Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis


Molecular Biotechnology Center (MBC), Department of Molecular Biotechnology and Health Sciences, University of Torino, 10126 Torino, Italy
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2020, 21(11), 3760; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113760
Submission received: 7 May 2020
/
Revised: 21 May 2020
/
Accepted: 22 May 2020
/
Published: 26 May 2020
(This article belongs to the Special Issue Molecular Mechanisms, Physiopathology and Therapeutic Management of Episodic Ataxia)
Abstract
:Heme and Fe-S clusters regulate a plethora of essential biological processes ranging from cellular respiration and cell metabolism to the maintenance of genome integrity. Mutations in genes involved in heme metabolism and Fe-S cluster biogenesis cause different forms of ataxia, like posterior column ataxia and retinitis pigmentosa (PCARP), Friedreich’s ataxia (FRDA) and X-linked sideroblastic anemia with ataxia (XLSA/A). Despite great efforts in the elucidation of the molecular pathogenesis of these disorders several important questions still remain to be addressed. Starting with an overview of the biology of heme metabolism and Fe-S cluster biogenesis, the review discusses recent progress in the understanding of the molecular pathogenesis of PCARP, FRDA and XLSA/A, and highlights future line of research in the field. A better comprehension of the mechanisms leading to the degeneration of neural circuity responsible for balance and coordinated movement will be crucial for the therapeutic management of these patients.
1. Introduction
Balance and coordinated movements of the body are the result of a complex neuronal network involving the basal ganglia, cerebellum and cerebral cortex as well as peripheral motor and sensory pathways. Alterations in any part of this circuitry lead to loss of coordination and balance, also termed ataxia [1]. The etiology of ataxias is complex and includes toxic, metabolic, immune and genetic causes [2,3,4,5]. Hereditary forms represent an important subgroup of the ataxic disorders with more than 70 different types described [1,6,7]. Unfortunately, only few types of ataxia are fully treatable, while the majority are still only symptomatically managed, mostly because the molecular pathogenesis of the disease is poorly understood [1]. Hereditary ataxias can be divided by mode of inheritance in autosomal dominant and autosomal recessive disorders, and by gene in which causative mutations occur [7]. A wide range of mutations have been described in different genes resulting in the alteration of distinct mechanisms as mitochondrial function, oxidative stress, DNA repair, protein folding, cytoskeletal proteins, heme metabolism and iron–sulfur [Fe-S] cluster biogenesis [1].
The present review focuses exclusively on those forms of hereditary ataxias caused by mutations in genes involved in heme metabolism or Fe-S cluster biogenesis (Table 1), the two major iron consuming processes in the cell. Despite recent progresses in elucidating the role of Fe-S clusters in the maintenance of the neuronal networks at the basis of coordination and balance, less is known about the role of heme in these processes. Overall, the pathological mechanisms at the basis of neurodegeneration in these forms of ataxia are still unclear. The aim of this review is to provide a detailed description of the biology of heme metabolism and Fe-S cluster biogenesis with a focus on the possible altered molecular mechanisms at the basis of ataxia.
2. Heme Metabolism and Fe-S Cluster Biogenesis
Heme and Fe-S clusters are iron-containing cofactors involved in a variety of processes indispensable for cell viability. Heme, a complex of iron and protoporphyrin IX, is required for oxygen transport, cell respiration, drug metabolism, ion channel function and gene regulation [15,16,17,18,19,20,21]. Fe-S clusters are a complex between iron and the inorganic sulfurs. The most frequent forms include [2Fe-2S], [3Fe-4S] and [4Fe-4S]. However, more complicated forms have been characterized [22]. Fe-S clusters are involved in cell respiration, cell metabolism, genome maintenance, ribosome biogenesis and antiviral defense [23,24,25,26].
Both heme and Fe-S clusters are endogenously synthesized through complex and highly regulated processes occurring between the mitochondria and cytosol. The scenario is complicated by the requirement for heme and Fe–S clusters in distinct subcellular compartments coupled to their hydrophobicity and potential cytotoxicity. Therefore, the intracellular delivery of these cofactors to target apoproteins requires the binding of heme and Fe-S clusters to specific cochaperone and chaperones systems [18,19,20,21,23,24,25,26]. All these fascinating mechanisms have been just recently elucidated, but several questions still remain unanswered.
The heme biosynthetic pathway involves eight enzymatic reactions that are compartmentalized between the mitochondrion and cytosol. Heme synthesis starts and terminates in the mitochondrial matrix but the intermediate enzymatic steps occur in the cytosol or in the intermembrane space of mitochondria, meaning that heme precursors must cross mitochondrial membranes.
The initial step of heme biosynthesis is the condensation of succinyl-CoA and glycine, by δ-aminolevulinic acid synthase (ALAS), to form δ-aminolevulinic acid (ALA) in the mitochondrial matrix. Notably two different isoforms of ALAS exists: the ubiquitously expressed ALAS1 and the erythroid-specific isoform ALAS2. Upon its synthesis, ALA is exported into the cytosol where it is converted into coproporphyrinogen III (CPgenIII) through four subsequent enzymatic reactions. CPgenIII is then translocated in the intermembrane space of the mitochondria where it is converted into protoporphyrin IX (PPIX) in two enzymatic steps. A detailed description of each of these reactions and the putative transporters involved in the translocation of heme precursors through mitochondrial membranes can be found elsewhere [17,27]. In the last step, ferrous iron is inserted into the protoporphyrin ring by ferrochelatase (FECH) to form heme (heme b). Iron delivery to mitochondria is ensured by the iron importer mitoferrin (MFRN) located in the inner mitochondrial membrane [28,29]. Independent studies showed that FECH interacts with other proteins involved in heme synthesis—such as MFRN [30], the ATP binding cassette proteins ABCB7 [31,32] and ABCB10 [30,32], ALAS2 [33], and enzymes involved in the succinyl-CoA production [34,35]—thus supporting the existence of a large complex mediating heme synthesis, also termed mitochondrial heme metabolism complex [33].
Upon its synthesis heme b and/or its derivatives heme a, heme o, and heme c are incorporated into target apoproteins that reside in different subcellular compartments (mitochondria, ER, golgi, nucleus, lysosomes and peroxisomes) [20]. Therefore, once synthesized, heme must first cross the two mitochondrial membranes and then reach specific organelles to be incorporated into apoproteins. How this is achieved is still a matter of debate. The mitochondrial isoform of feline leukemia virus subgroup C receptor 1 (FLVCR1), FLVCR1b, plays a role in the efflux of heme out of mitochondria [36]. However, alternative potential mechanisms of mitochondrial heme export have been proposed [17]. Due to the hydrophobic nature and cytotoxic properties of free-heme, the intracellular trafficking of heme to target subcellular compartments must be finely controlled as well [18,21,37]. This is likely achieved by heme binding to specific chaperones [38,39].
The bioavailability of free-heme is further controlled by heme catabolism, mediated by heme oxygenase 1 (HO1) [40], and heme trafficking across the plasma membrane. Notably, heme transporter HRG1 [41], heme transporter HRG4 [42] and feline leukemia Virus subgroup C receptor 2 (FLVCR2) [43] have been described as heme importers whereas FLVCR1 [44], the multidrug resistance protein MRP-5/ABCC5 [45] and the ATP binding cassette subfamily G member 2 (ABCG2) [46,47] as heme exporters. Although the physiological significance of heme export out of the cell is still not completely understood, several lines of evidence suggest that heme export out of the plasma membrane is coupled with heme synthesis. Indeed, the export of heme through the plasma membrane isoform of FLVCR1, FLVCR1a, was reported to be essential for the regulation of ALAS1 activity in hepatocytes [48]. Consistently, the transcription of Flvcr1a and Alas1 are upregulated upon the induction of heme synthesis [48]. Altogether, these data suggest that heme export is something more than a simple cell detoxifying mechanism, as initially proposed. Since heme inhibits its own synthesis by negatively regulating ALAS1 transcription, translation and stability [16], it is tempting to speculate that heme export out of the plasma membrane is necessary to sustain endogenous heme synthesis.
The biogenesis of Fe-S clusters and their insertion into apoproteins is another extremely complex process involving more than 30 known biogenesis factors located in mitochondria and cytosol. The mitochondrial iron-sulfur cluster assembly (ISC) machinery is responsible for the de novo synthesis of Fe-S clusters and their insertion into mitochondrial apoproteins. The first step involves the assembly of a [2Fe-2S] cluster on the scaffold protein ISCU2 from inorganic iron and sulfur. The sulfur is donated by a cysteine desulfurase complex named NFS1/ISD11/ACP1 [49]. The mechanism of iron insertion into the cluster has been long debated. Initial studies suggest FRATAXIN (FXN) as an iron donor for [2Fe-2S] clusters assembly [50,51,52]. However, subsequent investigations propose FXN as an allosteric regulator of the process, able to stimulate the rate of sulfur transfer to the nascent Fe-S cluster [53,54,55,56,57,58]. Nowadays, the preloading of ISCU2 with iron has been proposed as major mechanism [23].
Following the synthesis of the [2Fe-2S] cluster on the ISCU2 scaffold, the [2Fe-2S] cluster is delivered to glutaredoxin 5 (GLX5) [23,59] to be incorporated into target [2Fe-2S] proteins. Alternatively, the [2Fe-2S] cluster is converted into [4Fe-4S] cluster and incorporated into different mitochondrial apoproteins, including aconitase, lipoyl synthase and respiratory cytochromes [23].
The ISC machinery also plays an essential role in the synthesis of a sulfur-containing compound (X-S) that is exported from mitochondria by the inner membrane ABC transporter, ABCB7 [60,61].
In the cytosol, X-S is used to generate the Fe/S clusters for cytosolic and nuclear target proteins as well as for those associated to the endoplasmic reticulum. These steps are mediated by the cytosolic iron-sulfur protein assembly (CIA) machinery that comprises at least 11 proteins [23,24]. For additional details on the molecular mechanisms underlying Fe-S clusters biogenesis, the reader is referred to additional outstanding reviews [23,24,25,26].
Several lines of evidence indicate that heme metabolism and Fe-S cluster biogenesis are strongly interconnected (Figure 1). Interestingly, heme synthesis depends on Fe-S cluster biogenesis. As mentioned above, the synthesis of heme requires succinyl-CoA as a first substrate. Succinyl-CoA derives from the TCA cycle that is also dependent of Fe-S cluster proteins. Notably, the supply of acetyl-CoA to the tricarboxylic acid cycle (TCA) cycle is mediated by the pyruvate dehydrogenase complex (PDC), that converts pyruvate into acetyl-CoA. The synthesis of PDC requires lipoate that is generated by an Fe-S cluster enzyme, the lipoyl synthase (LIAS) [62]. Furthermore, two TCA cycle enzymes, succinate dehydrogenase B (SDHB) and aconitase 2 (ACO2) are Fe-S cluster proteins [63,64].
