Characterization and Analysis of the Mitochondrial Genome of Common Bean (Phaseolus vulgaris) by Comparative Genomic Approaches


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, DNA Extraction, and Sequencing
2.2. Mitogenome Assembly and Annotation
2.3. Selective Pressure Analysis
2.4. Prediction of RNA Editing Sites
2.5. Identification of Repeat Sequences in P. vulgaris Mitogenome
2.6. Phylogenetic Analyses
3. Results and Discussion
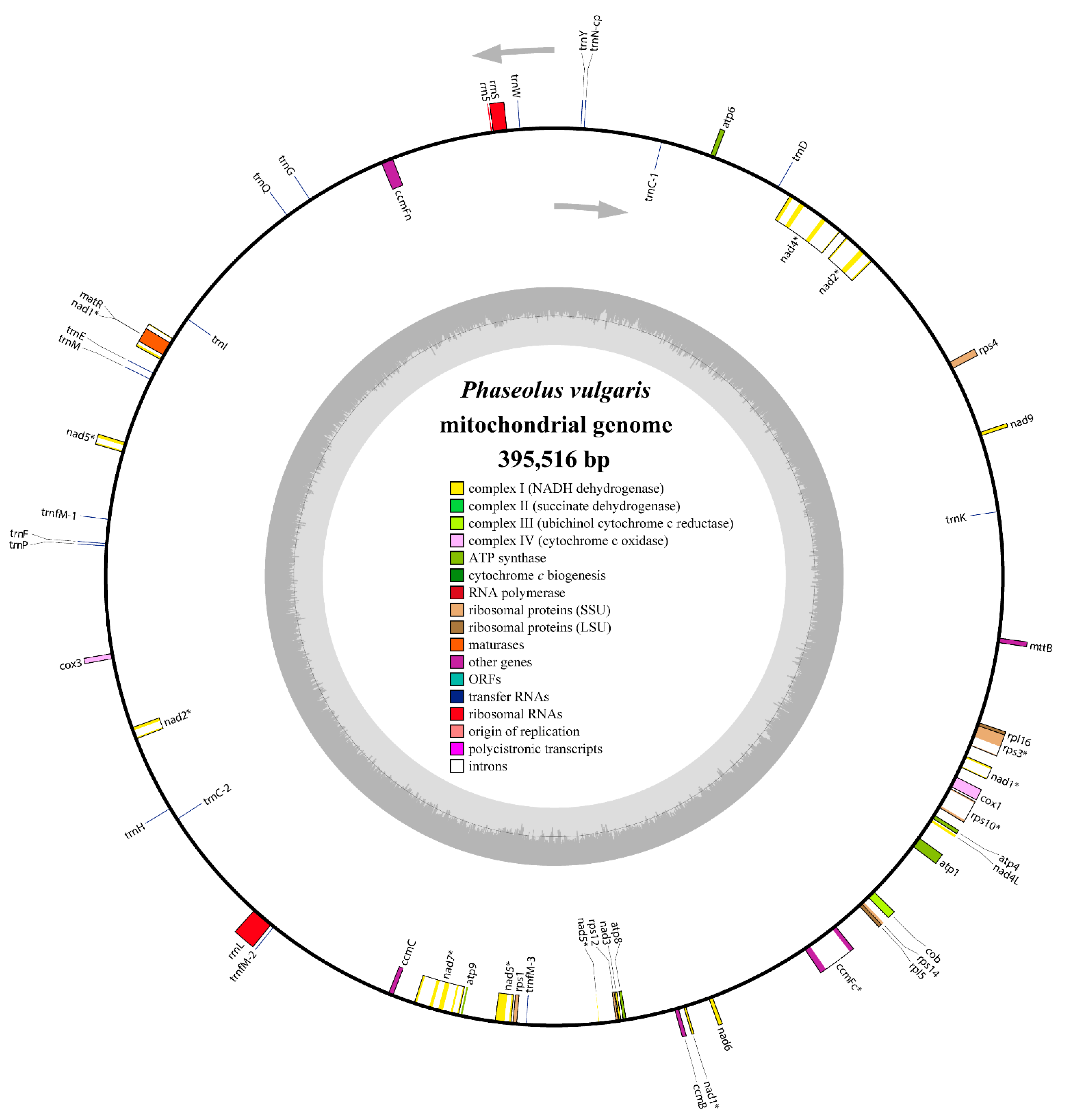
3.1. Genomic Features of the P. vulgaris Mitogenome
