Animal Models to Translate Phage Therapy to Human Medicine


1
Dipartimento di Biotecnologie Mediche e Medicina Traslazionale, Università degli Studi di Milano, LITA, Via Fratelli Cervi 93, Segrate, 20090 Milano, Italy
2
Dipartimento di Scienze Cliniche e Comunità, Università degli Studi di Milano, Via San Barnaba 8, 20122 Milano, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(10), 3715; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103715
Submission received: 13 April 2020
/
Revised: 21 May 2020
/
Accepted: 22 May 2020
/
Published: 25 May 2020
(This article belongs to the Section Molecular Microbiology)
Abstract
:Phagotherapy, the use of bacteriophages to fight bacterial infections as an alternative to antibiotic treatments, has become of increasing interest in the last years. This is mainly due to the diffusion of multi-drug resistant (MDR) bacterial infections that constitute a serious issue for public health. Phage therapy is gaining favor due to its success in agriculture and veterinary treatments and its extensive utilization for human therapeutic protocols in the Eastern world. In the last decades, some clinical trials and compassionate treatments have also been performed in the Western world, indicating that phage therapy is getting closer to its introduction in standard therapy protocols. However, several questions concerning the use of phages in human therapeutic treatments are still present and need to be addressed. In this review, we illustrate the state of art of phage therapy and examine the role of animal models to translate these treatments to humans.
1. Phages for Therapy: Positive and Negative Outcomes
Bacteriophages (phages) are viruses that specifically infect and multiply within the bacteria [1]. The use of phages to counteract bacterial infection dates to almost one century ago, and their use has never been abandoned completely, although it was mostly eclipsed by the advent of antibiotics. Nowadays, phages are regaining interest to overcome the development of antibiotic resistant bacteria [2]. However, phages are extremely appealing but also frightening in some aspects, particularly due to the incomplete knowledge of their mechanism of action. In the first part of this review, we describe phage characteristics, highlighting positive and negative aspects for their use in clinics (Figure 1).
The end point of phage infection is the death of the bacterium, usually through its lysis, and the release of progeny virions. Phages that have only this way of multiplication are called lytic phages and are suitable for phage therapy. Other phages, called lysogenic, may parasitize the host, leaving their genome inside the infected bacterium for generations [3]. In the lysogenic condition, the bacterium acquires immunity to superinfection of phages of the same type, a bad outcome for the purpose of phage therapy. Therefore, lysogenic phages are not used for therapy [4].
Another negative feature of several phages is their ability to transduce parts of the bacterial genome following the infection. This could cause the transmission of noxious or virulence genes in the bacterial population [5]. The possibility of the transducing ability of the phage used for therapy has to be checked. Moreover, some phages contain potentially harmful genes in their genomes. It is good practice to analyze the whole genome of the phage used for phage therapy to exclude it. Phage genome analyses could be a time-consuming technique incompatible with the need for urgent infection treatments in which phages are able to infect the bacteria of the patients and should be quickly identified and administered.
In phage therapy, it is appropriate to decide whether to use a single phage or a mix (cocktail) of phages with different characteristics. Given the high specificity of phage infection for a certain bacterial strain, the use of multiple phages is often better at containing an infection [6]. In particular, if the bacterial strain undergoes a mutation to resistance (e.g., mutation of the bacterial gene encoding the specific receptor necessary for phage adsorption or mutation in a bacterial function essential for phage reproduction), the presence of phages using different receptors or alternative functions will overcome the defeat of a single phage by the success of another. In addition, the use of phage cocktails that infect different bacterial species is often used in cases of skin infections in which different bacterial species are normally present [7].
Custom-designed phage cocktails are currently used for therapeutic treatments in countries of Eastern Europe, especially Georgia, where bacteriophages preparations are considered as pharmaceutics and prepared in authorized pharmacies. In Western countries, the situation is more complicated, mainly because no current rules for the use of phages as drugs have been developed until now. Indeed, as one of the main goals of phage therapy is the tailored treatment of an acute infection of an individual patient, it is difficult to apply the standard methods used for medical products (i.e., random and double-blinded clinical trials) for their commercialization. Moreover, non-engineered phages are natural compounds that cannot be patented, thus making their production of poor economic interest [8]. Recently, a few clinical trials have also been conducted in Western countries [9], and patient-tailored-phage therapy has been used for compassionate studies, such as to counteract Acinetobacter baumanii, Pseudomonas aeruginosa, and Mycobacterium abscessus infections [10,11,12]. Phage cocktails were prepared by combining the efforts of different laboratories and then intravenously injected in the patients and used in combination with antibiotics. All the published phage therapies were effective against the life-threatening disseminated infections of the patients.
Considering the time required for isolating phages from the environment, it would be of great interest to generate a phage bank containing libraries of characterized phages and a phage preparation storage at higher phage titer for rapid delivery, as is done in the Eastern countries [13]. One intriguing scenario could be the generation of a bank containing phages targeting all the multi-drug resistant (MDR) bacteria isolated from patients in each sanitary structure.
Phage preparation for human medical uses requires strict purification protocols to prevent endotoxin contamination. For studies in animal models, a sufficient degree of purification is achieved by CsCl gradient ultra-centrifugation [14] with subsequent endotoxin removal. Chromatographic methods can be also used for phage purification as well [15]. In chromatography-purified phages, endotoxin levels are decreased 10- to 30-fold with respect to the traditional method, but often the final phage titer is lower.
For human administration, the upper endotoxin (EU) threshold was defined at 5 EU/kg per h according to European Pharmacopeia regulations (FDA guideline, QAS11-452_FINAL_July12). Specialized institutes such as the Center for Phage Technology (CPT) or the Eliava Institute of Tblisi (Georgia) produce and provide large-scale, highly purified phages for clinical or research purposes [10,16,17,18,19,20]. Stability of phage preparations is essential to achieve efficient phage administration over time.
However, since each specific phage is different from another in its sensitivity to chemical and environmental factors, a universal strategy for their preparation is not possible yet. Usually, phages are resuspended in simple aqueous solutions. However, a gradual loss of phage activity can be observed during long-term storage of phage solutions, and, therefore, stabilizers must be added. Given the proteinaceous nature of phage capsids, protein stabilizers are usually added to phage preparations, including sugars (e.g., sucrose) and polymers (e.g., polyethylene glycol) [15]. Alternatively, phage solutions can be lyophilized and converted into powder with a high grade of stability [21].
2. Animal Models for Testing Phage Therapy
In the last years, several animal models of the most common and relevant human bacterial infections have been created and used to test newly isolated phages and their efficacy in fighting these pathogens in vivo [22]. Animal models of bacterial infection are necessary tools to (i) verify the efficacy of phage therapy in vivo, (ii) search for possible adverse effects, (iii) unravel interactions with the host (e.g., immune system activation). In the second part of this review, we describe how the generation of animal models of bacterial infections might help in the translation of phage therapy to human clinics.