The condensation of succinyl-CoA and glycine by ALAS represents the rate limiting step in heme biosynthesis and, in erythroid cells, it is dependent on Fe-S clusters levels. The cytosolic Fe-S cluster protein aconitase is a bifunctional enzyme and, following the loss of its Fe-S cluster (in iron deficiency), functions as an iron responsive protein (IRP) [64,65] inhibiting the translation of ALAS2 [66,67,68]. Moreover, GRX5 deficiency reduces heme synthesis in human erythroblasts by affecting Fe-S cluster biogenesis and IRPs activity [69].
In every cell type, the last step of heme synthesis is dependent on Fe-S cluster biogenesis. Notably, FECH contains an [2Fe-2S] cluster that does not participate in catalysis [70,71,72]. Reduced maturation and stabilization of FECH was observed in conditions of decreased availability of Fe-S clusters, like in ISCU miopathy [73], frataxin deficiency [74] or sideroflexin 4 deficiency [75].
Furthermore, as previously mentioned, FECH is part of a large complex of proteins, including the Fe-S cluster protein ABCB7 [32]. The downmodulation of ABCB7 results in low Fe-S clusters availability, decreased FECH expression and reduced heme levels [32].
Collectively, these observations suggest that the alteration of Fe-S cluster biogenesis may impact on heme levels by affecting multiple steps of its biosynthetic pathway, the supply of succinyl-CoA substrate as well as the expression of the first and last enzymes involved in heme synthesis. Whether alteration of heme biosynthesis affects Fe-S cluster biogenesis is not clear.
Since heme synthesis and Fe-S clusters biogenesis represent the two major Fe consuming processes in mitochondria, the two pathways may compete for iron acquisition. Studies in differentiating erythroid cells suggested that FXN may act as a metabolic switch in the diversion of iron between Fe-S cluster biogenesis and heme synthesis. Indeed, protoporphyrin IX (PPIX) accumulation results in downregulation of FXN and in the redirection of iron from Fe-S clusters biogenesis toward heme biosynthesis [76].
Finally, it is interesting to note that heme and Fe-S clusters are both necessary for oxidative phosphorylation. Indeed, heme is a cofactor for cytochromes c and cytochromes in complexes II-III-IV of the mitochondrial electron transport chain (ETC) [77,78], whereas Fe-S clusters are cofactors for cytochromes in complexes I-III [23]. The observation that both heme and Fe-S clusters are required for ETC further suggests that the two pathways must be tightly co-regulated to ensure energy production.
3. Posterior Column Ataxia and Retinitis Pigmentosa (PCARP)
PCARP is a rare autosomal recessive neurodegenerative syndrome characterized by sensory ataxia and retinitis pigmentosa [8]. The disease begins in infancy with areflexia and signs of retinal degeneration that worsen over time with a progressive restriction of the visual field and loss of the retina function. Ataxia is clinically evident in the second decade of life. It is due to the progressive degeneration of the posterior columns of the spinal cord resulting in the loss of proprioceptive sensations. Affected individuals have no clinical or radiological evidence of cerebral or cerebellar involvement. Additional features of the disease include scoliosis, camptodactyly, achalasia and gastrointestinal dysmotility [9,79].
The disease-causing gene was first mapped to chromosome 1q31-q32 [8] and then identified as FLVCR1 [79]. FLVCR1 gene codes for two heme export proteins: the plasma membrane isoform FLVCR1a and the mitochondrial isoform FLVCR1b [36]. Both homozygous missense mutations and compound heterozygous mutations have been described in several families [79,80,81,82,83]. Interestingly, mutations in the same gene have been identified in patients with non-syndromic retinitis pigmentosa [84,85,86,87,88] or in patients with pain insensitivity [89,90,91]. All the FLVCR1 mutations identified to date in PCARP patients are reported in Table 2.
The majority of mutations identified so far in PCARP occur in the first exon of the FLVCR1 gene (Table 2) [36]. As FLVCR1b protein arises from an alternative transcription start site located in the second exon of the FLVCR1 gene, the majority of identified mutations likely affect only FLVCR1a. However, the impact of the mutations on the expression and function of the two FLVCR1 isoforms still remains to be investigated in detail. The observation that loss of Flvcr1 is incompatible with life in animal models [36,92,93] strongly suggests that the identified mutations in FLVCR1 did not completely abrogate the expression of the transporter. This statement is also supported by data obtained in cells expressing the FLVCR1 mutations identified in patients with pain insensitivity [89,90].
The pathophysiology of the disease is still completely unknown. FLVCR1 is a ubiquitously expressed heme exporter belonging to the major facilitator superfamily (MFS) of transporter [44,94,95]. Initial studies focused on the role of FLVCR1 isoforms during erythroid differentiation [36,92,93] and erythroid diseases [96,97]. Then, heme efflux through FLVCR1a has been involved in the regulation of hepatic P450 cytochromes [48], the maintenance of endothelial integrity [98] as well as the regulation of cell proliferation [99,100]. These data are consistent with a role for heme in multiple essential biological processes. Curiously, almost no information is available regarding the function of FLVCR1 in the nervous system. The reported animal models of FLVCR1 deficiency are not viable thus limiting the possibility to investigate the role of FLVCR1 in neurodegeneration, specifically in the retina and in the peripheral nervous system. Information concerning the disease pathogenesis now available have been obtained in cell lines overexpressing the pathogenetic mutations. It has been shown that FLVCR1 mutations cause the mislocalization of FLVCR1 protein and consequently loss of its heme export function [101]. Therefore, it has been proposed that loss of FLVCR1 function may cause heme accumulation and cellular damage by heme toxicity in the cells of PCARP patients. Increased oxidative stress and enhanced susceptibility to programmed cell death have been observed also in neuroblastoma cells upon FLVCR1a downmodulation [89]. However, these experiments are not exhaustive and studies in appropriate in vitro and in vivo models of the disease are absolutely required. As discussed in the previous paragraph, several data support the idea that the export of heme is co-regulated with heme synthesis, thus suggesting that defective heme export may lead to decreased heme synthesis. Furthermore, defective heme export may also induce different compensatory mechanisms including the induction of HO1 with the release of carbon monoxide, iron and bilirubin, that could compromise the function and survival of proprioceptive neurons. Furthermore, the alteration of heme metabolism may affect additional pathways—including proteostasis, energetic metabolism and/or ion channel function [102]—leading to neurodegeneration. Future research should be focused on the generation of good in vitro and in vivo models of the diseases in order to definitely understand the consequences of defective heme export and its contribution to the disease pathogenesis.
4. Friedreich’s Ataxia (FRDA)
Friedreich’s ataxia (FRDA) is the most common form of recessive inherited ataxia in the Caucasian population, accounting for 75% of ataxias with onset prior to 25 years of age [103]. Symptoms usually manifest during childhood, and in 10 years patients lose the ability to walk, stand or sit up unassisted [10,11]. To note, in a small portion of cases, FRDA develops later on and it is characterized by a milder phenotype and a slower disease progression [12]. FRDA patients present a complex set of clinical features throughout the disease, including a mixed spinocerebellar and sensory ataxia, dysarthria, muscular weakness and absence of tendon reflexes with deep sensory loss [104].These neurological signs are the results of pathological changes that disturb both the central and peripheral nervous systems. Neurodegeneration firstly occurs in dorsal root ganglia (DRG) with progressive loss of large sensory neurons and their axonal projection in the posterior columns, followed by degeneration in the spinocerebellar and corticospinal tracts of the spinal cord [105,106]. Satellite cells and Schwann cells also seem to be affected in FRDA [107] but there is no clear evidence whether the observed phenotype may result from abnormal satellite cell or Schwann cell-neuron interactions. Both lack of myelination of large sensory neurons axons in the DRG and axonal degeneration have been described [108] and axonopathy has been associated with a dying-back mechanism [109]. Moreover, lesions in dentate nuclei and Purkinje cellsin cerebellum have been observed and account for the cerebellar phenotype [107]. Degeneration of enolase (NSE)-reactive neurons and of large inhibitory GABAergic terminals, as well as alterations in axonal connections of these neurons with Purkinje cells have also been described [110]. In addition to central and peripheral nervous systems degeneration, patients show primary non-neurological features as cardiomyopathy and increased incidence of diabetes mellitus [111]. Heart failure and supraventricular arrhythmias resulting from cardiomyopathy are the most common cause of death in FRDA [112].
FRDA is caused by homozygous or compound heterozygous mutations in the FXN gene [113,114]. Specifically, 96% of patients are homozygous for an expanded GAA trinucleotide repeat in intron 1 of the gene. Normally, there are fewer than 36 GAA repeats while those affected have between 56 and 1300 repeats. The remaining 4% of patients are compound heterozygous for a GAA expansion in one allele and a different pathogenic mutation in the other allele. More than 60 different mutations have been identified and structural modeling analysis showed that mutations in the hydrophobic core of FXN alter protein stability, while mutations occurring on surface residues affect interaction with ISC machinery and heme biosynthetic proteins [115]. In most cases, compound heterozygous patients are clinically identical to patients homozygous for the GAA expansion, but few missense mutations cause an atypical or milder clinical presentation [115,116,117,118]. Specifically, a milder phenotype is observed in patients carrying missense mutations G130V, D122Y, L106S [117,118]. G130V and D122Y mutations alter FXN interaction with the ISC machinery complex, while L106S mutation alters protein stability [115]. Even if these mutations alter FXN function in different ways, it is important to note that all occur in the amino terminal half of FXN protein. Interestingly, the length of GAA expansion correlates negatively with the expression of the encoded protein and positively with the age of onset and severity of the disease [119].
The first molecular consequence of the GAA expansion is the decreased transcription of the FXN gene that leads to the expression of lower levels of a structurally and functionally normal frataxin, up to 5–30% if compared to healthy individuals [114]. The reduction in FXN gene transcription in FRDA is mediated by two different mechanisms. Firstly, the expanded GAA repeat leads to both histone methylation and hypoacetylation, resulting in gene silencing [120,121]. Secondly, the GAA repeat stimulates the formation of R-loop structures, DNA-RNA hybrid, and of sticky DNA [122] that block the progress of RNA polymerase II. It is worth noting that R-loops are also known to mediate the formation of repressive chromatin and therefore the presence of the R-loop may lead to both repressive chromatin formation and to RNA-polymerase II block [123].