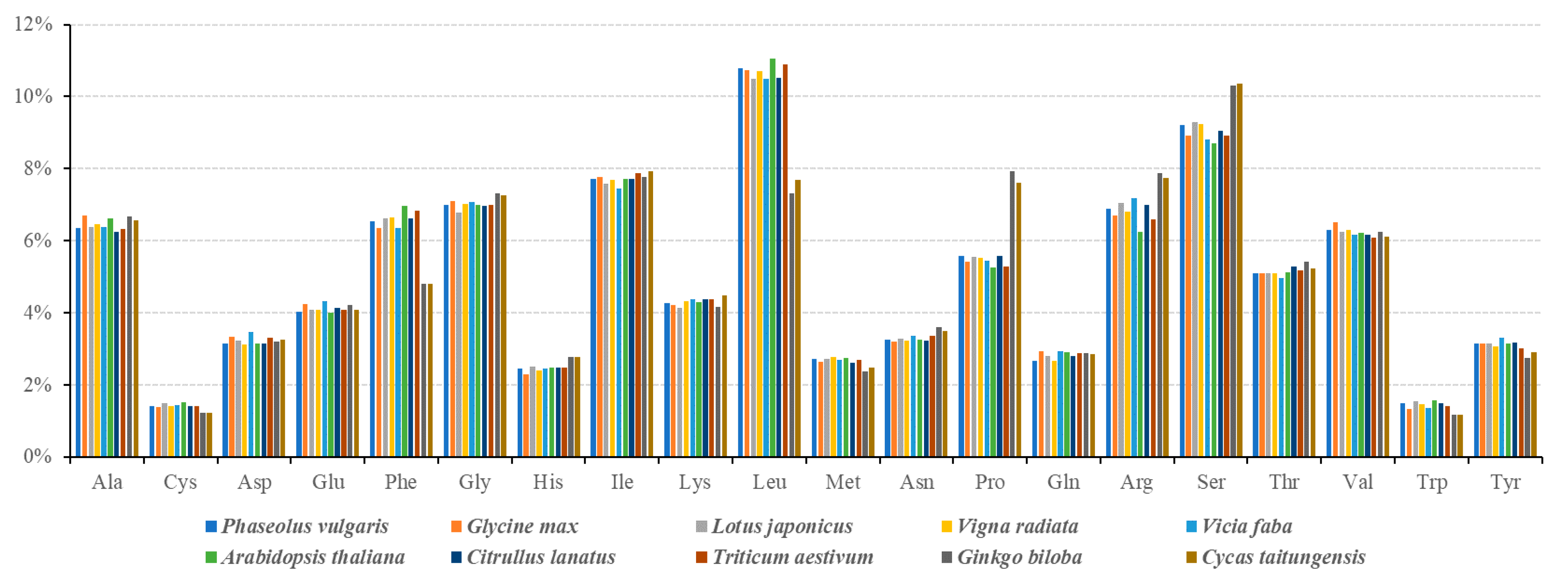
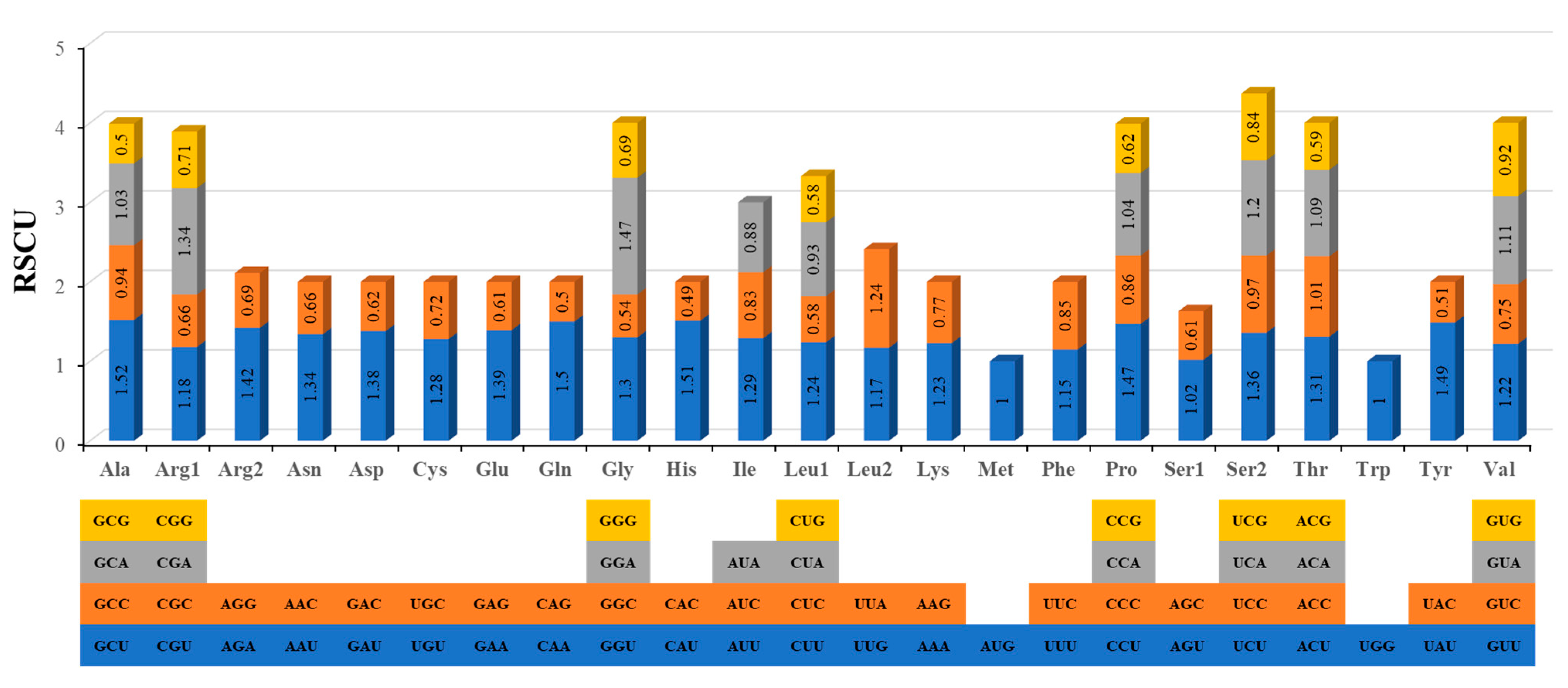
3.2. Codon Usage Analysis of PCGs
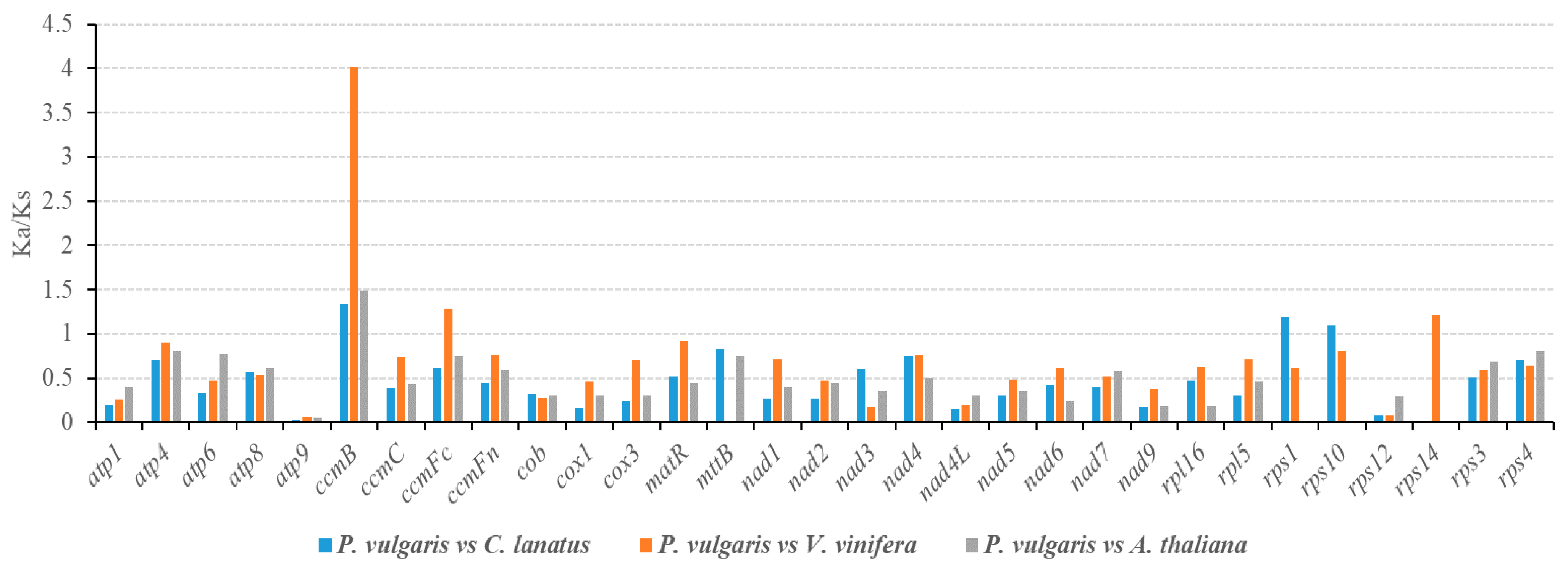