2.1. Phage Therapy and Antimicrobial Action Using Invertebrate and Vertebrate Animal Models
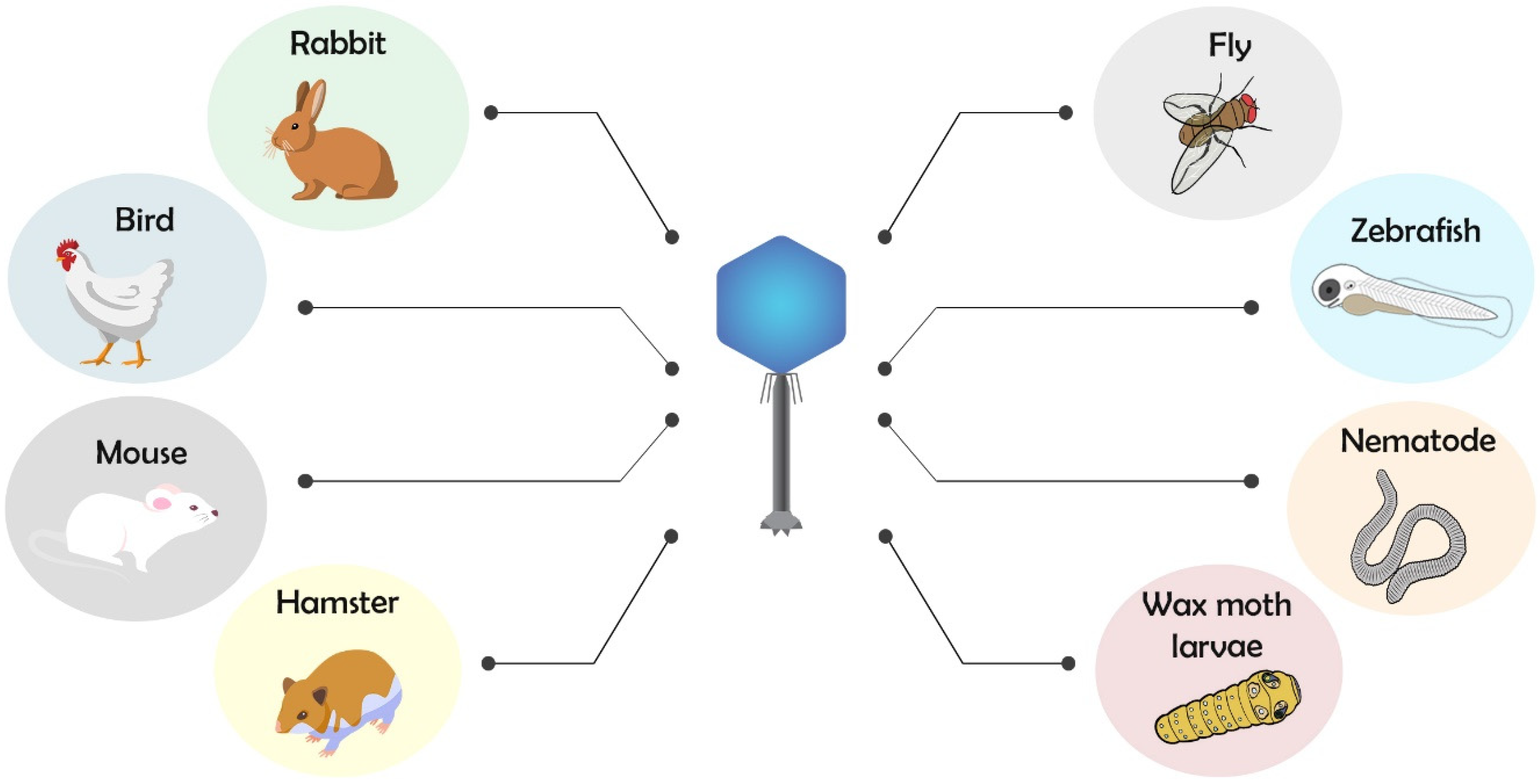
Among the main used invertebrate or lower vertebrate models for phagotherapy, there are nematode (Caenorhabditis elegans), common fruit fly (Drosophila melanogaster), wax moth (Galleria mellonella), and zebrafish (Danio rerio), while for higher vertebrate, there are chicken (Gallus gallus), rabbit (Oryctolagus cuniculus), hamster (Mesocricetus auratus), and mouse (Mus musculus) models (Figure 2).
C. elegans is a small-size nematode (1 mm in length) that can be easily infected by bacteria, fungi, and virions inducing lethality of non-lethal infections [23,24]. The long list of pathogens infecting C. elegans also includes common human bacteria such as P. aeruginosa. Moreover, bacterial virulence factors that induce lethality in nematode are conserved in mammals, opening new opportunities in the use of C. elegans for large screening studies. While avoiding professional immune cells, in C. elegans, the defense to pathogens is mediated by epithelial cells that activate autophagy and the immune system though the production of antimicrobial proteins, peptides (AMPs), and p38 pathway activation [25]. The infection in nematodes can be easily achieved, as their nutritional source is the bacteria, thus pathogens primarily colonize the intestine, and phages can be delivered via the same route of administration. Augustine et al. (2014) and Glowacka-Rutkowska et al. (2019) [26,27] established C. elegans models for Salmonella enteritidis and Staphylococcus aureus infections and phage therapy application. In both cases, the bacteriophage administration resulted in a considerable increase in the survival of infected larvae. Remarkably, the healthy state of the recovered nematodes was confirmed by the fact that they produced healthy progeny after 100 h after phage treatment. Although these two studies take into account the mortality as a unique parameter for testing a phage’s efficacy and effects, the results indicated that C. elegans can be a useful animal model for these studies.
Among non-vertebrate infection models, insects have a strong potential due to their complex innate immune system, which shows high similarity to those of mammals [28,29]. Moreover, they are considered suitable alternative models to larger mammals for bacterial colonization studies and excellent tools for pharmacokinetic studies of antimicrobials [28,30,31]. In two different studies, D. melanogaster was used to evaluate the therapeutic effect of phages against P. aeruginosa infections. In the first study done by Lindberg et al. (2014) [32], the authors investigated the pharmacokinetics and the possible toxicity of phages by themselves. Phage solutions were mixed to corn meal-dextrose medium and administered to healthy flies. The presence of live bacteriophages in the flys’ lysates at different time points after treatment demonstrated that phages survived and were not degraded in the gastrointestinal system. This suggests that oral administration can be successfully studied in animal models, highlighting the interesting possibility of using D. melanogaster to test oral administration of phages. Moreover, the absence of lethality after phage administration indicates that phage treatments are safe and free of toxicity. In the second work done by Heo et al. (2009) [33], the authors compared the effects of P. aeruginosa infection and phage administration in mice and D. melanogaster. The use of two infection models is important to confirm the antibacterial activity of phages against P. aeruginosa that activates different virulence factors depending on the host. Given the promising potential of D. melanogaster as a simple, rapid and cheap animal model to conduct studies on bacterial infection and phage therapy, a guided protocol has recently been set up to evaluate the antibacterial efficacy of new bacteriophages against P. aeruginosa infection in this model [34].
Another invertebrate used for microbial infection and phage therapy is G. mellonella. In a study done by Seed et al. (2009) [35], different bacteriophages were efficiently administrated in G. mellonella larvae to treat Burkholderia cepacia infection. The authors also addressed if the protective effect observed in the treated larvae was due to bacteriophages’ action rather than to the reaction of the immune system of the host triggered by the phage injection. They found that heat-inactivated phages activated the immune system but did not improve larvae survival, indicating that the antibacterial action depended on active phage multiplication. Interestingly, two different studies in wax moth larvae also reported a prophylactic efficacy when phage cocktail was injected or orally administrated. In the first study done by Nale et al. (2016) [36], a four-phage cocktail able to disrupt C. difficile biofilm was effective in increasing survival when added to the larvae food, preventing bacterial colonization. In a second study done by Forti et al. (2018) [37], the six-phage cocktail initially used to prevent P. aeruginosa infections in G. mellonella efficiently counteracted lung infections in the mouse. This result showed that the same bacteriophages can function in different species, both invertebrates and vertebrates.
Zebrafish is gaining favor as a model for the study of host-bacterial interactions, especially in its embryonic stage [38,39,40]. The presence of a developed innate immune system, genetic tractability, and optical transparency of the embryos make it useful for studying aspects of infectious diseases not accessible in traditional animal models. Recently, some zebrafish models were set up to study bacterial infections such as Enterococcus faecalis and P. aeruginosa for phage therapy application [41,42]. Systemic infection in zebrafish embryos is performed through the injection of bacteria in the circulation followed by phage administration via the same route. The success of phage therapy treatment was demonstrated by an increased survival of the infected zebrafish embryos, their recovery from the altered morphology caused by bacterial infection, and decreased bacterial burden after plating homogenized embryos. This vertebrate model validates the efficiency of phage therapy in a quick (five days) and cheap way and demonstrates the survival and the efficacy of phages delivered in the blood with an aquatic model.
The use of invertebrates and lower vertebrates such as zebrafish presents several advantages for the research, such as reduced cost and experimental procedure time. However, to translate phage therapy to humans, it is also necessary to use higher vertebrate models. For instance, in birds, oral phage administration was applied as prophylaxis or post-infection treatment to counteract salmonellosis, colibacillosis, and campylobacteriosis infections that represent a worldwide economic and health problem in poultry [43,44]. Some studies also investigated the use of encapsulated phages of part of the virion, such as the tailspike domain, to improve phage therapy in chickens [45,46,47]. Given the importance of phage therapy application using birds as animal models, a procedure to test phage efficacy using a chicken embryo of colibacillosis infection was recently described [48].