As previously mentioned, FXN is a mitochondrial protein involved in the biogenesis of Fe-S clusters. Even if some literature data indicate that FXN might be a multifunctional protein involved in different mitochondrial pathways, only the role of FXN in Fe-S cluster biogenesis has been convincingly and extensively proved [124].
Correct Fe-S cluster biogenesis in mitochondria is intimately linked to cellular iron homeostasis and failure to assemble mitochondrial Fe-S proteins upon FXN deficiency results in increased iron import and availability in mitochondria through the activation of IRP1 (iron regulatory protein 1). IRP1 binds to IRE elements in the 5’ and 3’ UTR of the mRNAs of iron metabolism-related proteins, resulting in iron import and accumulation in mitochondria [125]. Pathological mitochondrial iron accumulation has been reported in the heart, liver and spleen of FRDA patients [126,127] and in several animal models [128,129,130]. However, the presence of iron deposits in neurons has not been fully proved yet. Indeed, different studies showed no iron accumulation in the dentate nuclei and DRG of patients [105,127,131]. On the other hand, a recent study involving a higher number of FRDA subjects [132] found a significant increase in iron concentration in the dentate nucleus and red nucleus in the midbrain. In agreement with this observation, a relocation of iron from neurons to glia cells in the dentate nucleus and from neurons to satellites cells in DRGs has been observed [132,133], thus suggesting a redistribution of iron from dying neurons to satellite/Schwann cells. The generation of different animal models did not fully clarify this issue. Indeed, no mitochondrial iron accumulation has been reported in conditional knockout models with FXN reduction in DRG or cerebellum [128], but it was described in the brain of a FRDA model of D. melanogaster [134]. Therefore, the presence of iron deposits in the nervous system in FRDA still remains a controversial topic.
Iron could be highly toxic mainly because it mediates the catalysis of reactive oxygen species (ROS) by Fenton reaction. Formation of ROS results in glutathione depletion, lipid peroxidation and cell death [12]. Increased ROS levels have been described in different tissues of mice lacking FXN, including neurons [130]. However, other studies suggest ROS independent mechanisms at the basis of neurodegeneration. The most interesting one showed that loss of FXN in nervous system in flies and mice results in iron accumulation, increased sphingolipids synthesis, and in the activation of the PDK1/Mef2 (3-phosphoinositide dependent protein kinase-1/myocyte enhancer factor-2) pathway [135,136]. Mef2 is an important transcription factor that modulates the expression of multiple genes and its activity is regulated post-transcriptionally by the activity of different kinases, including PDK1 [137]. Mef2 participates in essential molecular events in the central nervous system, as neuronal development and survival and synaptic plasticity, by modulating the expression of genes involved in the regulation of neuronal activity as well as genes involved in energy storage and immune response [138]. Mutations in Mef2 have also been described in Rett-like disorder that is characterized by motor abnormalities and abnormal hand movements [139].
Despite several evidences showing the pathological role of iron accumulation in mitochondria, its implications in the pathophysiology of FRDA still remain elusive. Indeed, many studies suggest that iron accumulation represents only a late event in FRDA [128,140,141] and accumulation of iron has surprisingly been proposed as a protective mechanism in the liver, where iron is used to sustain defective heme synthesis and Fe-S cluster biogenesis occurring upon loss of FXN [125]. Moreover, as discussed above, the presence of iron deposits in neurons has not been fully proved yet. Therefore, more studies are needed to clarify the role of iron accumulation in the pathogenesis of FRDA neurodegeneration.
Fe-S clusters are involved in a plethora of pathways in cells and therefore abnormal FXN function can cause multiple and cumulative cellular dysregulations, independently of iron accumulation in mitochondria. Indeed, deficits in Fe-S cluster-containing enzymes, including TCA cycle enzymes (aconitase) and mitochondrial respiratory complexes I, II and III have been observed in different cellular and animal models of FXN deficiency. The reduced activity of these enzymes leads to changes in metabolic flow and to reduced mitochondrial ATP production. Mitochondrial dysfunctions associated with reduced ATP levels have been described in patient-derived cells [142], and in neurons [135,143,144] and other tissues [145,146] from multiple models of FRDA. Consistently, overexpression of FXN causes upregulation of TCA flux, respiration and ATP content [147]. Notably, reduction of Fe-S cluster-containing enzymes has not always been observed in FRDA models. Indeed, in patient derived DRGs samples a slight decrease of aconitase only—but not in respiratory complexes I, II and III—has been described [127]. Therefore, whether FXN deficiency always results in Fe-S cluster-containing enzymes deficits still need to be fully elucidated. Neurons are highly energy demanding cells and it is tempting to speculate that they could be specifically sensitive to cellular energy dysregulation. However, the reason why only large sensory neurons and neurons in the dentate nuclei in the cerebellum are involved still remains an intriguing unsolved question.
Another important Fe-S cluster containing enzymes is FECH, the last enzyme involved in heme biosynthesis. Deregulated heme synthesis and decreased heme levels have been observed in FXN deficient models but it was never reported in FRDA neurons [102,129,148]. Alteration of heme metabolism has been proposed as a pathogenetic event in other types of ataxias suggesting that a similar phenotype could be observed in FRDA. However more studies are needed to clarify the involvement of heme in FRDA.
Calcium homeostasis is an additional processes linked to FXN deficiency in neurons. Interestingly, altered calcium levels lead to failure of retrograde axonal transport along DRG axons in mice and flies [13]. Autophagy in neurons is highly dependent on axonal transport and a decrease in autophagic flux associated with high levels of ubiquitinylated proteins and damaged mitochondria has also been observed in DRGs [13]. Therefore, loss of FXN might result in calcium dyshomeostasis, altered axonal transport and aberrant autophagy. To note, other works suggest that FXN loss results instead in increased autophagy. However, in this case, if increased autophagy is a neurodegenerative mechanism [141] or a defense mechanism against oxidative stress [144] is yet to be clarified.
Alterations in actin cytoskeleton have been observed in FXN deficient DRGs. Specifically, loss of FXN results in altered actin dynamics leading to aberrant changes in the growth cone of neurons. Interestingly, hyperactive cofilin, that is an actin binding protein, emerges as the link between frataxin deficiency and actin cytoskeleton alterations [14].
In conclusion, different mechanisms have been proposed to be at the basis of neurodegeneration in FRDA and an overview is shown in Figure 2. However, which of them is the primary event triggering the neurological phenotype is still unclear. Due to the complexity of FRDA pathophysiology, probably more than one mechanism is involved. To note, it has been demonstrated that FXN deficiency causes tissue-specific changes in several metabolic pathways [149].
Analysis of FRDA patients clearly reveals a selective neurological and pathological pattern in FRDA in which specific populations of neurons are more susceptible to FXN deficiency. Indeed, neurodegeneration specifically involves large sensory neurons in the DRG, neurons of the posterior column of the spinal cord, neurons of the dentate nucleus and connections between these neurons and Purkinje cells in cerebellum. The identification of this specific susceptibility to FXN deficiency will definitely help to identify the precise molecular mechanism and to define therapeutic approaches to FRDA.
5. X-Linked Sideroblastic Anemia with Ataxia (XLSA/A)
Sideroblastic anemias are a group of disorders mainly characterized by hypochromic microcytic erythrocytes and mitochondrial iron accumulation in erythrocyte precursors [150]. X-linked sideroblastic anemia with ataxia (XLSA/A) is a rare form of inherited sideroblastic anemia characterized by an X-linked mode of inheritance [151]. Interestingly, in addition to anemia [6], XLSA/A patients present with motor delay and evidence of spinocerebellar dysfunction, including an early onset ataxia associated with severe cerebellar hypoplasia [61,152]. Intention tremor and dysarthria might also be present [6].
XLSA/A is due to mutations in the ABCB7 gene [151] that encodes for an ABC transporter located in the inner membrane of mitochondria. ABC family of transporters is a large family of multiple transmembrane proteins involved in energy-dependent transport of a wide range of substrates across membranes [153]. Four distinct mutations have been reported in four different XLSA/A families. All described variants are missense mutations and occur in a region thought to be involved in binding of the transported substrate [154,155,156]. Phenotypic complementation assays in yeast showed that these mutations have mild phenotypic effects, suggesting that they are partial loss-of-function mutations in vivo [61] and that a more severe allele would be lethal in humans. Indeed, study of Abcb7-deficient mice showed that the protein is essential during development. Moreover, systemic and tissue-specific deletion of Abcb7 in most organs, including central nervous system and bone marrow, was lethal, with the exception of liver and endothelial cells [60].
The molecular mechanisms linking ABCB7 mutations to ataxia are poorly understood. As mentioned before, ABCB7 transports a sulfur-containing compound (X-S) from mitochondria to the cytosol used to generate the Fe/S clusters for cytosolic and nuclear target proteins as well as for those associated to the endoplasmic reticulum. Recent studies suggested that a glutathione-coordinated [2Fe-2S] cluster is the substrate of the transporter [157]. Indeed, the analysis of different animal and cellular models showed a selective deficiency of cytosolic, but not mitochondrial, Fe-S proteins upon ABCB7 downregulation [158]. Reduction of cytosolic Fe-S proteins may result in the alteration of a plethora of cellular mechanisms as altered DNA repair, glycolysis, fatty acid synthesis, purine catabolism and ribosome biogenesis [159]. To note, loss of ABCB7 in HeLa cells reduces the activity of aconitase [160], suggesting that the alteration of energetic metabolism may play a role in the disease pathogenesis.
An additional mechanism that may contribute to the ataxic phenotype in XLSA/A is represented by defective heme synthesis. Indeed, loss of ABCB7 alters the activity of IRP1, which is responsive to the availability of cytosolic Fe-S clusters. This results in the inhibition of the expression of ALAS2 that is involved in heme synthesis, impaired cellular iron homeostasis and mitochondrial iron overload [60,161]. The accumulated mitochondrial iron has been shown not to be available to mitochondrial ferritin and so, it is not readily usable for heme synthesis [160]. Moreover, the interaction between ABCB7 and FECH, the last enzyme in heme biosynthesis, has been demonstrated and ABCB7 positively regulates the activity of this enzyme [31]. Therefore, loss of ABCB7 directly or indirectly inhibits heme synthesis.
Although the majority of these studies have been performed in erythroid cells, it is likely that defects in energetic metabolism and/or heme synthesis may contribute to the degeneration of neural circuity regulating balance and coordination of movement as well.
