P.C.; Holmskov, U.; Anders, E.M. Antiviral activity of bovine collectins against rotaviruses. J. Gen. Virol. 1998, 79 Pt 9, 2255–2263. [Google Scholar] [CrossRef] [Green Version]
- Perino, J.; Thielens, N.M.; Crouch, E.; Spehner, D.; Crance, J.M.; Favier, A.L. Protective effect of surfactant protein d in pulmonary vaccinia virus infection: Implication of A27 viral protein. Viruses 2013, 5, 928–953. [Google Scholar] [CrossRef] [Green Version]
- Favier, A.L.; Reynard, O.; Gout, E.; van Eijk, M.; Haagsman, H.P.; Crouch, E.; Volchkov, V.; Peyrefitte, C.; Thielens, N.M. Involvement of Surfactant Protein D in Ebola Virus Infection Enhancement via Glycoprotein Interaction. Viruses 2018, 11, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, J.P.; Xu, Y.; Na, C.L.; Wong, H.R.; Weaver, T.E. Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J. Cell Biol. 2006, 172, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Numata, M.; Chu, H.W.; Dakhama, A.; Voelker, D.R. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection. Proc. Natl. Acad. Sci. USA 2010, 107, 320–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, M.; Kandasamy, P.; Nagashima, Y.; Fickes, R.; Murphy, R.C.; Voelker, D.R. Phosphatidylinositol inhibits respiratory syncytial virus infection. J. Lipid Res. 2015, 56, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Numata, M.; Mitchell, J.R.; Tipper, J.L.; Brand, J.D.; Trombley, J.E.; Nagashima, Y.; Kandasamy, P.; Chu, H.W.; Harrod, K.S.; Voelker, D.R. Pulmonary surfactant lipids inhibit infections with the pandemic H1N1 influenza virus in several animal models. J. Biol. Chem. 2020, 295, 1704–1715. [Google Scholar] [CrossRef]
- Numata, M.; Nagashima, Y.; Moore, M.L.; Berry, K.Z.; Chan, M.; Kandasamy, P.; Peebles, R.S., Jr.; Murphy, R.C.; Voelker, D.R. Phosphatidylglycerol provides short-term prophylaxis against respiratory syncytial virus infection. J. Lipid Res. 2013, 54, 2133–2143. [Google Scholar] [CrossRef] [Green Version]