Rabbits were also used as a model of S. aureus wound infection followed by phages administration [49]. As with humans, but contrary to mice, rabbits naturally suffer for S. aureus infections, representing a suitable animal model to study the invasion of these pathogens. The bacterial infection was performed by subcutaneous injections that generated abscesses. Phage administration was performed simultaneously to bacteria or immediately after but in the same location. To evaluate the efficacy of phage therapy, animals were killed four or six days after infection, and the bacterial load in the abscesses area was evaluated. In another study presented by Kishor et al. (2016) [50] and commented upon by Abedon (2016) [51], phage therapy was tested in a rabbit model of S. aureus infection. Although authors demonstrated the feasibility of phage therapy to cure bacterial infection, the rabbit model was different from the patient’s situation in which bacterial infection was chronic, and phage therapy was applied after the failure of conventional approaches. A prophylactic effect of phages to prevent or reduce bacterial infection was demonstrated in new-born mouse and rabbit models infected by Vibrio cholerae by Yen et al. (2017) [52].
An interesting work by Nale et al. (2016) [53] demonstrated that hamsters infected with Clostridium difficile and orally administered with phage cocktail showed increased survival rate. Due to the lack of virulent lytic phages infecting C. difficile, in this study, temperate phages were used. Thus, it was unsuitable for therapeutic treatment. However, the authors showed how the combination of multiple phage types might limit their harmful impact.
Among mammals, murine models are the most frequently used to study phage therapy. Given their high similarity with humans, they have been used not only to demonstrate the efficacy of the classical phage therapy [37,54,55,56,57] but also to investigate the interactions between phages and the host immune system. This second issue is treated in detail in the following chapter.
In Table 1, we resume the studies on animal models and antibacterial activity of phages.
2.2. Phages and Immune System Interactions Studies Using Animal Models
Mouse models of bacterial infection and phage treatment have also been used to investigate different aspects of phage activity in vivo, such as the interaction between phages, bacteria, and the host immune system. For example, Abd El-Aziz and colleagues (2019) [63] used a mouse model of P. aeruginosa infection to investigate the synergism between phages and the innate immunity of the host considering the activity of phages in serum. When the serum and the phages were added to the bacterial culture, an increased antimicrobial activity of phages was achieved. On the contrary, when heat-inactivated serum was added, the phage antimicrobial activity was not increased. This might explain why phages are more efficient in counteracting bacterial infection when administered intravenously than in lungs of infected mice.
Roach et al. (2017) [64] used immunodeficient mice Myd88−/−, Rag−/−, Il2rg−/−, and a neutrophil-depleted line to dissect the contribution of immunity cells to phage–host synergy. Upon P. aeruginosa infection, only the neutrophils-depleted mice were completely unresponsive to phage treatment. Rag−/− and Il2rg−/− mice lacking two key genes for lymphocyte function behaved as the wild-type. An intermediate situation with an initial response followed by the proliferation of phage-resistant bacteria was achieved when Myd88, the main adaptor for Toll-like Receptor (TLR) pathway, was depleted. Both studies suggest an interplay between phages and innate immune response involving complement cascade pathway and neutrophils activity—an important aspect to consider in view of phage therapy application to immunodeficient patients. Another study done by Trigo et al. (2013) [60] investigated the immune response in a mouse footpad infected by Mycobacterium ulcerans. Subcutaneous phage administration reduced bacterial proliferation both in the skin and in the draining lymph nodes, thus preventing ulcerations. Moreover, histopathological analyses of the necrotic tissues of phage-treated mice showed extended macrophages and lymphocytic infiltrates, which colocalize with few residual bacteria, indicating a phage-mediated activation of phagocytes and adaptative response.
Immune response and phage therapy were addressed also by Cafora et al. (2019) [42] in a zebrafish model of cystic fibrosis (CF). A P. aeruginosa systemic infection was generated in CF zebrafish embryos and treated effectively by phage administration. In addition, a decrease of pro-inflammatory cytokine levels was observed in phage-treated infected embryos. Interestingly, CF zebrafish embryos showed a basal inflammatory status in the absence of bacterial infection similar to the high inflammation presented in human CF patients. Phage administration in CF embryos reduced the levels of pro-inflammatory cytokines, suggesting that phages might modulate the innate immune system and therefore opening the possibility to use bacteriophages as anti-inflammatory agents in conditions of constitutive inflammation.
Another important aspect is phage immunogenicity, which is the aptitude of phages to induce specific immune responses with the production of specific antibodies against phage antigens. Opinions and data collected about phage immunogenicity in humans are few and contradictory. Importantly, some clinical outcomes indicate that phages widely vary in their immunogenicity depending on phage-type, dose, route of administration, and host immune status. In general, no strict dependence between phage treatment efficacy and level of antiphage-antibodies emerged [65,66,67,68]. Animal models contributed to shed light on this debatable question. The dynamics of phage immunogenicity was studied by Capparelli et al. (2007) [61] in a mouse model in which a dose of 107 plaque forming units (pfu) of S. aureus phages were intravenously administrated at intervals of two weeks. Phage presence persisted in the blood circulation for approximately 21–25 days and, although antibodies against phages were present, they did not neutralize phage-antibacterial activity. Similar immunogenicity tolerance was obtained by Roach et al. (2017) [64] in a mouse model intranasally inoculated with a single dose of approximately 109 pfu of P. aeruginosa phages; a high degree of phage persistence was observed in the airways of all tested mice in the four days after the administration. Another study in mice was performed by Majewska et al. (2015) [69] in the absence of bacterial infection to analyze the immunological response of long-term exposure of T4 phage (100 days of continuous administration). The T4 phage was administrated in the drinkable water at a concentration of 4 × 109 pfu/mL, and the evaluation of Immunoglobulin (Ig)M, IgG, and secretory IgA production in serum and gut was correlated to the microbiological profile of the mouse at day 240. IgM induction was detected only after IgG boost that started from 36 days and remained sustained even after phage removal from the diet. IgA secretion was detected after 79 days and gradually decreased after T4 removal. After 100 days, the production of IgG was dependent on the phage titer administration (higher dose 4 × 109 pfu/mL, lower dose 4 × 108 pfu/mL). These studies using animal models suggest that specific phage-humoral responses could occur, but it might be dependent on route, dose, and time of administration.
An interesting point concerns the T-cell proliferative rate in response to phages. In a study done by Kim et al. (2008) [70], explanted murine T-cells exposed to salmonella phages showed a higher proliferative response when the donor mice were pre-exposed in vivo to salmonella phages in comparison to T-cells isolated from non-pre-treated mice, suggesting that phages might activate per se the host immune system. Phage proteins differ in their capacity to activate humoral responses. Indeed, Dabrowska et al. (2014) [71] showed that a pre-immunization of mice with purified T4 phage capsid proteins gp23, gp24, Hoc, or Soc impaired the antimicrobial activity of T4 when mice were infected with Escherichia coli and administered with the phages. These important results obtained in vivo in an animal model of infection demonstrated that phages may have reduced activity to counteract bacteria as they are subjected to specific immunization due to phage capsid proteins.
2.3. Route of Phage Administration in Animal Models
Phagotherapy is efficient only when a sufficient amount of phages are able to reach the bacteria and kill them [72]. Three main routes of phage administration have been tested in human clinical trials or case reports depending on the infection type and the localization: topical, intravenous, and oral. Each of these routes presents some complications that still must be solved before translation of phage therapy in human standard medical procedures. Animal models represent a useful tool to investigate these aspects and to optimize phage application in humans.
Topical phage application has been largely used for the treatment of bacterial infections associated with ulcers, surgical wounds, or burns [73]. Recently, several cases of compassionate bacteriophage treatments of diabetic foot ulcers have been reported as successful [18,19]. This is of particular importance, as diabetic patients are commonly immunocompromised with nephropathy and hepatic insufficiency, and prolonged antibiotic treatments are not well tolerated. No animal models of diabetic ulcers are available to test phage therapy to date, but several mouse models of skin ulcers, burn wounds, and infections were topically treated with phages [59,60,74] safely and without adverse effects. These data in mice suggest that ineffective results in two small human burn trials [75,76] are probably linked to an incorrect combination of modality, doses, and times of application rather than to ineffectiveness of phage therapy.
Besides treatment of epithelial lesions, topical phage therapy has been successfully used to treat infections of specific tissues or organs. For example, inhalation of phage solutions has proven to be effective in counteracting MDR bacterial lung infections both in humans [77] and in mouse models [62,78].
Although the size and the immune systems of animal models are different from humans, a pre-screening of phage compound in animals might at least optimize treatment schedules and anticipate any adverse/toxic effects possibly elicited by topical phage administration.