6. Therapeutic Approaches
There are no therapeutic options that actually halt the progression of PCARP, FRDA and XLSA/A. Therefore, the management of these patients is mainly symptomatic. In particular, FRDA has a complex and variable clinical phenotype and therefore it requires a broad multidisciplinary approach to manage [12].
Physiotherapy provides an important tool for the maintenance of balance, flexibility, strength and accuracy of limb movements, all of which can help to improve the functional consequences of gait and limb ataxia. Occupational therapy is also important and allows for the assessment and optimization of functional status, thereby reducing the impediments to activities of daily living.
In FRDA, the heart requires special attention because hearth failure is a common cause of mortality and therefore, electrocardiogram and echocardiography should be performed at diagnosis. Many patients show impaired glucose tolerance or diabetes and should be counseled on the importance of dietary changes and physical exercise [12].
Progresses in the identification of novel strategies to cure or limit the disease progression has been made only for FRDA. This is likely due to a better understanding of the molecular pathogenesis and the higher frequency of the diseases allowing the design of clinical trials. A number of molecules targeting specific pathological processes have been evaluated in clinical trials. Earlier clinical trials attempted to resolve oxidative stress and iron accumulation by using antioxidant and iron chelation agents, both as monotherapy and in combination, but failed to give consistent results [162,163]. However, recently the antioxidant drug omaveloxolone (RTA408), an activator of the anti-oxidant nuclear factor, erythroid 2 like 2 protein (Nrf2), has shown promising results as a treatment for FRDA [164]. A Phase II study is currently evaluating the safety, pharmacodynamics and efficacy of omaveloxolone [162].
To date, heterozygous carriers with almost 50% level of FXN transcript remain asymptomatic [123]. This observation indicates that even a small increase in FXN transcript levels is likely to be therapeutic. Therefore, another possible approach is treating FRDA by restoring FXN levels. For example, erythropoietin has been shown to increase FXN levels in FRDA models, but clinical trials failed to demonstrate clinical efficacy of this molecule [165,166]. In general, these compounds produce small increases in FXN protein levels. A greater increase can be achieved by gene therapy. Gene therapy requires the administration of adeno-associated virus (AAV) carrying human FXN. Their administration in two different mouse models of FRDA, both before and after the insurgence of heart failure or neurological dysfunctions, was sufficient to inhibit or complete revers cardiomyopathy or sensory neuropathy, respectively [167,168]. Different clinical trials are now underway for the treatment of FRDA using gene therapy. However, some immunological complications are present. Interestingly, synthetic lipid nanoparticles have been used to deliver FXN mRNA and this approach has the potential to overcome some of the immunological limitations associated with viral delivery systems [162,169].
More recent approaches focused on epigenetic elements. For example, inhibition of histone deacetylase (HDAC) has emerged as a promising therapeutic strategy [170]. An alternative strategy uses oligonucleotides to disrupt R-loop formation, and also showed partial reversal of FXN gene silencing [171]. Similarly, dimethyl fumarate reduces the formation of R-loop structures and increases transcript of FXN [172]. Moreover, syn-TEF1, a synthetic transcription factor, specifically reversed the block of polymerase II and showed partial reactivation FXN gene transcription [173].
Finally, thanks to recent advances in genome editing, deleting, or at least reducing the expanded GAA repeat might represent another possible therapeutic approach.
7. Concluding Remarks
In the present review, we discussed current knowledge on the molecular mechanisms underlying PCARP, FRDA and XLSA/A. Although the three forms of ataxia are clinically heterogeneous, they are undoubtedly caused by mutations in genes involved in strongly interconnected pathways. As already discussed, heme synthesis depends on Fe-S cluster availability and we cannot exclude that heme metabolism may influence Fe-S cluster levels.
Strong progresses in the understanding of the molecular pathogenesis of FRDA ataxia has been made in recent years, whereas less is known about PCARP and XLSA/A. Due to the involvement of heme and Fe-S clusters in multiple essential processes, it is conceivable that multiple mechanisms could be active in these disorders. However, the observation that heme and Fe-S clusters regulate common pathways, like energetic cell metabolism, suggest the possibility that similar pathogenetic mechanisms may be responsible for XLSA/A and PCARP. Eventually, the identification of common pathways altered in these forms of ataxia could be important for drug repurposing strategies. Future research should be directed to a better elucidation of the molecular mechanisms underlying these rare diseases and to the identification of novel therapeutic options.
Author Contributions
Conceptualization: D.C. and F.B.; Writing—original draft: D.C. and F.B.; Writing—review and editing: D.C. and E.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was founded by the Italian Regenerative Medicine Infrastructure (IRMI) and the Interacademic Consortium for Biotechnology (CIB) to Fiorella Altruda.
Acknowledgments
We thank the Italian Regenerative Medicine Infrastructure (IRMI) and the “Consorzio Interuniversitario per le Biotecnologie” for supporting this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Akbar, U.; Ashizawa, T. Ataxia. Neurol. Clin. 2015, 33, 225–248. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, J.A.; Luce, J.K. Cerebellar ataxia with weekly 5-fluorouracil administration. Lancet 1971, 1, 138–139. [Google Scholar] [CrossRef]
- Diener, H.C.; Dichgans, J.; Bacher, M.; Guschlbauer, B. Improvement of ataxia in alcoholic cerebellar atrophy through alcohol abstinence. J. Neurol. 1984, 231, 258–262. [Google Scholar] [CrossRef]
- Matthews, B.R.; Jones, L.K.; Saad, D.A.; Aksamit, A.J.; Josephs, K.A. Cerebellar ataxia and central nervous system whipple disease. Arch. Neurol. 2005, 62, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, L.A.; Jarius, S.; Pellkofer, H.L.; Schueller, M.; Krumbholz, M.; Koenig, F.; Johannis, W.; la Fougere, C.; Newman, T.; Vincent, A.; et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J. Neurol. Neurosurg. Psychiatry 2008, 79, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Stephens, K.; Amemiya, A. GeneReviews; Gene Reviews: Seattle, WA, USA, 1993. [Google Scholar]
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.J.; Morton, D.H.; Patronas, N.; Nee, L.E. An autosomal recessive disorder with posterior column ataxia and retinitis pigmentosa. Neurology 1997, 49, 1717–1720. [Google Scholar] [CrossRef]
- Higgins, J.J.; Kluetzman, K.; Berciano, J.; Combarros, O.; Loveless, J.M. Posterior column ataxia and retinitis pigmentosa: A distinct clinical and genetic disorder. Mov. Disord. 2000, 15, 575–578. [Google Scholar] [CrossRef]
- Li, K. Iron Pathophysiology in Friedreich’s Ataxia. Adv. Exp. Med. Biol. 2019, 1173, 125–143. [Google Scholar] [CrossRef]
- De Michele, G.; Perrone, F.; Filla, A.; Mirante, E.; Giordano, M.; De Placido, S.; Campanella, G. Age of onset, sex, and cardiomyopathy as predictors of disability and survival in Friedreich’s disease: A retrospective study on 119 patients. Neurology 1996, 47, 1260–1264. [Google Scholar] [CrossRef]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef]
- Mollá, B.; Muñoz-Lasso, D.C.; Riveiro, F.; Bolinches-Amorós, A.; Pallardó, F.V.; Fernandez-Vilata, A.; de la Iglesia-Vaya, M.; Palau, F.; Gonzalez-Cabo, P. Reversible Axonal Dystrophy by Calcium Modulation in Frataxin-Deficient Sensory Neurons of YG8R Mice. Front. Mol. Neurosci. 2017, 10, 264. [Google Scholar] [CrossRef]
- Muñoz-Lasso, D.C.; Mollá, B.; Calap-Quintana, P.; García-Giménez, J.L.; Pallardo, F.V.; Palau, F.; Gonzalez-Cabo, P. Cofilin dysregulation alters actin turnover in frataxin-deficient neurons. Sci. Rep. 2020, 10, 5207. [Google Scholar] [CrossRef] [Green Version]
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Reddi, A.R.; Hamza, I. Heme Mobilization in Animals: A Metallolipid’s Journey. Acc. Chem. Res. 2016, 49, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Hanna, D.A.; Harvey, R.M.; Martinez-Guzman, O.; Yuan, X.; Chandrasekharan, B.; Raju, G.; Outten, F.W.; Hamza, I.; Reddi, A.R. Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. USA 2016, 113, 7539–7544. [Google Scholar] [CrossRef] [Green Version]
- Schultz, I.J.; Chen, C.; Paw, B.H.; Hamza, I. Iron and porphyrin trafficking in heme biogenesis. J. Biol. Chem. 2010, 285, 26753–26759. [Google Scholar] [CrossRef] [Green Version]
- Severance, S.; Hamza, I. Trafficking of heme and porphyrins in metazoa. Chem. Rev. 2009, 109, 4596–4616. [Google Scholar] [CrossRef] [Green Version]
- Beinert, H.; Holm, R.H.; Münck, E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020. [Google Scholar] [CrossRef]
- Lill, R.; Dutkiewicz, R.; Freibert, S.A.; Heidenreich, T.; Mascarenhas, J.; Netz, D.J.; Paul, V.D.; Pierik, A.J.; Richter, N.; Stümpfig, M.; et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell Biol. 2015, 94, 280–291. [Google Scholar] [CrossRef]
- Rouault, T.A.; Maio, N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Rouault, T.A. Iron-sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 2015, 1853, 1493–1512. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef]
- Taketani, S.; Kakimoto, K.; Ueta, H.; Masaki, R.; Furukawa, T. Involvement of ABC7 in the biosynthesis of heme in erythroid cells: Interaction of ABC7 with ferrochelatase. Blood 2003, 101, 3274–3280. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756–1767. [Google Scholar] [CrossRef]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the Mitochondrial Heme Metabolism Complex. PLoS ONE 2015, 10, e0135896. [Google Scholar] [CrossRef] [Green Version]
- Furuyama, K.; Sassa, S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J. Clin. Invest. 2000, 105, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.F.; Tchaikovskii, V.; Hoffbrand, A.V.; Fraser, M.E.; Margolis, S. X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the β-subunit of succinyl-CoA synthetase (SUCLA2). J. Biol. Chem. 2012, 287, 28943–28955. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Invest. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [Green Version]