- Hartshorn, K.L.; White, M.R.; Tecle, T.; Holmskov, U.; Crouch, E.C. Innate defense against influenza A virus: Activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J. Immunol. 2006, 176, 6962–6972. [Google Scholar] [CrossRef]
- Doss, M.; White, M.R.; Tecle, T.; Gantz, D.; Crouch, E.C.; Jung, G.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Hartshorn, K.L. Interactions of alpha-, beta-, and theta-defensins with influenza A virus and surfactant protein D. J. Immunol. 2009, 182, 7878–7887. [Google Scholar] [CrossRef] [Green Version]
- White, M.R.; Crouch, E.; Vesona, J.; Tacken, P.J.; Batenburg, J.J.; Leth-Larsen, R.; Holmskov, U.; Hartshorn, K.L. Respiratory innate immune proteins differentially modulate the neutrophil respiratory burst response to influenza A virus. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L606–L616. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Ariki, S.; Sohma, H.; Nishitani, C.; Inoue, K.; Ebata, N.; Takahashi, M.; Hasegawa, Y.; Kuronuma, K.; Takahashi, H.; et al. Pulmonary surfactant protein A protects lung epithelium from cytotoxicity of human beta-defensin 3. J. Biol. Chem. 2012, 287, 15034–15043. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.P.; Tuder, R.M. Role of Apoptosis in Amplifying Inflammatory Responses in Lung Diseases. J. Cell Death 2010, 3, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Galani, V.; Tatsaki, E.; Bai, M.; Kitsoulis, P.; Lekka, M.; Nakos, G.; Kanavaros, P. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): An up-to-date cell-specific review. Pathol. Res. Pract. 2010, 206, 145–150. [Google Scholar] [CrossRef]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Drakopanagiotakis, F.; Xifteri, A.; Polychronopoulos, V.; Bouros, D. Apoptosis in lung injury and fibrosis. Eur. Respir. J. 2008, 32, 1631–1638. [Google Scholar] [CrossRef] [Green Version]