Intravenous (IV) administration consists of injecting the phage preparations directly in the bloodstream, an ideal route for the treatment of bacteremia or widespread infections. The study of Speck and Smithyman (2015) [79] presented IV phage therapy as safe and effective, but some aspects have been questioned. One of the most common objections is that the phage’s lytic activity could lead to the release of great amounts of endotoxins directly in the bloodstream, triggering a strong immune response and anaphylaxis. Notably, Duplessis et al. (2008) [80] reported an anaphylactic response in a pediatric case after IV phage administration, and the authors did not exclude a link to endotoxin release.
Moreover, phages could be rapidly cleared from the blood, losing their effectiveness. These concerns must be deeply investigated prior to introducing IV phage therapy in standard medical procedures. Therefore, several studies about the pharmacokinetics of phages administered with IV injection have been conducted in rodents. Dąbrowska (2016) [81] demonstrated that, after IV injection, phages are not simply diluted in the body volume but are probably neutralized by anti-phage antibodies and removed from the bloodstream by the phagocyte system.
Oral administration of bacteriophages involves gastro-intestinal transit and might have implications for the entire organism. Two main issues must be considered: first, endotoxins released by lysed bacteria can be absorbed and enter the blood stream, leading to systemic inflammation; second, the endogenous microbiome, which is crucial for digestive processes and defense against pathogens, can be altered.
Several safety tests have been done to verify the efficacy of oral administration and exclude possible adverse effects due to the presence of the phage itself [16,68,82]. These studies also reported that the overall composition of the microbiota remains quite stable upon phage administration. Although active phages were found in fecal samples after oral administration, it is not clear if phages can bypass the gastric environment without being killed massively by the acid solutions. Again, in vivo studies using Drosophila as an animal model provide important evidence that phages can be orally administered with food and survive through the gastro-intestinal tract [33]. Another study on pigs done by Jamalludeen and colleagues (2009) [58] showed that an anti-acid pre-treatment with sodium bicarbonate significantly increases the number of active phages in feces and reduces the severity of diarrheal symptoms. Alternatively, alginate/CaCO3 microencapsulation decreases phage degradation in the gastric environment, thus ameliorating phage efficacy as reported by Colom and colleagues using a chicken model of S. enterica infection [45].
2.4. Methods to Improve Phage Therapy Using Animal Models
One of the most promising aspects of phage therapy is the possibility to perform combined treatments with antibiotics commonly used to treat bacterial infection. This is of particular interest, as it reduces the time and the doses of antibiotic administration, diminishing the development of antibiotic-resistant bacteria and adverse effects of the drugs on the host (i.e., microbiome destruction). A successful combined treatment of phages and antibiotics was described by Cafora et al. (2019) [42] in the zebrafish CF model with P. aeruginosa infection. The reduction in mortality rate following phage cocktail administration was greater when phages were combined with ciprofloxacin. Moreover, when bacteria mutate and become phage resistant, it is possible to isolate new phages using the phage-resistant mutants. Although it is time consuming, this method could be used to overcome resistance in bacteria. Another important consideration is that the bacteria that become phage-resistant display increased sensitivity to the antibiotic treatment that previously failed [83]. For instance, Schooley et al. (2017) [10] showed that a minocycline isolate of Acinetobacter baumannii from a patient with severe pancreatitis displayed reduced minocycline resistance after phage therapy.
Phages may also be considered for prophylaxis to prevent or reduce infections of different bacterial species, as demonstrated in several animal models [36,37,43,54,56,58].
A further interesting possibility to improve phage therapy is the use of liposomes and transfersomes for phage delivery [84]. These vesicles act by creating a broader distribution, preventing rapid degradation and enhancing cellular uptake [85,86]. This possibility was explored in a study done by Chhibber and colleagues (2017) [87] in a rat model of acute skin and soft tissue infection of S. aureus in which transfersomes-entrapped phages performed a faster rescue than free phages. A similar effect of improved phage efficacy was achieved in Colom et al. [46] by using liposome-entrapped phages in a chicken model of S. enterica infection. Therefore, liposomes and transfersomes could be a useful tool to potentiate the efficacy of phage activity.
Last but not least, many studies successfully demonstrated the antibacterial action of some phage enzymes, such as endolysins, virion-associated lysins, and capsular depolymerases [88]. Several studies about phage-derived lysins have been concluded both in animal models and in humans [89], and a recent clinical trial demonstrated the safety of endolysin intravenous administration [90]. In contrast to lysins, depolymerases do not lyse bacterial cells, thereby preventing endotoxin release and inflammation. Moreover, after capsule removal by depolymerases, bacteria are directly exposed to the host immune system and can be more easily killed and removed. The efficacy of depolymerase treatment was demonstrated in vivo in a mouse model of E. coli infection [91]. The same treatment successfully resulted in the rescue of both immunocompetent and leukopenic mice, with a higher efficiency when the enzyme was administrated shortly after infection [92]. One major limitation in the use of depolymerases is that, while phages can replicate autonomously in bacterial cells, enzymes cannot, and therefore the administration of a single dose could not resolve the infections.
3. Conclusions
In conclusion, phages proved time and again to have the appropriate characteristics to be introduced in human clinical treatments. Although several concerns are still present in Western countries, a progressive improvement of their use in clinical trials or for compassionate studies has been achieved. Before translating phage therapy to human clinics, some points still need to be clarified and excluded, as described in this review.
Animal models are an important tool to further understand the mechanisms and the efficacies of bacteriophage action in vivo. Both invertebrates and vertebrates demonstrate the success of such treatments in a way that is cheaper, faster, and more ethical than human clinical trials. The generation of vertebrate animal models to test phages may give a more comprehensive understanding of the mechanisms triggering host immune and inflammatory response to phages, one of the most important concerns of phage therapy application to humans. Several strategies to improve and potentiate phage activity have been tested, underlining the promising strength of bacteriophages or their enzymes in human therapies. Among these, we cited the possibility to improve antibacterial action by enhancing phage delivery, the use of phages mixed in cocktails, the combination of phages with antibiotics, and the possibility to use phages for prophylactic treatments. All these studies can be easily (and cheaply) achieved in animal models before translation to humans.
Moreover, considering that one of the most important goals of modern phage therapy is to rapidly identify phages able to counteract bacterial infection in compassionate studies, animals can be used to indicate the safety of select phages before patient treatment. Since the custom-made use of phages cannot be regulated right now, phage pre-screening in animals for personalized therapies could at least limit some of the concerns [22].
For all these reasons, animal models are a key tool in investigating potential phage therapies and introducing them to human medicine.
Author Contributions
Conceptualization, A.B. and A.P.; writing—original draft preparation, A.B., M.C., M.A. and A.P.; writing—review and editing, supervision, A.P.; project administration, A.P. and M.A.; funding acquisition, A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondazione Ricerca Fibrosi Cistica, FFC#23/2019, “Un respiro in più Onlus, La mano tesa Onlus”.