- Hanna, D.A.; Martinez-Guzman, O.; Reddi, A.R. Heme Gazing: Illuminating Eukaryotic Heme Trafficking, Dynamics, and Signaling with Fluorescent Heme Sensors. Biochemistry 2017, 56, 1815–1823. [Google Scholar] [CrossRef]
- Galmozzi, A.; Kok, B.P.; Kim, A.S.; Montenegro-Burke, J.R.; Lee, J.Y.; Spreafico, R.; Mosure, S.; Albert, V.; Cintron-Colon, R.; Godio, C.; et al. PGRMC2 is an intracellular haem chaperone critical for adipocyte function. Nature 2019, 576, 138–142. [Google Scholar] [CrossRef]
- Sweeny, E.A.; Singh, A.B.; Chakravarti, R.; Martinez-Guzman, O.; Saini, A.; Haque, M.M.; Garee, G.; Dans, P.D.; Hannibal, L.; Reddi, A.R.; et al. Glyceraldehyde-3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Biol. Chem. 2018, 293, 14557–14568. [Google Scholar] [CrossRef] [Green Version]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.D.; Paw, B.H.; et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.P.; Shing, J.; Saraon, P.; Berger, L.C.; Eiden, M.V.; Wilde, A.; Tailor, C.S. The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol. Cell Biol. 2010, 30, 5318–5324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolnek, T.; Zhang, J.; Beardsley, S.; Scheffer, G.L.; Hamza, I. Control of metazoan heme homeostasis by a conserved multidrug resistance protein. Cell Metab. 2014, 19, 1008–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desuzinges-Mandon, E.; Arnaud, O.; Martinez, L.; Huché, F.; Di Pietro, A.; Falson, P. ABCG2 transports and transfers heme to albumin through its large extracellular loop. J. Biol. Chem. 2010, 285, 33123–33133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, P.; Ross, D.D.; Nakanishi, T.; Bailey-Dell, K.; Zhou, S.; Mercer, K.E.; Sarkadi, B.; Sorrentino, B.P.; Schuetz, J.D. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004, 279, 24218–24225. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; Ingoglia, G.; Chiabrando, D.; Mercurio, S.; Turco, E.; Silengo, L.; Altruda, F.; Tolosano, E. Heme exporter FLVCR1a regulates heme synthesis and degradation and controls activity of cytochromes P450. Gastroenterology 2014, 146, 1325–1338. [Google Scholar] [CrossRef] [Green Version]
- Beilschmidt, L.K.; Puccio, H.M. Mammalian Fe-S cluster biogenesis and its implication in disease. Biochimie 2014, 100, 48–60. [Google Scholar] [CrossRef]
- Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. Interactions of iron-bound frataxin with ISCU and ferredoxin on the cysteine desulfurase complex leading to Fe-S cluster assembly. J. Inorg. Biochem. 2018, 183, 107–116. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J.A. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef]
- Layer, G.; Ollagnier-de Choudens, S.; Sanakis, Y.; Fontecave, M. Iron-sulfur cluster biosynthesis: Characterization of Escherichia coli CYaY as an iron donor for the assembly of [2Fe-2S] clusters in the scaffold IscU. J. Biol. Chem. 2006, 281, 16256–16263. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Barondeau, D.P. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Bridwell-Rabb, J.; Fox, N.G.; Tsai, C.L.; Winn, A.M.; Barondeau, D.P. Human frataxin activates Fe-S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 2014, 53, 4904–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autréaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Müller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Das, D.; Chakrabarti, M.; Lindahl, P.A.; Barondeau, D.P. Frataxin Accelerates [2Fe-2S] Cluster Formation on the Human Fe-S Assembly Complex. Biochemistry 2015, 54, 3880–3889. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef] [Green Version]
- Wingert, R.A.; Galloway, J.L.; Barut, B.; Foott, H.; Fraenkel, P.; Axe, J.L.; Weber, G.J.; Dooley, K.; Davidson, A.J.; Schmid, B.; et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 2005, 436, 1035–1039. [Google Scholar] [CrossRef]
- Pondarré, C.; Antiochos, B.B.; Campagna, D.R.; Clarke, S.L.; Greer, E.L.; Deck, K.M.; McDonald, A.; Han, A.P.; Medlock, A.; Kutok, J.L.; et al. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum. Mol. Genet. 2006, 15, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Bekri, S.; Kispal, G.; Lange, H.; Fitzsimons, E.; Tolmie, J.; Lill, R.; Bishop, D.F. Human ABC7 transporter: Gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 2000, 96, 3256–3264. [Google Scholar] [CrossRef]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; Del Toro, M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J.; et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Ghezzi, D.; Verrigni, D.; Rizza, T.; Bertini, E.; Martinelli, D.; Zeviani, M.; Singh, A.; Carrozzo, R.; Rouault, T.A. Disease-Causing SDHAF1 Mutations Impair Transfer of Fe-S Clusters to SDHB. Cell Metab. 2016, 23, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Volz, K. The functional duality of iron regulatory protein 1. Curr. Opin. Struct. Biol. 2008, 18, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Eisenstein, R.S. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu. Rev. Nutr. 2000, 20, 627–662. [Google Scholar] [CrossRef]
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Invest. 2010, 120, 1749–1761. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.K.; Dailey, H.A.; Rose, J.P.; Burden, A.; Sellers, V.M.; Wang, B.C. The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 2001, 8, 156–160. [Google Scholar] [CrossRef]
- Schneider-Yin, X.; Gouya, L.; Dorsey, M.; Rüfenacht, U.; Deybach, J.C.; Ferreira, G.C. Mutations in the iron-sulfur cluster ligands of the human ferrochelatase lead to erythropoietic protoporphyria. Blood 2000, 96, 1545–1549. [Google Scholar] [CrossRef]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Crooks, D.R.; Ghosh, M.C.; Haller, R.G.; Tong, W.H.; Rouault, T.A. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 2010, 115, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillon, B.; Bulteau, A.L.; Wattenhofer-Donzé, M.; Schmucker, S.; Friguet, B.; Puccio, H.; Drapier, J.C.; Bouton, C. Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe-S proteins. FEBS J. 2009, 276, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Tesfay, L.; Winkler, C.R.; Torti, F.M.; Torti, S.V. Sideroflexin 4 affects Fe-S cluster biogenesis, iron metabolism, mitochondrial respiration and heme biosynthetic enzymes. Sci. Rep. 2019, 9, 19634. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.M.; Greer, J.M.; Ponka, P.; Richardson, D.R. Erythroid differentiation and protoporphyrin IX down-regulate frataxin expression in Friend cells: Characterization of frataxin expression compared to molecules involved in iron metabolism and hemoglobinization. Blood 2002, 99, 3813–3822. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta 2012, 1823, 1604–1616. [Google Scholar] [CrossRef] [Green Version]
- Fiorito, V.; Chiabrando, D.; Tolosano, E. Mitochondrial Targeting in Neurodegeneration: A Heme Perspective. Pharmaceuticals (Basel) 2018, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Rajadhyaksha, A.M.; Elemento, O.; Puffenberger, E.G.; Schierberl, K.C.; Xiang, J.Z.; Putorti, M.L.; Berciano, J.; Poulin, C.; Brais, B.; Michaelides, M.; et al. Mutations in FLVCR1 cause posterior column ataxia and retinitis pigmentosa. Am. J. Hum. Genet. 2010, 87, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Beigi, F.; Del Pozo-Valero, M.; Martin-Merida, I.; Vahidi Mehrjardi, M.Y.; Manaviat, M.R.; Sherafat, A.; Ayuso, C.; Ghasemi, N. Posterior column ataxia with retinitis pigmentosa (PCARP) in an Iranian patient associated with the. Ophthalmic. Genet. 2020, 41, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Scanga, H.L.; Dansingani, K.K.; Taubenslag, K.J.; Zlotcavitch, L.; Chauhan, B.K.; Sylvester, C.L.; Morton, D.H.; Nischal, K.K. Clinical and imaging characteristics of posterior column ataxia with retinitis pigmentosa with a specific FLVCR1 mutation. Ophthalmic. Genet. 2018, 39, 735–740. [Google Scholar] [CrossRef]
- Shaibani, A.; Wong, L.J.; Wei Zhang, V.; Lewis, R.A.; Shinawi, M. Autosomal recessive posterior column ataxia with retinitis pigmentosa caused by novel mutations in the FLVCR1 gene. Int. J. Neurosci. 2015, 125, 43–49. [Google Scholar] [CrossRef]
- Ishiura, H.; Fukuda, Y.; Mitsui, J.; Nakahara, Y.; Ahsan, B.; Takahashi, Y.; Ichikawa, Y.; Goto, J.; Sakai, T.; Tsuji, S. Posterior column ataxia with retinitis pigmentosa in a Japanese family with a novel mutation in FLVCR1. Neurogenetics 2011, 12, 117–121. [Google Scholar] [CrossRef]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [Green Version]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, I.H.; Shanks, M.E.; Clouston, P.; MacLaren, R.E. A splice-site variant in FLVCR1 produces retinitis pigmentosa without posterior column ataxia. Ophthalmic. Genet. 2018, 39, 263–267. [Google Scholar] [CrossRef]
- Whelan, L.; Dockery, A.; Wynne, N.; Zhu, J.; Stephenson, K.; Silvestri, G.; Turner, J.; O’Byrne, J.J.; Carrigan, M.; Humphries, P.; et al. Findings from a Genotyping Study of Over 1000 People with Inherited Retinal Disorders in Ireland. Genes (Basel) 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Kuehlewein, L.; Schöls, L.; Llavona, P.; Grimm, A.; Biskup, S.; Zrenner, E.; Kohl, S. Phenotypic spectrum of autosomal recessive retinitis pigmentosa without posterior column ataxia caused by mutations in the FLVCR1 gene. Graefes. Arch. Clin. Exp. Ophthalmol. 2019, 257, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Castori, M.; di Rocco, M.; Ungelenk, M.; Gießelmann, S.; Di Capua, M.; Madeo, A.; Grammatico, P.; Bartsch, S.; Hübner, C.A.; et al. Mutations in the Heme Exporter FLVCR1 Cause Sensory Neurodegeneration with Loss of Pain Perception. PLoS Genet. 2016, 12, e1006461. [Google Scholar] [CrossRef] [PubMed]
- Bertino, F.; Firestone, K.; Bellacchio, E.; Jackson, K.E.; Asamoah, A.; Hersh, J.; Fiorito, V.; Destefanis, F.; Gonser, R.; Tucker, M.E.; et al. Heme and sensory neuropathy: Insights from novel mutations in the heme exporter FLVCR1. Pain 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castori, M.; Morlino, S.; Ungelenk, M.; Pareyson, D.; Salsano, E.; Grammatico, P.; Tolosano, E.; Kurth, I.; Chiabrando, D. Posterior column ataxia with retinitis pigmentosa coexisting with sensory-autonomic neuropathy and leukemia due to the homozygous p.Pro221Ser FLVCR1 mutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, S.; Petrillo, S.; Chiabrando, D.; Bassi, Z.I.; Gays, D.; Camporeale, A.; Vacaru, A.; Miniscalco, B.; Valperga, G.; Silengo, L.; et al. The heme exporter Flvcr1 regulates expansion and differentiation of committed erythroid progenitors by controlling intracellular heme accumulation. Haematologica 2015, 100, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Law, C.J.; Maloney, P.C.; Wang, D.N. Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Scietti, L.; Prajica, A.G.; Bertino, F.; Tolosano, E.; Magnani, F. Expression and purification of the heme exporter FLVCR1a. Protein. Expr. Purif. 2020, 172, 105637. [Google Scholar] [CrossRef]