- Djiadeu, P.; Farmakovski, N.; Azzouz, D.; Kotra, L.P.; Sweezey, N.; Palaniyar, N. Surfactant protein D regulates caspase-8-mediated cascade of the intrinsic pathway of apoptosis while promoting bleb formation. Mol. Immunol. 2017, 92, 190–198. [Google Scholar] [CrossRef]
- Janssen, W.J.; McPhillips, K.A.; Dickinson, M.G.; Linderman, D.J.; Morimoto, K.; Xiao, Y.Q.; Oldham, K.M.; Vandivier, R.W.; Henson, P.M.; Gardai, S.J. Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am. J. Respir. Crit. Care Med. 2008, 178, 158–167. [Google Scholar] [CrossRef]
- Legrand, N.; Huntington, N.D.; Nagasawa, M.; Bakker, A.Q.; Schotte, R.; Strick-Marchand, H.; de Geus, S.J.; Pouw, S.M.; Bohne, M.; Voordouw, A.; et al. Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 13224–13229. [Google Scholar] [CrossRef] [Green Version]
- White, C.W.; Greene, K.E.; Allen, C.B.; Shannon, J.M. Elevated expression of surfactant proteins in newborn rats during adaptation to hyperoxia. Am. J. Respir. Cell Mol. Biol. 2001, 25, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, Q.; Tan, F.; Zhang, J.; Zhu, W. Mechanism of IL-8-induced acute lung injury through pulmonary surfactant proteins A and B. Exp. Ther. Med. 2020, 19, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Hough, R.F.; Islam, M.N.; Gusarova, G.A.; Jin, G.; Das, S.; Bhattacharya, J. Endothelial mitochondria determine rapid barrier failure in chemical lung injury. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Spengler, D.; Winoto-Morbach, S.; Kupsch, S.; Vock, C.; Blochle, K.; Frank, S.; Rintz, N.; Diekotter, M.; Janga, H.; Weckmann, M.; et al. Novel therapeutic roles for surfactant-inositols and -phosphatidylglycerols in a neonatal piglet ARDS model: A translational study. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L32–L53. [Google Scholar] [CrossRef]
- Preuss, S.; Scheiermann, J.; Stadelmann, S.; Omam, F.D.; Winoto-Morbach, S.; Lex, D.; von Bismarck, P.; Adam-Klages, S.; Knerlich-Lukoschus, F.; Wesch, D.; et al. 18:1/18:1-Dioleoyl-phosphatidylglycerol prevents alveolar epithelial apoptosis and profibrotic stimulus in a neonatal piglet model of acute respiratory distress syndrome. Pulm. Pharmacol. Ther. 2014, 28, 25–34. [Google Scholar] [CrossRef]