Acknowledgments
The authors thank Daniela Ghisotti and Akash Singh for their precious help in extensive revision of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| MDR | Multi drug resistant |
| CF | Cystic fibrosis |
| EU | Endotoxin units |
| FDA | Food and drug administration |
| CPT | Center for Phage Technology |
| IV | Intravenous |
| PFU | Plaque forming units |
References
- Bordet, J.; Ciuca, M. Remarques sur l’historique de recherches concernant la lyse microbienne transmissible. Compt. Rend. Soc. Biol. 1921, 84, 745–747. [Google Scholar]
- Abedon, S.T. Use of phage therapy to treat long-standing, persistent, or chronic bacterial infections. Adv. Drug Deliv. Rev. 2019, 145, 18–39. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with ‘Lytic or lysogenic’. FEMS Microbiol. Lett. 2016, 363, fnw047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B.; Delattre, A.-S.; Lavigne, R. Learning from bacteriophages—Advantages and limitations of phage and phage-encoded protein applications. Curr. Protein Pept. Sci. 2012, 13, 699–722. [Google Scholar] [CrossRef] [Green Version]
- Goh, S. Phage transduction. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2016; Volume 1476, pp. 177–185. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Fauconnier, A. Phage therapy regulation: From night to dawn. Viruses 2019, 11, 352. [Google Scholar] [CrossRef] [Green Version]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [Green Version]
- Law, N.; Logan, C.; Furr, C.; Lehman, S.; Morales, S.; Rosas, F.; Gaidamaka, A.; Bilinsky, I.; Grint, P.; Schooley, R.; et al. Successful bacteriophage therapy for treatment of multidrug-resistant pseudomonas aeruginosa infection in a cystic fibrosis patient. J. Hear. Lung Transplant. 2019, 38, S38. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Médecine Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, P. Purification of bacteriophages and SDS-PAGE analysis of phage structural proteins from ghost particles. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2009; Volume 502, pp. 227–238. [Google Scholar]
- Gill, J.; Hyman, P. Phage choice, isolation, and preparation for phage therapy. Curr. Pharm. Biotechnol. 2010, 11, 2–14. [Google Scholar] [CrossRef] [PubMed]
- McCallin, S.; Alam Sarker, S.; Barretto, C.; Sultana, S.; Berger, B.; Huq, S.; Krause, L.; Bibiloni, R.; Schmitt, B.; Reuteler, G.; et al. Safety analysis of a Russian phage cocktail: From MetaGenomic analysis to oral application in healthy human subjects. Virology 2013, 443, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Gindin, M.; Febvre, H.P.; Rao, S.; Wallace, T.C.; Weir, T.L. Bacteriophage for gastrointestinal health (PHAGE) study: Evaluating the safety and tolerability of supplemental bacteriophage consumption. J. Am. Coll. Nutr. 2019, 38, 68–75. [Google Scholar] [CrossRef]
- Fish, R.; Kutter, E.; Bryan, D.; Wheat, G.; Kuhl, S. Resolving digital staphylococcal osteomyelitis using bacteriophage—A case report. Antibiotics 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Fish, R.; Kutter, E.; Wheat, G.; Blasdel, B.; Kutateladze, M.; Kuhl, S. Compassionate use of bacteriophage therapy for foot ulcer treatment as an effective step for moving toward clinical trials. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2018; Volume 1693, pp. 159–170. [Google Scholar] [CrossRef]
- Corbellino, M.; Kieffer, N.; Kutateladze, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Tsertsvadze, G.; Rimoldi, S.G.; Nizharadze, D.; Hoyle, N.; et al. Eradication of a multidrug-resistant, carbapenemase-producing Klebsiella pneumoniae isolate following oral and intra-rectal therapy with a custom made, lytic bacteriophage preparation. Clin. Infect. Dis. 2020, 70, 1998–2001. [Google Scholar] [CrossRef]
- Manohar, P.; Ramesh, N. Improved lyophilization conditions for long-term storage of bacteriophages. Sci. Rep. 2019, 9, 15242. [Google Scholar] [CrossRef] [Green Version]
- Melo, L.D.R.; Oliveira, H.; Pires, D.P.; Dabrowska, K.; Azeredo, J. Phage therapy efficacy: A review of the last 10 years of preclinical studies. Crit. Rev. Microbiol. 2020, 46, 78–99. [Google Scholar] [CrossRef]
- Cohen, L.B.; Troemel, E.R. Microbial pathogenesis and host defense in the nematode C. elegans. Curr. Opin. Microbiol. 2015, 23, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Pukkila-Worley, R.; Ausubel, F.M. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr. Opin. Immunol. 2012, 24, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewbank, J.J.; Zugasti, O. C. elegans: Model host and tool for antimicrobial drug discovery. Dis. Model. Mech. 2011, 4, 300–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, J.; Gopalakrishnan, M.V.; Bhat, S.G. Application of ΦSP-1 and ΦSP-3 as a therapeutic strategy against Salmonella Enteritidis infection using Caenorhabditis elegans as model organism. FEMS Microbiol. Lett. 2014, 356, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Głowacka-Rutkowska, A.; Gozdek, A.; Empel, J.; Gawor, J.; Żuchniewicz, K.; Kozińska, A.; Dębski, J.; Gromadka, R.; Łobocka, M. The ability of lytic staphylococcal podovirus vB_SauP_phiAGO1.3 to coexist in equilibrium with its host facilitates the selection of host mutants of attenuated virulence but does not preclude the phage antistaphylococcal activity in a nematode infection model. Front. Microbiol. 2019, 9, 3227. [Google Scholar] [CrossRef]
- Buchon, N.; Silverman, N.; Cherry, S. Immunity in Drosophila melanogaster—From microbial recognition to whole-organism physiology. Nat. Rev. Immunol. 2014, 14, 796–810. [Google Scholar] [CrossRef]
- Lemaitre, B.; Hoffmann, J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef] [Green Version]
- Hill, L.; Veli, N.; Coote, P.J. Evaluation of Galleria mellonella larvae for measuring the efficacy and pharmacokinetics of antibiotic therapies against Pseudomonas aeruginosa infection. Int. J. Antimicrob. Agents 2014, 43, 254–261. [Google Scholar] [CrossRef]
- Ramarao, N.; Nielsen-Leroux, C.; Lereclus, D. The insect Galleria mellonella as a powerful infection model to investigate bacterial pathogenesis. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, H.M.; McKean, K.A.; Wang, I.-N. Phage fitness may help predict phage therapy efficacy. Bacteriophage 2014, 4, e964081. [Google Scholar] [CrossRef]
- Heo, Y.-J.; Lee, Y.-R.; Jung, H.-H.; Lee, J.; Ko, G.; Cho, Y.-H. Antibacterial efficacy of phages against Pseudomonas aeruginosa infections in mice and Drosophila melanogaster. Antimicrob. Agents Chemother. 2009, 53, 2469–2474. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.-J.; Bae, H.-W.; Cho, Y.-H. Exploitation of Drosophila infection models to evaluate antibacterial efficacy of phages. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2019; Volume 1898, pp. 183–190. [Google Scholar]