- Mercurio, S.; Aspesi, A.; Silengo, L.; Altruda, F.; Dianzani, I.; Chiabrando, D. Alteration of heme metabolism in a cellular model of Diamond-Blackfan anemia. Eur. J. Haematol. 2015. [Google Scholar] [CrossRef]
- Rey, M.A.; Duffy, S.P.; Brown, J.K.; Kennedy, J.A.; Dick, J.E.; Dror, Y.; Tailor, C.S. Enhanced alternative splicing of the FLVCR1 gene in Diamond Blackfan anemia disrupts FLVCR1 expression and function that are critical for erythropoiesis. Haematologica 2008, 93, 1617–1626. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef]
- Fiorito, V.; Forni, M.; Silengo, L.; Altruda, F.; Tolosano, E. Crucial role of Flvcr1a in the maintenance of intestinal heme homeostasis. Antioxid. Redox. Signal 2015. [Google Scholar] [CrossRef]
- Fiorito, V.; Chiabrando, D.; Petrillo, S.; Bertino, F.; Tolosano, E. The Multifaceted Role of Heme in Cancer. Front. Oncol. 2019, 9, 1540. [Google Scholar] [CrossRef] [Green Version]
- Yanatori, I.; Yasui, Y.; Miura, K.; Kishi, F. Mutations of FLVCR1 in posterior column ataxia and retinitis pigmentosa result in the loss of heme export activity. Blood Cells Mol. Dis. 2012, 49, 60–66. [Google Scholar] [CrossRef]
- Chiabrando, D.; Fiorito, V.; Petrillo, S.; Tolosano, E. Unraveling the Role of Heme in Neurodegeneration. Front. Neurosci. 2018, 12, 712. [Google Scholar] [CrossRef]
- Cossée, M.; Schmitt, M.; Campuzano, V.; Reutenauer, L.; Moutou, C.; Mandel, J.L.; Koenig, M. Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: Founder effect and premutations. Proc. Natl. Acad. Sci. USA 1997, 94, 7452–7457. [Google Scholar] [CrossRef] [Green Version]
- Harding, A.E. Friedreich’s ataxia: A clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 1981, 104, 589–620. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256 (Suppl. 1), 3–8. [Google Scholar] [CrossRef]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dyck, P.J.; Lambert, E.H. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch. Neurol. 1968, 18, 603–618. [Google Scholar] [CrossRef]
- Said, G.; Marion, M.H.; Selva, J.; Jamet, C. Hypotrophic and dying-back nerve fibers in Friedreich’s ataxia. Neurology 1986, 36, 1292–1299. [Google Scholar] [CrossRef]
- González-Cabo, P.; Palau, F. Mitochondrial pathophysiology in Friedreich’s ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 53–64. [Google Scholar] [CrossRef]
- Harding, A.E.; Hewer, R.L. The heart disease of Friedreich’s ataxia: A clinical and electrocardiographic study of 115 patients, with an analysis of serial electrocardiographic changes in 30 cases. Q. J. Med. 1983, 52, 489–502. [Google Scholar]
- Tsou, A.Y.; Paulsen, E.K.; Lagedrost, S.J.; Perlman, S.L.; Mathews, K.D.; Wilmot, G.R.; Ravina, B.; Koeppen, A.H.; Lynch, D.R. Mortality in Friedreich ataxia. J. Neurol. Sci. 2011, 307, 46–49. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; et al. Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef]
- De Michele, G.; Filla, A.; Cavalcanti, F.; Tammaro, A.; Monticelli, A.; Pianese, L.; Di Salle, F.; Perreti, A.; Santoro, L.; Caruso, G.; et al. Atypical Friedreich ataxia phenotype associated with a novel missense mutation in the X25 gene. Neurology 2000, 54, 496–499. [Google Scholar] [CrossRef]
- Cossée, M.; Dürr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschütter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206. [Google Scholar] [CrossRef]
- Bartolo, C.; Mendell, J.R.; Prior, T.W. Identification of a missense mutation in a Friedreich’s ataxia patient: Implications for diagnosis and carrier studies. Am. J. Med. Genet. 1998, 79, 396–399. [Google Scholar] [CrossRef]
- Montermini, L.; Richter, A.; Morgan, K.; Justice, C.M.; Julien, D.; Castellotti, B.; Mercier, J.; Poirier, J.; Capozzoli, F.; Bouchard, J.P.; et al. Phenotypic variability in Friedreich ataxia: Role of the associated GAA triplet repeat expansion. Ann. Neurol. 1997, 41, 675–682. [Google Scholar] [CrossRef]
- Castaldo, I.; Pinelli, M.; Monticelli, A.; Acquaviva, F.; Giacchetti, M.; Filla, A.; Sacchetti, S.; Keller, S.; Avvedimento, V.E.; Chiariotti, L.; et al. DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet. 2008, 45, 808–812. [Google Scholar] [CrossRef]
- Evans-Galea, M.V.; Carrodus, N.; Rowley, S.M.; Corben, L.A.; Tai, G.; Saffery, R.; Galati, J.C.; Wong, N.C.; Craig, J.M.; Lynch, D.R.; et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 2012, 71, 487–497. [Google Scholar] [CrossRef]
- Sakamoto, N.; Chastain, P.D.; Parniewski, P.; Ohshima, K.; Pandolfo, M.; Griffith, J.D.; Wells, R.D. Sticky DNA: Self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol. Cell 1999, 3, 465–475. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Bidichandani, S.I. Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol. Dis. 2019, 132, 104606. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Napierala, M.; Puccio, H. Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis. Model. Mech. 2012, 5, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, A.; Schmucker, S.; Reutenauer, L.; Mathieu, J.R.R.; Peyssonnaux, C.; Karim, Z.; Puy, H.; Galy, B.; Hentze, M.W.; Puccio, H. Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell Metab. 2015, 21, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Lamarche, J.B.; Côté, M.; Lemieux, B. The cardiomyopathy of Friedreich’s ataxia morphological observations in 3 cases. Can. J. Neurol. Sci. 1980, 7, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Huang, M.L.; Becker, E.M.; Whitnall, M.; Suryo Rahmanto, Y.; Ponka, P.; Richardson, D.R. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. USA 2009, 106, 16381–16386. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The dentate nucleus in Friedreich’s ataxia: The role of iron-responsive proteins. Acta Neuropathol. 2007, 114, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue atrophy and elevated iron concentration in the extrapyramidal motor system in Friedreich ataxia: The IMAGE-FRDA study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, A.H.; Morral, J.A.; Davis, A.N.; Qian, J.; Petrocine, S.V.; Knutson, M.D.; Gibson, W.M.; Cusack, M.J.; Li, D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009, 118, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Llorens, J.V.; Blanco-Sobero, L.; Gutiérrez, L.; Calap-Quintana, P.; Morales, M.P.; Moltó, M.D.; Martínez-Sebastián, M.J. Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich’s ataxia. Gene 2013, 521, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Lin, G.; Haelterman, N.A.; Ho, T.S.; Li, T.; Li, Z.; Duraine, L.; Graham, B.H.; Jaiswal, M.; Yamamoto, S.; et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. Elife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Ho, T.S.; Lin, G.; Tan, K.L.; Rasband, M.N.; Bellen, H.J. Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. Elife 2016, 5. [Google Scholar] [CrossRef]
- Yin, Y.; She, H.; Li, W.; Yang, Q.; Guo, S.; Mao, Z. Modulation of Neuronal Survival Factor MEF2 by Kinases in Parkinson’s Disease. Front. Physiol. 2012, 3, 171. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, J.B. The MEF2 family and the brain: From molecules to memory. Cell Tissue Res. 2013, 352, 179–190. [Google Scholar] [CrossRef]
- Lambert, L.; Bienvenu, T.; Allou, L.; Valduga, M.; Echenne, B.; Diebold, B.; Mignot, C.; Héron, D.; Roth, V.; Saunier, A.; et al. MEF2C mutations are a rare cause of Rett or severe Rett-like encephalopathies. Clin. Genet. 2012, 82, 499–501. [Google Scholar] [CrossRef]
- Poburski, D.; Boerner, J.B.; Koenig, M.; Ristow, M.; Thierbach, R. Time-resolved functional analysis of acute impairment of frataxin expression in an inducible cell model of Friedreich ataxia. Biol. Open 2016, 5, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.; Seznec, H.; Gansmuller, A.; Carelle, N.; Weber, P.; Metzger, D.; Rustin, P.; Koenig, M.; Puccio, H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 2004, 24, 1987–1995. [Google Scholar] [CrossRef] [Green Version]
- Rötig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Magrane, J.; Rattelle, A.; Stepanova, A.; Galkin, A.; Clark, E.M.; Dong, Y.N.; Halawani, S.M.; Lynch, D.R. Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Dis. Model. Mech. 2017, 10, 1343–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolinches-Amorós, A.; Mollá, B.; Pla-Martín, D.; Palau, F.; González-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodi, R.; Cooper, J.M.; Bradley, J.L.; Manners, D.; Styles, P.; Taylor, D.J.; Schapira, A.H. Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc. Natl. Acad. Sci. USA 1999, 96, 11492–11495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutak, R.; Xu, X.; Whitnall, M.; Kashem, M.A.; Vyoral, D.; Richardson, D.R. Proteomic analysis of hearts from frataxin knockout mice: Marked rearrangement of energy metabolism, a response to cellular stress and altered expression of proteins involved in cell structure, motility and metabolism. Proteomics 2008, 8, 1731–1741. [Google Scholar] [CrossRef]
- Ristow, M.; Pfister, M.F.; Yee, A.J.; Schubert, M.; Michael, L.; Zhang, C.Y.; Ueki, K.; Michael, M.D.; Lowell, B.B.; Kahn, C.R. Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12239–12243. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, R.A.; Napoli, E.; Wong, A.; Zhan, S.; Reutenauer, L.; Morin, D.; Buckpitt, A.R.; Taroni, F.; Lonnerdal, B.; Ristow, M.; et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 2005, 14, 3787–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, G.; Marmolino, D.; Lu, D.; Wang, Q.; Cnop, M.; Rai, M.; Acquaviva, F.; Cocozza, S.; Pandolfo, M.; Geschwind, D.H. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich’s ataxia. Hum. Mol. Genet. 2009, 18, 2452–2461. [Google Scholar] [CrossRef] [Green Version]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. North. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef]
- Pagon, R.A.; Bird, T.D.; Detter, J.C.; Pierce, I. Hereditary sideroblastic anaemia and ataxia: An X linked recessive disorder. J. Med. Genet. 1985, 22, 267–273. [Google Scholar] [CrossRef]