- Mulugeta, S.; Maguire, J.A.; Newitt, J.L.; Russo, S.J.; Kotorashvili, A.; Beers, M.F. Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L720–L729. [Google Scholar] [CrossRef] [Green Version]
- McCubbrey, A.L.; Curtis, J.L. Efferocytosis and lung disease. Chest 2013, 143, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Ryter, S.W.; Choi, A.M. Functional significance of apoptosis in chronic obstructive pulmonary disease. Copd 2007, 4, 347–353. [Google Scholar] [CrossRef]
- Vandivier, R.W.; Ogden, C.A.; Fadok, V.A.; Hoffmann, P.R.; Brown, K.K.; Botto, M.; Walport, M.J.; Fisher, J.H.; Henson, P.M.; Greene, K.E. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: Calreticulin and CD91 as a common collectin receptor complex. J. Immunol. 2002, 169, 3978–3986. [Google Scholar] [CrossRef] [Green Version]
- Schagat, T.L.; Wofford, J.A.; Wright, J.R. Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. J. Immunol. 2001, 166, 2727–2733. [Google Scholar] [CrossRef] [Green Version]
- Palaniyar, N.; Clark, H.; Nadesalingam, J.; Hawgood, S.; Reid, K.B. Surfactant protein D binds genomic DNA and apoptotic cells, and enhances their clearance, in vivo. Ann. N. Y. Acad. Sci. 2003, 1010, 471–475. [Google Scholar] [CrossRef]
- Palaniyar, N.; Nadesalingam, J.; Clark, H.; Shih, M.J.; Dodds, A.W.; Reid, K.B. Nucleic acid is a novel ligand for innate, immune pattern recognition collectins surfactant proteins A and D and mannose-binding lectin. J. Biol. Chem. 2004, 279, 32728–32736. [Google Scholar] [CrossRef] [Green Version]
- Jakel, A.; Reid, K.B.; Clark, H. Surfactant protein A (SP-A) binds to phosphatidylserine and competes with annexin V binding on late apoptotic cells. Protein Cell 2010, 1, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Borron, P.; McCormack, F.X.; Elhalwagi, B.M.; Chroneos, Z.C.; Lewis, J.F.; Zhu, S.; Wright, J.R.; Shepherd, V.L.; Possmayer, F.; Inchley, K.; et al. Surfactant protein A inhibits T cell proliferation via its collagen-like tail and a 210-kDa receptor. Am. J. Physiol. 1998, 275, L679–L686. [Google Scholar] [CrossRef]