- Seed, K.D.; Dennis, J.J. Experimental bacteriophage therapy increases survival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepacia complex. Antimicrob. Agents Chemother. 2009, 53, 2205–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nale, J.Y.; Chutia, M.; Carr, P.; Hickenbotham, P.T.; Clokie, M.R.J. ‘Get in early’; biofilm and wax moth (Galleria mellonella) models reveal new insights into the therapeutic potential of Clostridium difficile bacteriophages. Front. Microbiol. 2016, 7, 1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L.; et al. Design of a broad-range bacteriophage cocktail that reduces Pseudomonas aeruginosa biofilms and treats acute infections in two animal models. Antimicrob. Agents Chemother. 2018, 62, e02573-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Chen, S.; Cao, Z.; Lin, Y.; Mo, D.; Zhang, H.; Gu, J.; Dong, M.; Liu, Z.; Xu, A. Acute phase response in zebrafish upon Aeromonas salmonicida and Staphylococcus aureus infection: Striking similarities and obvious differences with mammals. Mol. Immunol. 2007, 44, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Neely, M.N.; Pfeifer, J.D.; Caparon, M. Streptococcus-zebrafish model of bacterial pathogenesis. Infect. Immun. 2002, 70, 3904–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llamas, M.A.; van der Sar, A.M. Assessing Pseudomonas virulence with nonmammalian host: Zebrafish. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2014; pp. 709–721. [Google Scholar] [CrossRef]
- Al-Zubidi, M.; Widziolek, M.; Court, E.K.; Gains, A.F.; Smith, R.E.; Ansbro, K.; Alrafaie, A.; Evans, C.; Murdoch, C.; Mesnage, S.; et al. Identification of novel bacteriophages with therapeutic potential that target Enterococcus faecalis. Infect. Immun. 2019, 87, e00512-19. [Google Scholar] [CrossRef] [Green Version]
- Cafora, M.; Deflorian, G.; Forti, F.; Ferrari, L.; Binelli, G.; Briani, F.; Ghisotti, D.; Pistocchi, A. Phage therapy against Pseudomonas aeruginosa infections in a cystic fibrosis zebrafish model. Sci. Rep. 2019, 9, 1527. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, M.; Amir Karimi Torshizi, M.; Rahimi, S.; Dennehy, J.J. Prophylactic bacteriophage administration more effective than post-infection administration in reducing Salmonella enterica serovar enteritidis shedding in Quail. Front. Microbiol. 2016, 7, 1253. [Google Scholar] [CrossRef] [Green Version]
- Wernicki, A.; Nowaczek, A.; Urban-Chmiel, R. Bacteriophage therapy to combat bacterial infections in poultry. Virol. J. 2017, 14, 179. [Google Scholar] [CrossRef]
- Colom, J.; Cano-Sarabia, M.; Otero, J.; Aríñez-Soriano, J.; Cortés, P.; Maspoch, D.; Llagostera, M. Microencapsulation with alginate/CaCO3: A strategy for improved phage therapy. Sci. Rep. 2017, 7, 41441. [Google Scholar] [CrossRef]
- Colom, J.; Cano-Sarabia, M.; Otero, J.; Cortés, P.; Maspoch, D.; Llagostera, M. Liposome-encapsulated bacteriophages for enhanced oral phage therapy against Salmonella spp. Appl. Environ. Microbiol. 2015, 81, 4841–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waseh, S.; Hanifi-Moghaddam, P.; Coleman, R.; Masotti, M.; Ryan, S.; Foss, M.; MacKenzie, R.; Henry, M.; Szymanski, C.M.; Tanha, J. Orally administered P22 phage tailspike protein reduces Salmonella colonization in chickens: Prospects of a novel therapy against bacterial infections. PLoS ONE 2010, 5, e13904. [Google Scholar] [CrossRef] [PubMed]
- Trotereau, A.; Schouler, C. Use of a chicken embryo lethality assay to assess the efficacy of phage therapy. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2019; Volume 1898, pp. 199–205. [Google Scholar] [CrossRef]
- Wills, Q.F.; Kerrigan, C.; Soothill, J.S. Experimental bacteriophage protection against Staphylococcus aureus abscesses in a rabbit model. Antimicrob. Agents Chemother. 2005, 49, 1220–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishor, C.; Mishra, R.; Saraf, S.; Kumar, M.; Srivastav, A.; Nath, G. Phage therapy of staphylococcal chronic osteomyelitis in experimental animal model. Indian J. Med. Res. 2016, 143, 87. [Google Scholar] [CrossRef]
- Abedon, S.T. Commentary: Phage therapy of staphylococcal chronic osteomyelitis in experimental animal model. Front. Microbiol. 2016, 7, 1251. [Google Scholar] [CrossRef] [Green Version]
- Yen, M.; Cairns, L.S.; Camilli, A. A cocktail of three virulent bacteriophages prevents Vibrio cholerae infection in animal models. Nat. Commun. 2017, 8, 14187. [Google Scholar] [CrossRef] [Green Version]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepiński, P.; Douce, G.R.; Clokie, M.R.J. Bacteriophage combinations significantly reduce Clostridium difficile growth in vitro and proliferation in vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef] [Green Version]
- Pabary, R.; Singh, C.; Morales, S.; Bush, A.; Alshafi, K.; Bilton, D.; Alton, E.W.F.W.; Smithyman, A.; Davies, J.C. Antipseudomonal bacteriophage reduces infective burden and inflammatory response in murine lung. Antimicrob. Agents Chemother. 2016, 60, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Alvi, I.A.; Asif, M.; Tabassum, R.; Aslam, R.; Abbas, Z.; Rehman, S.u. RLP, a bacteriophage of the family Podoviridae, rescues mice from bacteremia caused by multi-drug-resistant Pseudomonas aeruginosa. Arch. Virol. 2020, 165, 1289–1297. [Google Scholar] [CrossRef]
- Debarbieux, L.; Leduc, D.; Maura, D.; Morello, E.; Criscuolo, A.; Grossi, O.; Balloy, V.; Touqui, L. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J. Infect. Dis. 2010, 201, 1096–1104. [Google Scholar] [CrossRef] [Green Version]
- Leshkasheli, L.; Kutateladze, M.; Balarjishvili, N.; Bolkvadze, D.; Save, J.; Oechslin, F.; Que, Y.-A.; Resch, G. Efficacy of newly isolated and highly potent bacteriophages in a mouse model of extensively drug-resistant Acinetobacter baumannii bacteraemia. J. Glob. Antimicrob. Resist. 2019, 19, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Jamalludeen, N.; Johnson, R.P.; Shewen, P.E.; Gyles, C.L. Evaluation of bacteriophages for prevention and treatment of diarrhea due to experimental enterotoxigenic Escherichia coli O149 infection of pigs. Vet. Microbiol. 2009, 136, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Chadha, P.; Katare, O.P.; Chhibber, S. In vivo efficacy of single phage versus phage cocktail in resolving burn wound infection in BALB/c mice. Microb. Pathog. 2016, 99, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Trigo, G.; Martins, T.G.; Fraga, A.G.; Longatto-Filho, A.; Castro, A.G.; Azeredo, J.; Pedrosa, J. Phage therapy is effective against infection by Mycobacterium ulcerans in a murine footpad model. PLoS Negl. Trop. Dis. 2013, 7, e2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capparelli, R.; Parlato, M.; Borriello, G.; Salvatore, P.; Iannelli, D. Experimental phage therapy against Staphylococcus aureus in mice. Antimicrob. Agents Chemother. 2007, 51, 2765–2773. [Google Scholar] [CrossRef] [Green Version]