- Allikmets, R.; Raskind, W.H.; Hutchinson, A.; Schueck, N.D.; Dean, M.; Koeller, D.M. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum. Mol. Genet. 1999, 8, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.; Hellier, K.; Hammans, S.; May, A. X-linked cerebellar ataxia and sideroblastic anaemia associated with a missense mutation in the ABC7 gene predicting V411L. Br. J. Haematol. 2001, 115, 910–917. [Google Scholar] [CrossRef]
- Protasova, M.S.; Grigorenko, A.P.; Tyazhelova, T.V.; Andreeva, T.V.; Reshetov, D.A.; Gusev, F.E.; Laptenko, A.E.; Kuznetsova, I.L.; Goltsov, A.Y.; Klyushnikov, S.A.; et al. Whole-genome sequencing identifies a novel ABCB7 gene mutation for X-linked congenital cerebellar ataxia in a large family of Mongolian ancestry. Eur. J. Hum. Genet. 2016, 24, 550–555. [Google Scholar] [CrossRef] [PubMed]
- D’Hooghe, M.; Selleslag, D.; Mortier, G.; Van Coster, R.; Vermeersch, P.; Billiet, J.; Bekri, S. X-linked sideroblastic anemia and ataxia: A new family with identification of a fourth ABCB7 gene mutation. Eur. J. Paediatr. Neurol. 2012, 16, 730–735. [Google Scholar] [CrossRef]
- Li, J.; Cowan, J.A. Glutathione-coordinated [2Fe-2S] cluster: A viable physiological substrate for mitochondrial ABCB7 transport. Chem. Commun. (CAMB) 2015, 51, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Kispal, G.; Csere, P.; Prohl, C.; Lill, R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999, 18, 3981–3989. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A.; Tong, W.H. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008, 24, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Cavadini, P.; Biasiotto, G.; Poli, M.; Levi, S.; Verardi, R.; Zanella, I.; Derosas, M.; Ingrassia, R.; Corrado, M.; Arosio, P. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 2007, 109, 3552–3559. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Rouault, T.A. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar] [CrossRef]
- Zhang, S.; Napierala, M.; Napierala, J.S. Therapeutic Prospects for Friedreich’s Ataxia. Trends. Pharmacol. Sci. 2019, 40, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M.; Arpa, J.; Delatycki, M.B.; Le Quan Sang, K.H.; Mariotti, C.; Munnich, A.; Sanz-Gallego, I.; Tai, G.; Tarnopolsky, M.A.; Taroni, F.; et al. Deferiprone in Friedreich ataxia: A 6-month randomized controlled trial. Ann. Neurol. 2014, 76, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Nachbauer, W.; Hering, S.; Seifert, M.; Steinkellner, H.; Sturm, B.; Scheiber-Mojdehkar, B.; Reindl, M.; Strasak, A.; Poewe, W.; Weiss, G.; et al. Effects of erythropoietin on frataxin levels and mitochondrial function in Friedreich ataxia--a dose-response trial. Cerebellum 2011, 10, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, C.; Nachbauer, W.; Panzeri, M.; Poewe, W.; Taroni, F.; Boesch, S. Erythropoietin in Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 80–87. [Google Scholar] [CrossRef]
- Perdomini, M.; Belbellaa, B.; Monassier, L.; Reutenauer, L.; Messaddeq, N.; Cartier, N.; Crystal, R.G.; Aubourg, P.; Puccio, H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 2014, 20, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Piguet, F.; de Montigny, C.; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. 2018, 26, 1940–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, J.F.; Wood, K.M.; Rao, V.P.; Morin, J.; Bhamidipaty, S.; LaBranche, T.P.; Gooch, R.L.; Bozal, F.; Bulawa, C.E.; Guild, B.C. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci. Rep. 2016, 6, 20019. [Google Scholar] [CrossRef] [Green Version]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Matsui, M.; Corey, D.R. Activating frataxin expression by repeat-targeted nucleic acids. Nat. Commun. 2016, 7, 10606. [Google Scholar] [CrossRef]
- Jasoliya, M.; Sacca, F.; Sahdeo, S.; Chedin, F.; Pane, C.; Brescia Morra, V.; Filla, A.; Pook, M.; Cortopassi, G. Dimethyl fumarate dosing in humans increases frataxin expression: A potential therapy for Friedreich’s Ataxia. PLoS ONE 2019, 14, e0217776. [Google Scholar] [CrossRef] [PubMed]
- Erwin, G.S.; Grieshop, M.P.; Ali, A.; Qi, J.; Lawlor, M.; Kumar, D.; Ahmad, I.; McNally, A.; Teider, N.; Worringer, K.; et al. Synthetic transcription elongation factors license transcription across repressive chromatin. Science 2017, 358, 1617–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Heme metabolism and Fe-S cluster biogenesis are interconnected. The ISC machinery (composed by ISCU2, FXN, and other 16 proteins) is responsible for the generation of Fe-S clusters necessary for several mitochondrial proteins (in orange), including those of the electron transport chain (ETC), some enzymes of the TCA cycle (ACO2 and SDHB) and LIAS. The ISC machinery is also responsible for the generation of a X-S compound that is exported in the cytosol by ABCB7 and used by the CIA machinery for the generation of Fe-S clusters to be incorporated into non-mitochondrial proteins. Heme is synthesized through eight enzymatic reactions occurring between mitochondria and cytosol. The availability of free heme is further controlled at the level of heme catabolism (HO1) and heme export (FLVCR1a and FLVCR1b). FLVCR1b has been drawn on both mitochondrial membranes, however its specific subcellular localization still remains to be determined. Although not shown in the picture other heme importers and exporters have been identified (see the text for details). Both heme and Fe-S clusters are necessary for the ETC activity. Interestingly, the biosynthetic pathways of the two cofactors are strongly interconnected. Indeed, heme synthesis depends on Fe-S clusters for succinyl-CoA supply, for the expression of ALAS1 and FECH. Whether Fe-S clusters biogenesis depends on heme is less clear.
Figure 1.
Heme metabolism and Fe-S cluster biogenesis are interconnected. The ISC machinery (composed by ISCU2, FXN, and other 16 proteins) is responsible for the generation of Fe-S clusters necessary for several mitochondrial proteins (in orange), including those of the electron transport chain (ETC), some enzymes of the TCA cycle (ACO2 and SDHB) and LIAS. The ISC machinery is also responsible for the generation of a X-S compound that is exported in the cytosol by ABCB7 and used by the CIA machinery for the generation of Fe-S clusters to be incorporated into non-mitochondrial proteins. Heme is synthesized through eight enzymatic reactions occurring between mitochondria and cytosol. The availability of free heme is further controlled at the level of heme catabolism (HO1) and heme export (FLVCR1a and FLVCR1b). FLVCR1b has been drawn on both mitochondrial membranes, however its specific subcellular localization still remains to be determined. Although not shown in the picture other heme importers and exporters have been identified (see the text for details). Both heme and Fe-S clusters are necessary for the ETC activity. Interestingly, the biosynthetic pathways of the two cofactors are strongly interconnected. Indeed, heme synthesis depends on Fe-S clusters for succinyl-CoA supply, for the expression of ALAS1 and FECH. Whether Fe-S clusters biogenesis depends on heme is less clear.

Figure 2.
Altered molecular mechanisms in FRDA affected neurons. Frataxin (FXN) deficiency in FRDA results in the degeneration of specific neuron types leading to the ataxia phenotype. Different hypotheses have been postulated regarding the pathophysiological mechanism at the basis of neurodegeneration. FXN is a mitochondrial protein involved in Fe-S clusters biogenesis and alterations in FXN activity results in mitochondria dysfunctions and iron accumulation. The presence of iron deposits in mitochondria has been observed in patient-derived samples; however, whether iron accumulation occurs in neurons is less clear. Iron might be toxic and cause cellular dysfunctions by mediating the catalysis of ROS via Fenton reaction. However, a ROS-independent mechanism of iron toxicity has also been proposed. In this model, neurodegeneration arises from increased sphingolipids synthesis and activation of PDK1/Mef2 pathway occurring upon iron accumulation. Independently of iron accumulation, FXN deficiency results in reduced activity of Fe-S cluster containing enzymes leading to alteration in metabolic flow (aconitase), energetic failure (ETC), and altered heme synthesis (FECH). Moreover, a role of FXN in the regulation of calcium homeostasis has been described and linked to failure of retrograde axonal transport along axons and autophagy deregulation. Aberrant autophagy levels have been also reported in other independent works. However, whether autophagy is a neurodegenerative mechanism or a defense mechanism still need to be clarified. Finally, FXN deficiency may control actin dynamics and the growth cone of neurons. All hypotheses have been well described. However, which of them is the primary event triggering the neurological phenotype in FRDA is still unknown.
Figure 2.
Altered molecular mechanisms in FRDA affected neurons. Frataxin (FXN) deficiency in FRDA results in the degeneration of specific neuron types leading to the ataxia phenotype. Different hypotheses have been postulated regarding the pathophysiological mechanism at the basis of neurodegeneration. FXN is a mitochondrial protein involved in Fe-S clusters biogenesis and alterations in FXN activity results in mitochondria dysfunctions and iron accumulation. The presence of iron deposits in mitochondria has been observed in patient-derived samples; however, whether iron accumulation occurs in neurons is less clear. Iron might be toxic and cause cellular dysfunctions by mediating the catalysis of ROS via Fenton reaction. However, a ROS-independent mechanism of iron toxicity has also been proposed. In this model, neurodegeneration arises from increased sphingolipids synthesis and activation of PDK1/Mef2 pathway occurring upon iron accumulation. Independently of iron accumulation, FXN deficiency results in reduced activity of Fe-S cluster containing enzymes leading to alteration in metabolic flow (aconitase), energetic failure (ETC), and altered heme synthesis (FECH). Moreover, a role of FXN in the regulation of calcium homeostasis has been described and linked to failure of retrograde axonal transport along axons and autophagy deregulation. Aberrant autophagy levels have been also reported in other independent works. However, whether autophagy is a neurodegenerative mechanism or a defense mechanism still need to be clarified. Finally, FXN deficiency may control actin dynamics and the growth cone of neurons. All hypotheses have been well described. However, which of them is the primary event triggering the neurological phenotype in FRDA is still unknown.