- Minutti, C.M.; Jackson-Jones, L.H.; Garcia-Fojeda, B.; Knipper, J.A.; Sutherland, T.E.; Logan, N.; Ringqvist, E.; Guillamat-Prats, R.; Ferenbach, D.A.; Artigas, A.; et al. Local amplifiers of IL-4Ralpha-mediated macrophage activation promote repair in lung and liver. Science 2017, 356, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Marjon, K.D.; Zhu, F.; Weissman-Tsukamoto, R.; Levett, A.; Sullivan, K.; Kao, K.S.; Markovic, M.; Bump, P.A.; Jackson, H.M.; et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat. Commun. 2018, 9, 3194. [Google Scholar] [CrossRef]
- Willems, C.H.; Urlichs, F.; Seidenspinner, S.; Kunzmann, S.; Speer, C.P.; Kramer, B.W. Poractant alfa (Curosurf(R)) increases phagocytosis of apoptotic neutrophils by alveolar macrophages in vivo. Respir. Res. 2012, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Craig, J.M.; Scott, A.L.; Mitzner, W. Immune-mediated inflammation in the pathogenesis of emphysema: Insights from mouse models. Cell Tissue Res. 2017, 367, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Hesse, M.; Sandler, N.G.; Kaviratne, M.; Hoffmann, K.F.; Chiaramonte, M.G.; Reiman, R.; Cheever, A.W.; Sypek, J.P.; Mentink-Kane, M.M. P-selectin suppresses hepatic inflammation and fibrosis in mice by regulating interferon gamma and the IL-13 decoy receptor. Hepatology 2004, 39, 676–687. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Bazzan, E.; Turato, G.; Tine, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar] [CrossRef]