- Anand, T.; Virmani, N.; Kumar, S.; Mohanty, A.K.; Pavulraj, S.; Bera, B.C.; Vaid, R.K.; Ahlawat, U.; Tripathi, B.N. Phage therapy for treatment of virulent Klebsiella pneumoniae infection in a mouse model. J. Glob. Antimicrob. Resist. 2020, 21, 34–41. [Google Scholar] [CrossRef]
- Abd El-Aziz, A.M.; Elgaml, A.; Ali, Y.M. Bacteriophage therapy increases complement-mediated lysis of bacteria and enhances bacterial clearance after acute lung infection with multidrug-resistant Pseudomonas aeruginosa. J. Infect. Dis. 2019, 219, 1439–1447. [Google Scholar] [CrossRef]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 2017, 22, 38–47.e4. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Żaczek, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Letkiewicz, S.; Fortuna, W.; Rogóż, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; Olchawa, E.; et al. Antiphage activity of sera during phage therapy in relation to its outcome. Future Microbiol. 2017, 12, 109–117. [Google Scholar] [CrossRef]
- Żaczek, M.; Łusiak-Szelachowska, M.; Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Owczarek, B.; Kopciuch, A.; Fortuna, W.; Rogóż, P.; Górski, A. Antibody production in response to staphylococcal MS-1 phage cocktail in patients undergoing phage therapy. Front. Microbiol. 2016, 7, 1681. [Google Scholar] [CrossRef] [Green Version]
- Łusiak-Szelachowska, M.; Żaczek, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Kłak, M.; Fortuna, W.; Letkiewicz, S.; Rogóż, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; et al. Phage neutralization by sera of patients receiving phage therapy. Viral Immunol. 2014, 27, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruttin, A.; Brüssow, H. Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewska, J.; Beta, W.; Lecion, D.; Hodyra-Stefaniak, K.; Kłopot, A.; Kaźmierczak, Z.; Miernikiewicz, P.; Piotrowicz, A.; Ciekot, J.; Owczarek, B.; et al. Oral application of T4 phage induces weak antibody production in the gut and in the blood. Viruses 2015, 7, 4783–4799. [Google Scholar] [CrossRef]
- Kim, K.-P.; Cha, J.-D.; Jang, E.-H.; Klumpp, J.; Hagens, S.; Hardt, W.-D.; Lee, K.-Y.; Loessner, M.J. PEGylation of bacteriophages increases blood circulation time and reduces T-helper type 1 immune response. Microb. Biotechnol. 2008, 1, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Kaźmierczak, Z.; Letarov, A.; Górski, A. Immunogenicity studies of proteins forming the T4 phage head surface. J. Virol. 2014, 88, 12551–12557. [Google Scholar] [CrossRef] [Green Version]
- Dąbrowska, K.; Abedon, S.T. Pharmacologically aware phage therapy: Pharmacodynamic and pharmacokinetic obstacles to phage antibacterial action in animal and human bodies. Microbiol. Mol. Biol. Rev. 2019, 83. [Google Scholar] [CrossRef]
- Morozova, V.V.; Vlassov, V.V.; Tikunova, N.V. Applications of bacteriophages in the treatment of localized infections in humans. Front. Microbiol. 2018, 9, 1696. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S. Evidence to support the therapeutic potential of bacteriophage Kpn5 in burn wound infection caused by Klebsiella pneumoniae in BALB/c mice. J. Microbiol. Biotechnol. 2010, 20, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Rose, T.; Verbeken, G.; De Vos, D.; Merabishvili, M.; Vaneechoutte, M.; Lavigne, R.; Jennes, S.; Zizi, M.; Pirnay, J.-P. Experimental phage therapy of burn wound infection: Difficult first steps. Int. J. Burns Trauma 2014, 4, 66–73. [Google Scholar]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Hoyle, N.; Zhvaniya, P.; Balarjishvili, N.; Bolkvadze, D.; Nadareishvili, L.; Nizharadze, D.; Wittmann, J.; Rohde, C.; Kutateladze, M. Phage therapy against Achromobacter xylosoxidans lung infection in a patient with cystic fibrosis: A case report. Res. Microbiol. 2018, 169, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.M.; Neill, D.R.; Kaman, B.; Sahota, J.S.; Clokie, M.R.J.; Winstanley, C.; Kadioglu, A. Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax 2017, 72, 666–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speck, P.; Smithyman, A. Safety and efficacy of phage therapy via the intravenous route. FEMS Microbiol. Lett. 2016, 363, 242. [Google Scholar] [CrossRef] [PubMed]
- Duplessis, C.; Biswas, B.; Hanisch, B.; Perkins, M.; Henry, M.; Quinones, J.; Wolfe, D.; Estrella, L.; Hamilton, T. Refractory pseudomonas bacteremia in a 2-year-old sterilized by bacteriophage therapy. J. Pediatric Infect. Dis. Soc. 2018, 7, 253–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dąbrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.A.; McCallin, S.; Barretto, C.; Berger, B.; Pittet, A.-C.; Sultana, S.; Krause, L.; Huq, S.; Bibiloni, R.; Bruttin, A.; et al. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 2012, 434, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, W.N.; Concepción-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and order effects of antibiotics and phages in killing Pseudomonas aeruginosa biofilms. PLoS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef] [Green Version]
- Abu Lila, A.S.; Ishida, T. Liposomal delivery systems: Design optimization and current applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Pradhan, M.; Nag, M.; Singh, M.R. Vesicular system: Versatile carrier for transdermal delivery of bioactives. Artif. Cells Nanomed. Biotechnol. 2015, 43, 282–290. [Google Scholar] [CrossRef]
- Chhibber, S.; Shukla, A.; Kaur, S. Transfersomal phage cocktail is an effective treatment against methicillin-resistant Staphylococcus aureus-mediated skin and soft tissue infections. Antimicrob. Agents Chemother. 2017, 61, e02146-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.; Abedon, S. Bacteriophages and their enzymes in biofilm control. Curr. Pharm. Des. 2014, 21, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V. Development of phage lysins as novel therapeutics: A historical perspective. Viruses 2018, 10, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, S.Y.; Jang, I.J.; Yoon, S.; Jang, K.; Yu, K.-S.; Cho, J.Y.; Seong, M.-W.; Jung, G.M.; Yoon, S.J.; Kang, S.H. Pharmacokinetics and tolerance of the phage endolysin-based candidate drug SAL200 after a single intravenous administration among healthy volunteers. Antimicrob. Agents Chemother. 2017, 61, e02629-16. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Paff, M.L.; Molineux, I.J.; Bull, J.J. Therapeutic application of phage capsule depolymerases against K1, K5, and K30 capsulated E. coli in mice. Front. Microbiol. 2017, 8, 2257. [Google Scholar] [CrossRef]
- Lin, H.; Paff, M.; Molineux, I.; Bull, J. Antibiotic therapy using phage depolymerases: Robustness across a range of conditions. Viruses 2018, 10, 622. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Positive and negative outcomes of phages.
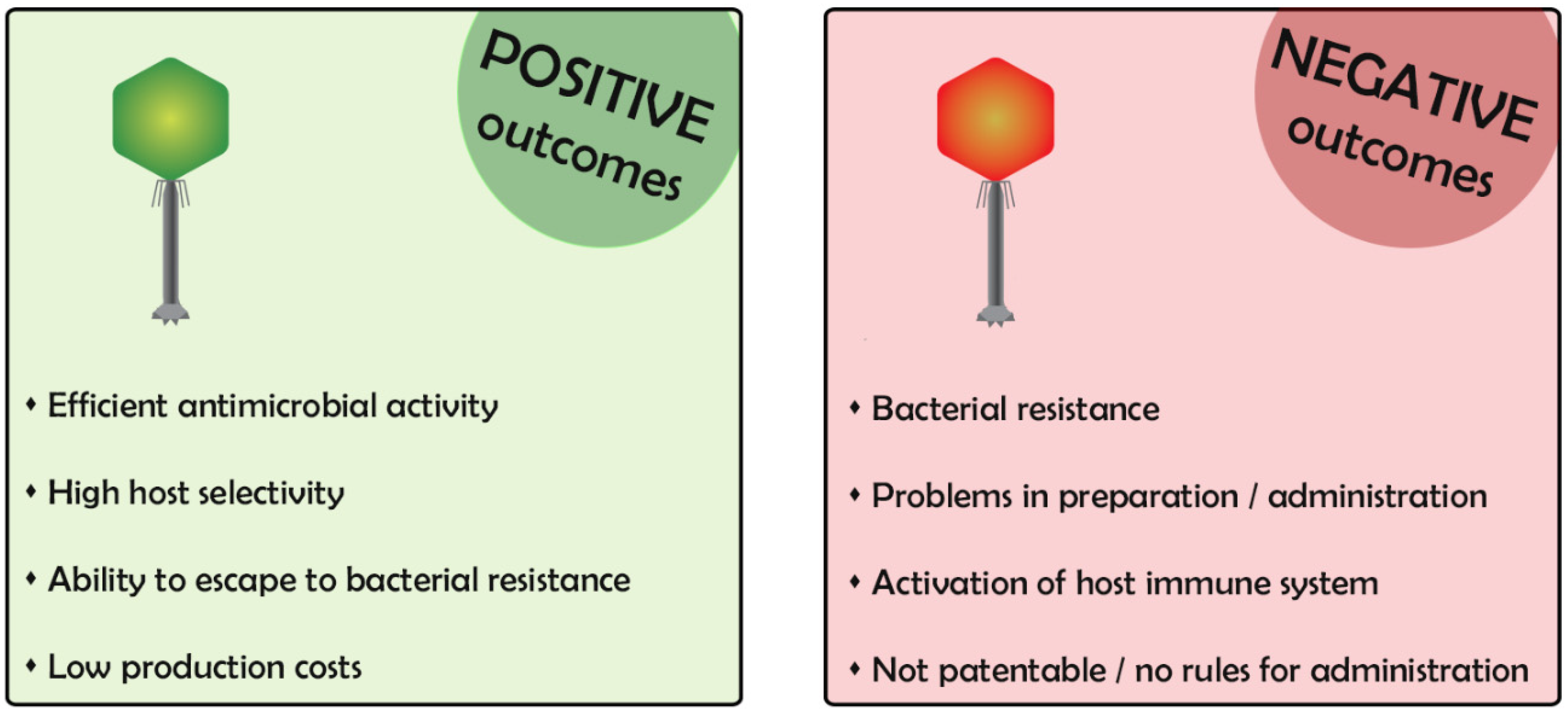
Figure 2.
Animal models used for bacterial infection and phage therapy application.


{kind=link}
{kind=link}
Table 1.
Animal models of human phage therapy for common human pathogens.