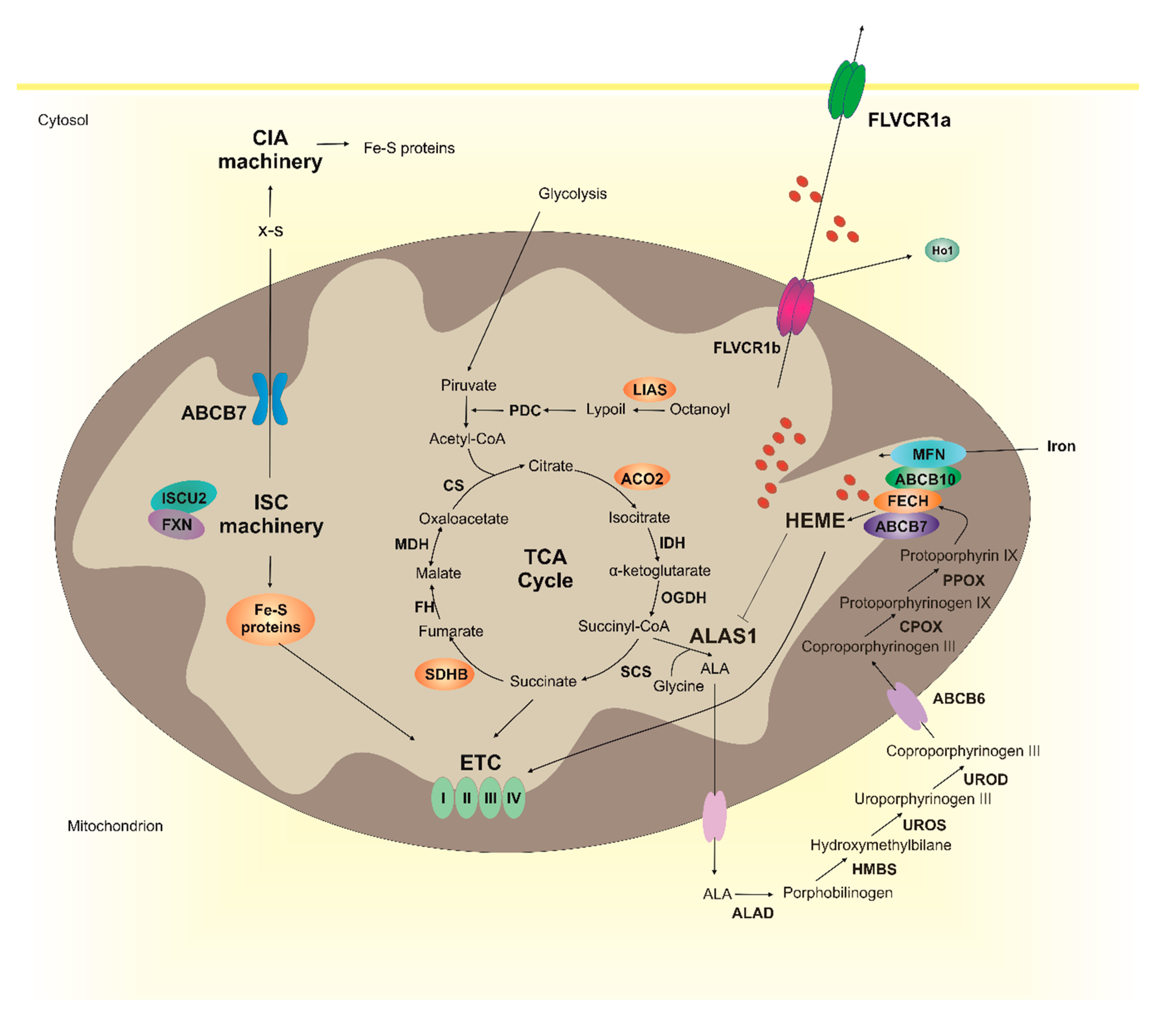{kind=link}
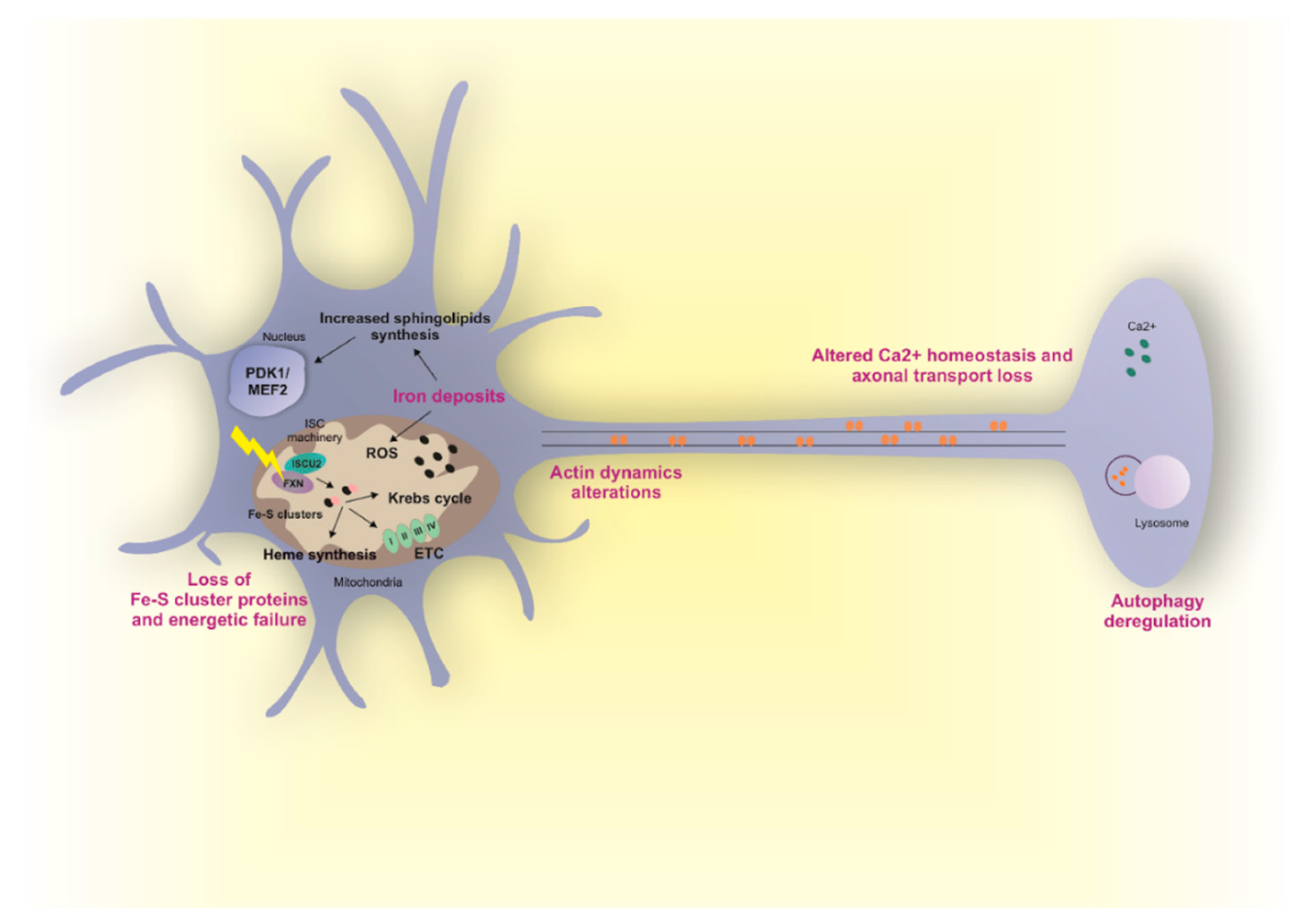{kind=link}
Table 1.
Main clinical features and sites of pathology of PCARP, FRDA and XLSA/A.
| Gene | Function | Neurological Clinical Features | Sites of Pathology | Ref. | |
|---|---|---|---|---|---|
| PCARP | FLVCR1 | Heme export | Sensory ataxia Retinitis pigmentosa | Posterior columns of spinal cord Retina | [8,9] |
| FRDA | FXN | Fe-S cluster biogenesis | Mixed spinocerebellar and sensory ataxiaDysarthriaMuscular weakness Absence of tendon reflexes with deep sensory loss | Large sensory neuronsPosterior columns of spinal cordDentate nucleus of cerebellum | [10,11,12] |
| XLSA/A | ABCB7 | Export of S-X compound from mitochondria | Spinocerebellar ataxia Intention tremor Dysarthria | Cerebellum | [13,14] |
Table 2.
FLVCR1 mutations reported in PCARP patients so far.
| Mutation(s) | Zygosity | Types of Mutation | Exon/Intron | Ref. |
|---|---|---|---|---|
| c.361A>G (p.Asn121Asp) | Homozygous | Missense | Exon 1 | [79,81] |
| c.721G>A (p.Ala241Thr) | Homozygous | Missense | Exon 1 | [79] |
| c.574T>C (p.Cys192Arg) | Homozygous | Missense | Exon 1 | [79] |
| c.1477G>C (Gly493Arg) | Homozygous | Missense | Exon 8 | [83] |
| c.1547G>A (p.Arg516Gln) c.1593+5 +8delGTAA | Compound Heterozygous | Missense Splicing | Exon 9 Intron 9–10 | [82] |
| c.596T>C (p.Leu199Pro) | Homozygous | Missense | Exon 1 | [80] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chiabrando, D.; Bertino, F.; Tolosano, E. Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis. Int. J. Mol. Sci. 2020, 21, 3760. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113760
AMA Style
Chiabrando D, Bertino F, Tolosano E. Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis. International Journal of Molecular Sciences. 2020; 21(11):3760. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113760
Chicago/Turabian StyleChiabrando, Deborah, Francesca Bertino, and Emanuela Tolosano. 2020. "Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis" International Journal of Molecular Sciences 21, no. 11: 3760. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113760
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.