- Willems, C.H.; Zimmermann, L.J.; Kloosterboer, N.; Kramer, B.W.; van Iwaarden, J.F. Surfactant protein A binds TGF-beta1 with high affinity and stimulates the TGF-beta pathway. Innate Immun. 2014, 20, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Thawer, S.; Auret, J.; Schnoeller, C.; Chetty, A.; Smith, K.; Darby, M.; Roberts, L.; Mackay, R.M.; Whitwell, H.J.; Timms, J.F.; et al. Surfactant Protein-D Is Essential for Immunity to Helminth Infection. PLoS Pathog. 2016, 12, e1005461. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Carrillo, M.; Wu, Y.M.; DiAngelo, S.L.; Silveyra, P.; Umstead, T.M.; Halstead, E.S.; Davies, M.L.; Hu, S.; Floros, J.; et al. SP-R210 (Myo18A) Isoforms as Intrinsic Modulators of Macrophage Priming and Activation. PLoS ONE 2015, 10, e0126576. [Google Scholar] [CrossRef] [Green Version]
- Minutti, C.M.; Garcia-Fojeda, B.; Saenz, A.; de Las Casas-Engel, M.; Guillamat-Prats, R.; de Lorenzo, A.; Serrano-Mollar, A.; Corbi, A.L.; Casals, C. Surfactant Protein A Prevents IFN-gamma/IFN-gamma Receptor Interaction and Attenuates Classical Activation of Human Alveolar Macrophages. J. Immunol. 2016, 197, 590–598. [Google Scholar] [CrossRef] [Green Version]
- Ota, C.; Yamada, M.; Fujino, N.; Motohashi, H.; Tando, Y.; Takei, Y.; Suzuki, T.; Takahashi, T.; Kamata, S.; Makiguchi, T.; et al. Histone deacetylase inhibitor restores surfactant protein-C expression in alveolar-epithelial type II cells and attenuates bleomycin-induced pulmonary fibrosis in vivo. Exp. Lung Res. 2015, 41, 422–434. [Google Scholar] [CrossRef]
- Nureki, S.I.; Tomer, Y.; Venosa, A.; Katzen, J.; Russo, S.J.; Jamil, S.; Barrett, M.; Nguyen, V.; Kopp, M.; Mulugeta, S.; et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Investig. 2018, 128, 4008–4024. [Google Scholar] [CrossRef] [Green Version]
- Grutters, J.C.; du Bois, R.M. Genetics of fibrosing lung diseases. Eur. Respir. J. 2005, 25, 915–927. [Google Scholar] [CrossRef] [Green Version]





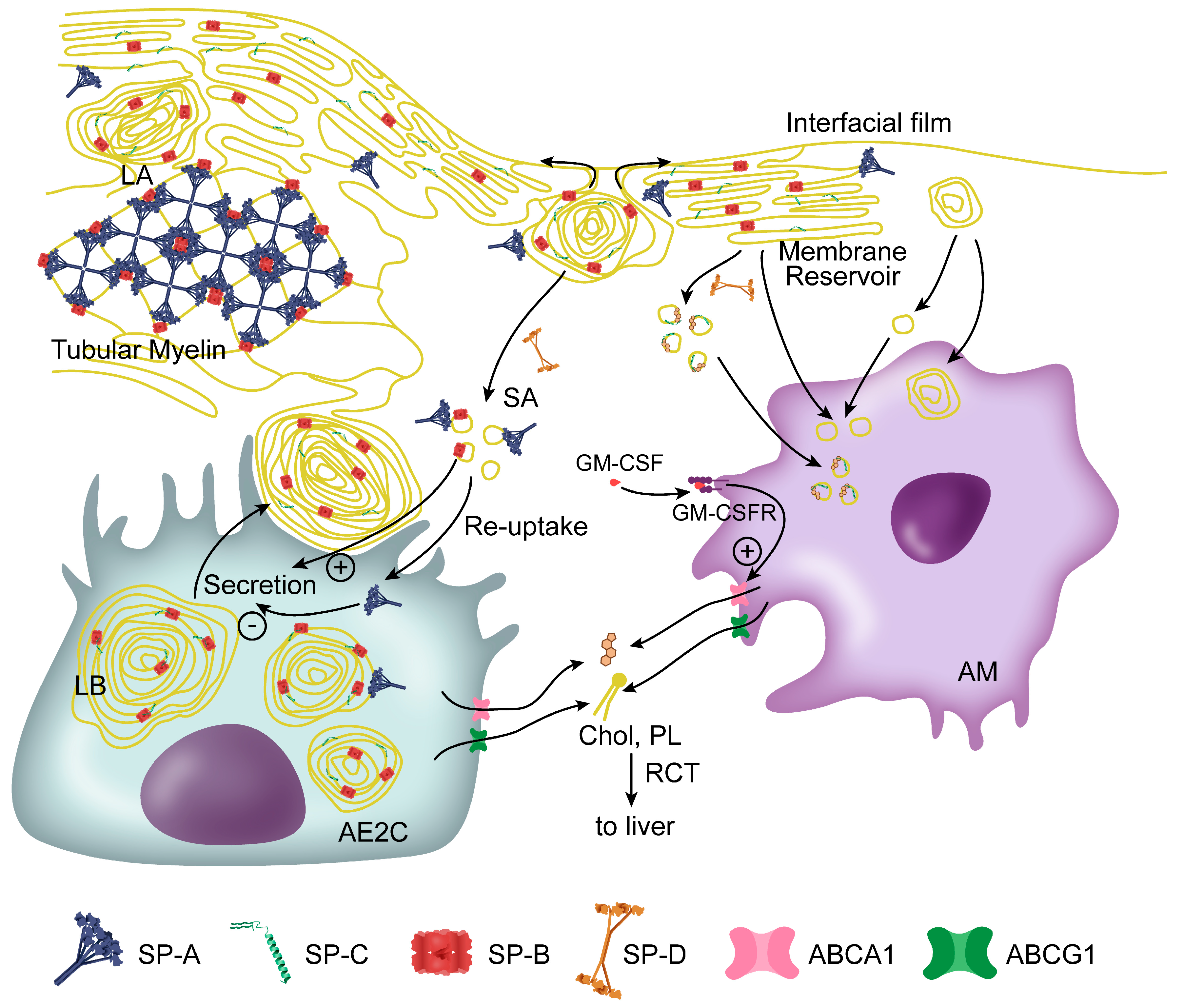{kind=link}
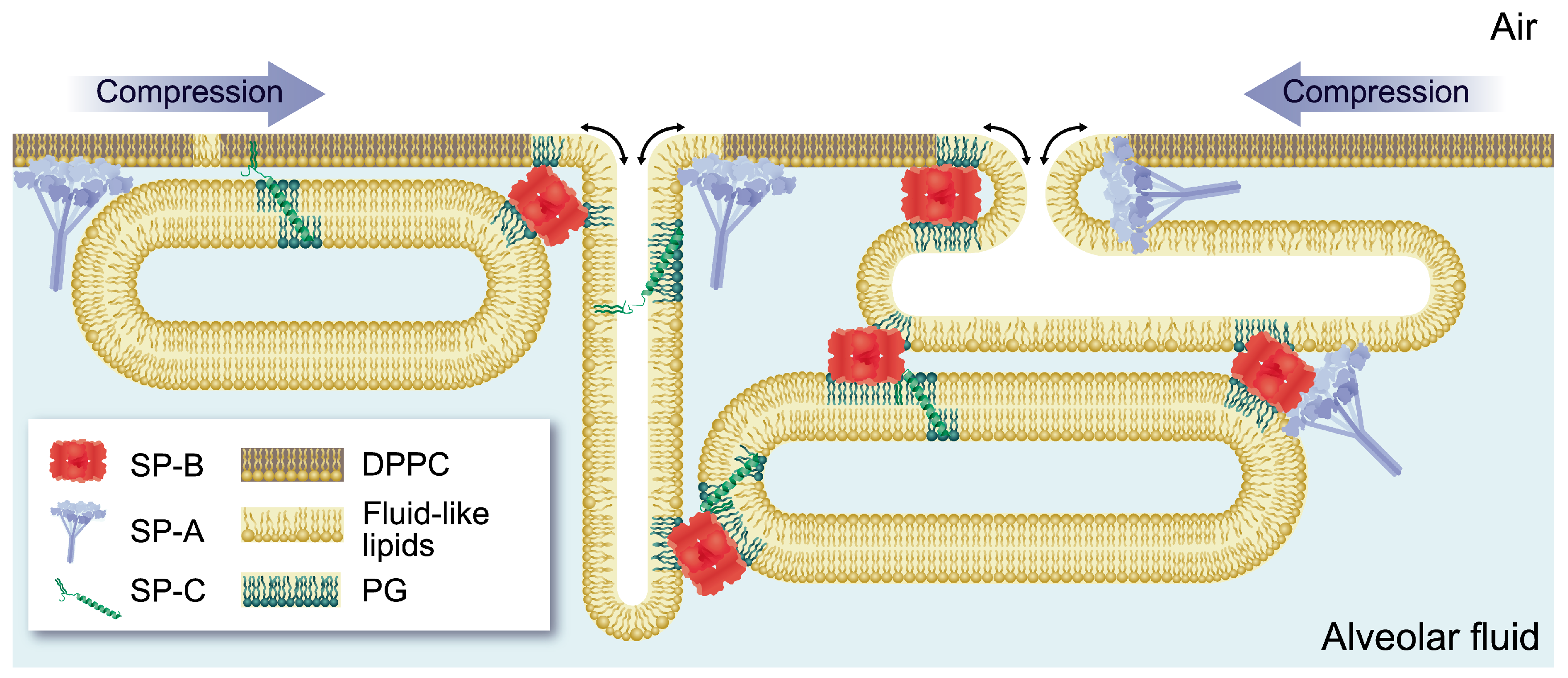{kind=link}
{kind=link}
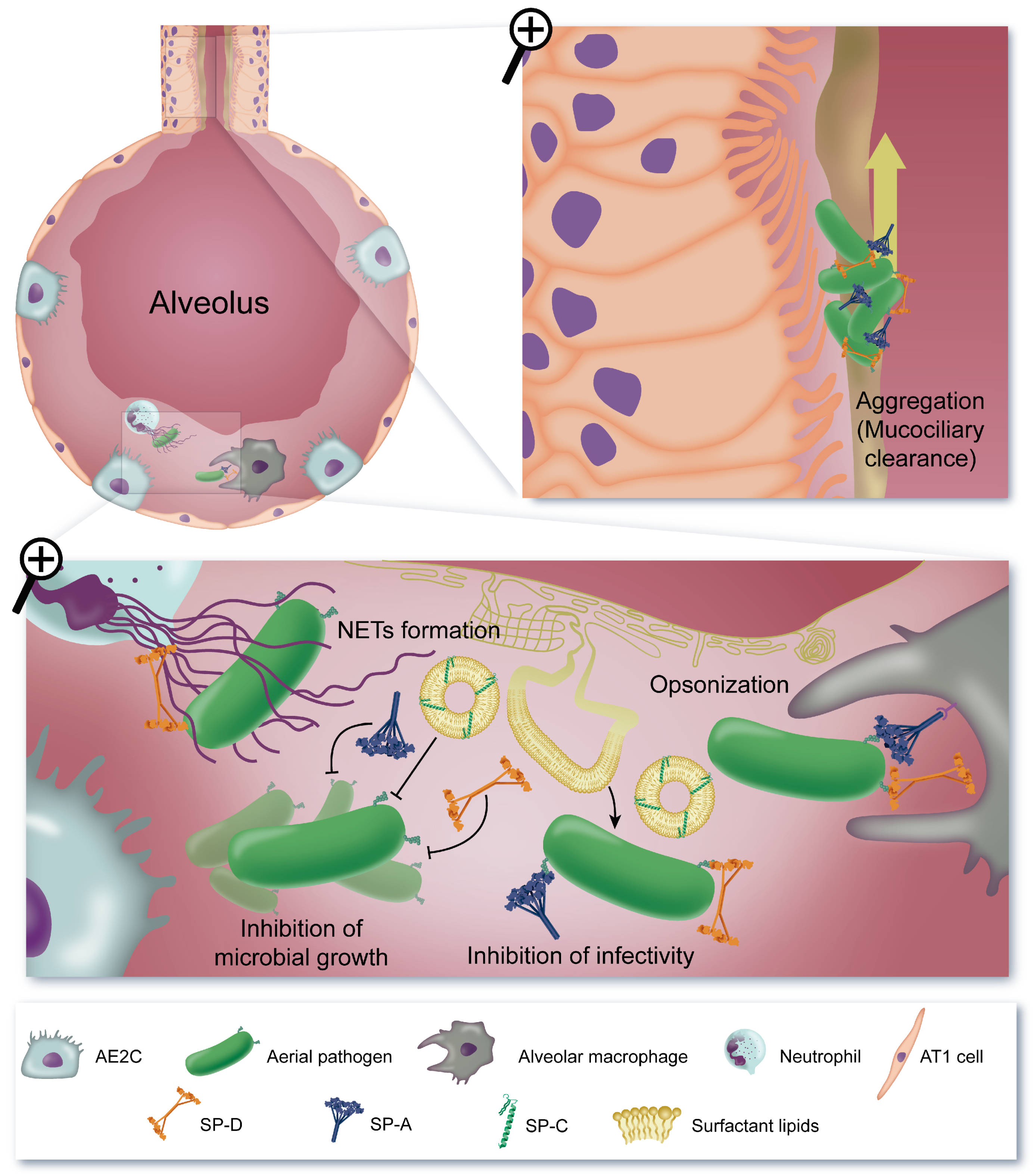{kind=link}
| Surfactant Protein | Interacting Protein or Lipid | Functional Role | References |
|---|---|---|---|