| Animal Model | Challenge (Pathogen) | Condition | Phage Treatment | Route of Administration | Results Summary | Reference |
|---|---|---|---|---|---|---|
| C. elegans | Salmonella enterica; spread on agar plate | lethal systemic infection | mono-phage, delay (24 hpi); 5 × 109–1× 1010 pfu | in growth medium | >survival rate | Augustine et al., 2014 [26] |
| C. elegans | Staphylococcus aureus; spread on agar plate | lethal systemic infection | mono-phage, delay (24 hpi); 109 pfu/ml | in growth medium | >survival rate | Glowacka-Rutkowska et al., 2019 [27] |
| D. melanogaster | Pseudomonas aeruginosa; intrathorax injection of 103 cfu/fly | lethal systemic infection | mono-phage, delay (6 hpi); 104 pfu/fly | intrathorax injection | >survival rate | Lindberg et al., 2014 [32] |
| D. melanogaster | Pseudomonas aeruginosa; intrathorax injection of 50–200 cfu/fly | lethal systemic infection | mono-phage, co-adm; 106 pfu/fly | oral (force feed) | >survival rate; <BB | Heo et al., 2009 [33] |
| G. mellonella | Clostridium difficile; oral administration 105 cfu/larva | lethal systemic infection | 4-phage cocktail: proph (2 hbi), delay (2 hpi) or co-adm; 1 to 4 doses of 106 pfu/larva | oral | reduced mortality (100% in proph); dose-dependence | Nale et al., 2016 [36] |
| G. mellonella | Burkholderia cepacia; injection of 2.5 × 103 cfu/larva | lethal bacteremia | mono-phage, delay (6 or 12 hpi); 2.5 × 103 pfu/larva | injection | >survival rate; <BB | Seed et al., 2009 [35] |
| G. mellonella | Pseudomonas aeruginosa (lab and clinical strains); injection of 30 cfu/larva | lethal bacteremia | 6-phage cocktail: proph (1 hbi) or delay (1 hpi); 1.5 to 4.5 × 103 pfu/larva | injection | prolonged survival time after infection | Forti et al., 2018 [37] |
| G. mellonella | Acinetobacter baumanii (XDR); injection of 5 × 105 cfu/larva | lethal bacteremia | 2-phage cocktail or mono-phage, delay (0.5 hpi); 5 × 107 pfu/larva | injection | >survival rate (≥80%) | Leshkasheli et al., 2019 [57] |
| Zebrafish | Enterococcus faecalis (clinical strain); injection in circulation of 3 × 104 cfu/embryo | lethal systemic infection | mono-phage, delay (2 hpi); 6 × 105 pfu/embryo in 2 nL | injection in circulation | >survival rate (of 57%); >healthy state | Al-Zubidi et al., 2019 [41] |
| Zebrafish | Pseudomonas aeruginosa; injection in circulation of 30 cfu/embryo | lethal systemic infection | 4-phage cocktail, delay (0.5 or 7 hpi); 500–1000 pfu/embryo in 2 nL | injection in circulation | >survival rate (of about 30%); <BB; reduced inflammatory response | Cafora et al., 2019 [42] |
| Quail | Salmonella enterica (Enteriditis); oral administration of 1.2 × 108 cfu/quail | gastrointestinal infection | mono-phage, proph or delay *; 105 pfu/mL, 3 doses daily | oral (oral gavage or vent lip) | <BB in cecal tonsils | Ahmadi et al., 2016 [43] |
| Chicken | Salmonella enterica (Typhimurium); oral administration of 107 cfu/chicken | gastrointestinal infection | 3-phage cocktail (liposome/alginate encapsulated), delay (24 hpi); 109/1010 pfu/chicken, 8 doses daily | oral | <BB in cecum (of 1.5–3.9 Log10 cfu) | Colom et al., 2015, 2017 [45,46] |
| Rabbit | Staphylococcus aureus; subcutaneous injection of 8 × 107 cfu/rabbit | local infection (abscess) | mono-phage, co-adm or delay (6, 12 or 24 hpi); 2 × 109 pfu/rabbit | subcutaneous injection | <BB of infected area and abscesses prevention in co-adm (no effect in delay) | Wills et al., 2005 [49] |
| Rabbit | Staphylococcus aureus (MRSA); Intramedullary injection of ≤5 × 106 cfu/rabbit (*) | chronic osteomyelitis | 7-phage cocktail, delay (21, or 42 dpi); 5 × 1011 pfu/rabbit, 4 doses total every 2 days | Intralesional injection | cure of infection in 21 dpf treatment | Kishor et al, 2016 [50] |
| Rabbit | Vibrio cholerae; oral administration of 5 × 108 cfu/rabbit | gastrointestinal infection | 3-phage cocktail: proph (3 or 24 hbi); 4–8 × 109 pfu/rabbit | oral | prevention of diarrheal symptoms; <BB in intestine (of 1–4 Log10 cfu) | Yen et al., 2017 [52] |
| Hamster | Clostridium difficile; oral administration of 2 × 103 spores/hamster | gastrointestinal infection | 2,3,4-phage cocktails or mono-phage, delay *; 8 × 107 pfu/mL, every 8 h × 36 hpi | oral | <BB in cecum and colon (of 2 Log10 cfu) | Nale et al., 2017 [53] |
| Pig | Escherichia coli (ETEC); oral administration of 1010 cfu/pig | gastrointestinal infection | 2,3-phage cocktail or mono-phage, proph (0.25 hbi, 3 × 109–1010 pfu/pig) or delay (24 hpi, 6 doses every 3 h, 108 pfu/pig) | oral | diarrhea symptoms ameliorate | Jamalludeen et al., 2009 [58] |
| Murine | Pseudomonas aeruginosa, intranasal injection of 1 × 107 cfu/mouse | lethal respiratory infection | 6-phage cocktail, delay (2 hpi); 107 pfu/mouse | intranasal injection | 100% reduced mortality; <BB (about 3 Log10 times) | Forti et al., 2018 [37] |
| Murine | Pseudomonas aeruginosa, intranasal injection of 2.5 × 107 cfu/mouse | respiratory infection | 3-phage cocktail: proph (48 hbi), co-adm or delay (24 hpi); 1.24 × 109 pfu/mouse | intranasal injection | >survival rate; bacterial clearance in BALs (proph 71%, co-adm 100% and delay 86%); reduced inflammatory response | Pabary et al., 2016 [54] |
| Murine | Pseudomonas aeruginosa (MDR), intraperitoneal injection of 107 cfu/mouse | lethal bacteremia | mono-phage, co-adm; 1 ×109 pfu/mouse | intraperitoneal injection | 85% reduced mortality; bacterial clearance in blood; reduced inflammatory response | Alvi et al., 2020 [55] |
| Murine | Pseudomonas aeruginosa (clinical strain), intranasal injection of 107 cfu/mouse | lethal lung infection | mono-phage, proph (24 hbi) or delay (2, 4, 6 hpi); 108 pfu/mouse | intranasal injection | >survival rate: delay-dependent (from 100% in 2 hpi to 25% in 6 hpi) and 100% in proph; reduced inflammatory response | Debarbieux et al., 2010 [56] |
| Murine | Acinetobacter baumanii (XDR), intraperitoneal injection of 6 × 107 cfu/mouse | lethal bacteremia | 2-phage cocktail or mono-phage, delay (2 hpi); 6 × 109 pfu/mouse | intraperitoneal injection | >survival rate (≥80%) | Leshkasheli et al., 2019 [57] |
| Murine | Klebsiella pneumoniae, topical administration 50 ul of 108 cfu/mL | burn wound infection | 5-phage cocktail or mono-phage, delay (6 hpi); 50 uL of 108 pfu/ml | topical | <BB in skin tissue; faster wound healing; reduced inflammatory response | Chadha et al., 2016 [59] |
| Murine | Mycobacterium ulcerans, subcutaneous injection of 105.5 afb | local infection (ulceration) | mono-phage, delay (33 dpi); 108 pfu/mouse | subcutaneous injection | <BB in skin tissue; prevent ulceration | Trigo et al., 2013 [60] |
| Murine | Staphylococcus aureus (MRSA), subcutaneous injection of 107 cfu/mouse | local infection (abscess) | mono-phage, co-adm or delay (4 dpi); 109 pfu/mouse | subcutaneous injection | prevent/ameliorate abscess development | Capparelli et al., 2007 [61] |
| Murine | Staphylococcus aureus (MRSA), intravenous injection of 108 cfu/mouse | systemic infection | mono-phage, co-adm; 109 pfu/mouse | intravenous injection | 97% reduced mortality; bacterial clearance in blood | Capparelli et al., 2007 [61] |
| Murine | Klebsiella pneumoniae, intranasal instillation of 109 cfu/mL | Lung infection | mono-phage, delay (2 hpi); 109 pfu/mouse | intranasal instillation | <BB in lung and serum; prevent severe lung lesions | Anand et al., 2019 [62] |
hbi = hours before initial infection; hpi = hours post initial infection; dpi = days post initial infection; cfu = colony-forming-units; pfu = plaque-forming-units; afb = acid fast bacilli; BB = bacterial burden (or bacterial load); delay = delayed treatment; co-adm = co-administration; proph = prophylactic treatment; MDR = multi drug resistant; XDR = extensively drug resistant; MRSA = methicillin-resistant Staphylococcus aureus; ETEC = enterotoxigenic E. coli; BAL = bronchoalveolar lavage; n° of doses = 1 if not differently indicated; * = not described.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Brix, A.; Cafora, M.; Aureli, M.; Pistocchi, A. Animal Models to Translate Phage Therapy to Human Medicine. Int. J. Mol. Sci. 2020, 21, 3715. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103715
AMA Style
Brix A, Cafora M, Aureli M, Pistocchi A. Animal Models to Translate Phage Therapy to Human Medicine. International Journal of Molecular Sciences. 2020; 21(10):3715. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103715
Chicago/Turabian StyleBrix, Alessia, Marco Cafora, Massimo Aureli, and Anna Pistocchi. 2020. "Animal Models to Translate Phage Therapy to Human Medicine" International Journal of Molecular Sciences 21, no. 10: 3715. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103715
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.