| SP-A | SP-B | Interfacial adsorption Secretion regulation * Cohesivity of multilayer reservoir * | [43,80,81,86] [30] [80] |
| SP-B/PG | Interfacial adsorption Tubular myelin formation | [89] [24] | |
| DPPC | Lipid re-uptake to AE2C | [90] | |
| Cholesterol | Protection against inhibition | [83] | |
| PLA2 | Lipid degradation inhibition | [91] | |
| SP-B | SP-B | Interfacial adsorption (lipid transfer) Permeability Cohesivity (membrane stacking) LB assembly (membrane stacking) | [22,45,46,53] [59,66] [46,52] [52] |
| SP-C | Interfacial adsorption Modulation of permeability and lipid transfer Pro SP-C processing * | [22,23] [59,66,68] [68] | |
| PG | Interfacial adsorption and film stability | [45,58,59,92] | |
| Cholesterol * | Unknown | [45] | |
| SP-C | PG | Interfacial adsorption | [58] |
| Cholesterol | Cholesterol removal from interfacial film (refining) Cholesterol removal from alveolar spaces * Crosstalk AE2C-AM | [47,69,77] [69] [78] | |
| SP-D | PI | Regulation of re-uptake to AE2C | [27,93,94] |
| GPR116 * | Regulation of secretion and re-uptake * | [95] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cañadas, O.; Olmeda, B.; Alonso, A.; Pérez-Gil, J. Lipid–Protein and Protein–Protein Interactions in the Pulmonary Surfactant System and Their Role in Lung Homeostasis. Int. J. Mol. Sci. 2020, 21, 3708. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103708
Cañadas O, Olmeda B, Alonso A, Pérez-Gil J. Lipid–Protein and Protein–Protein Interactions in the Pulmonary Surfactant System and Their Role in Lung Homeostasis. International Journal of Molecular Sciences. 2020; 21(10):3708. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103708
Chicago/Turabian StyleCañadas, Olga, Bárbara Olmeda, Alejandro Alonso, and Jesús Pérez-Gil. 2020. "Lipid–Protein and Protein–Protein Interactions in the Pulmonary Surfactant System and Their Role in Lung Homeostasis" International Journal of Molecular Sciences 21, no. 10: 3708. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103708