3.3. Selective Pressure Analysis
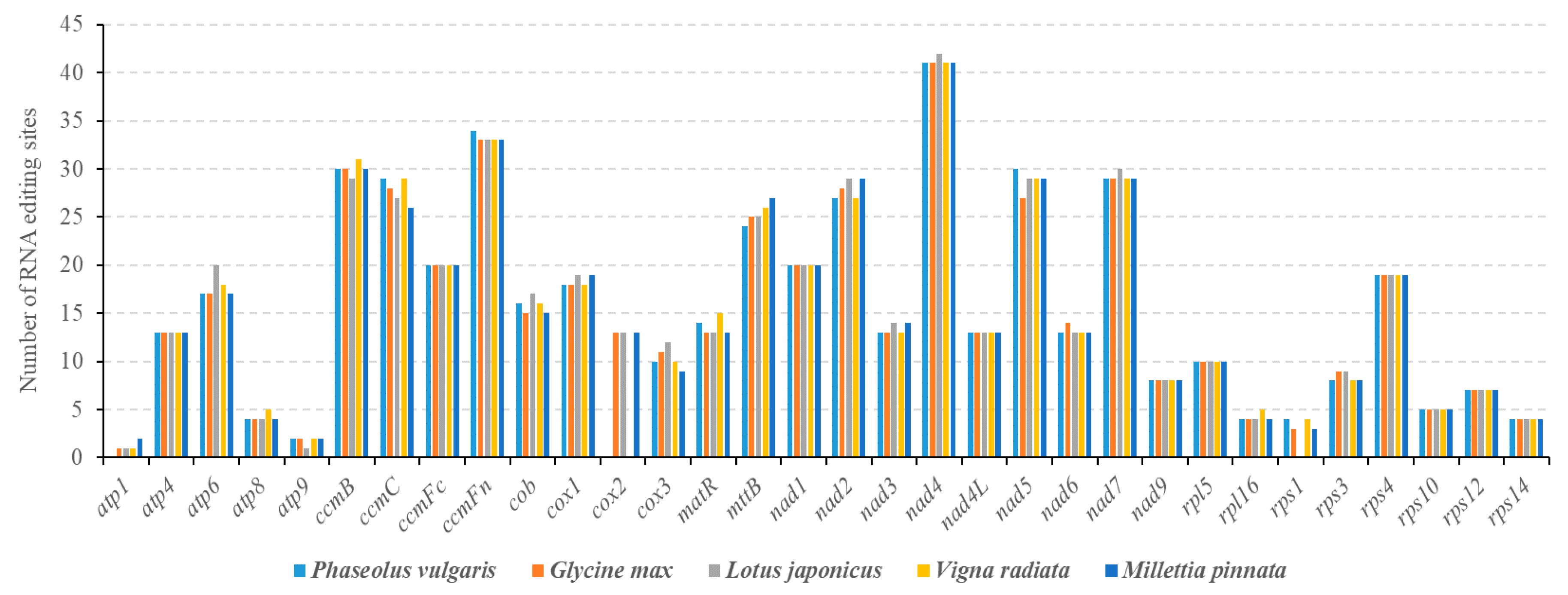
3.4. Prediction of RNA Editing Sites in PCGs
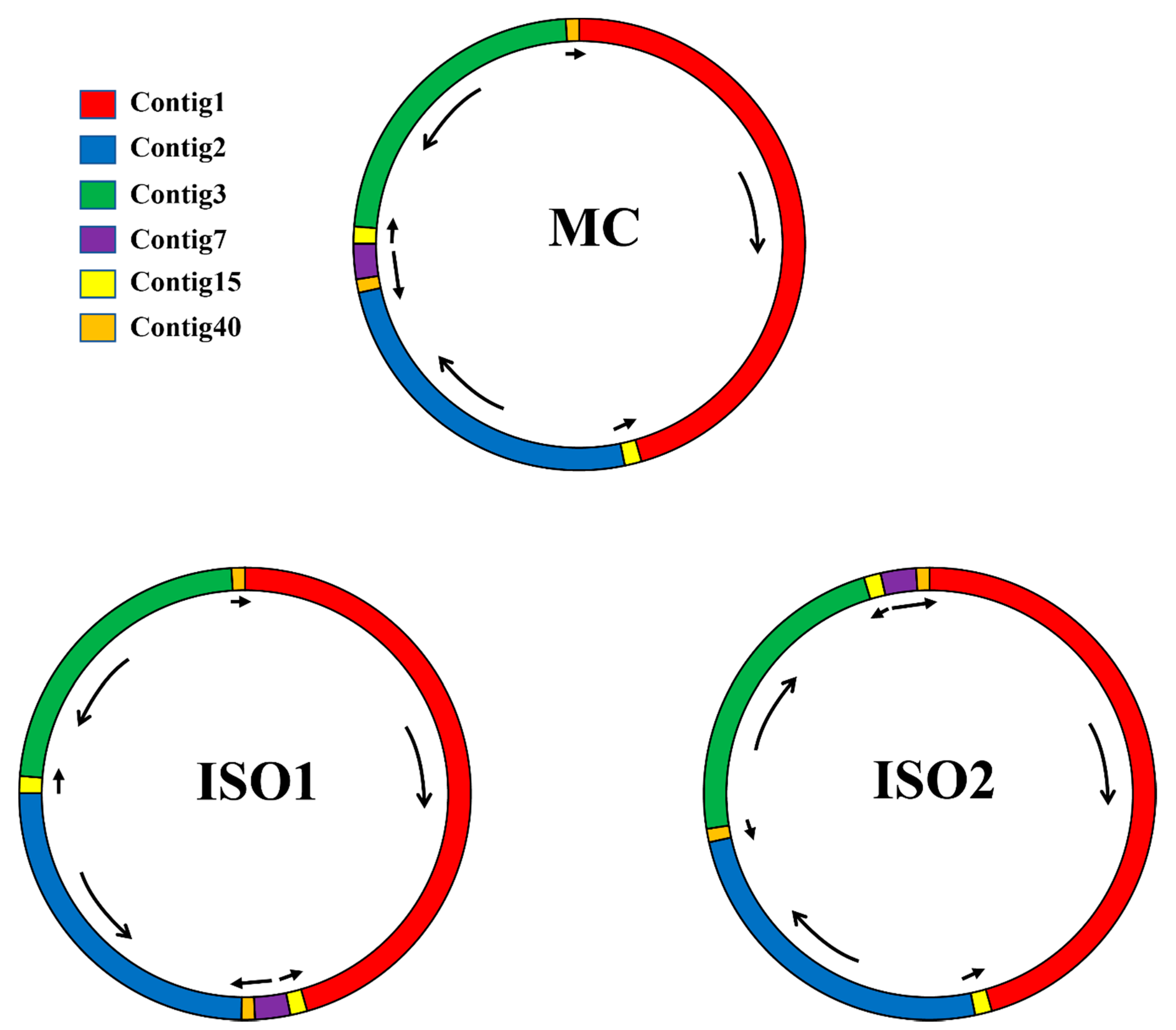
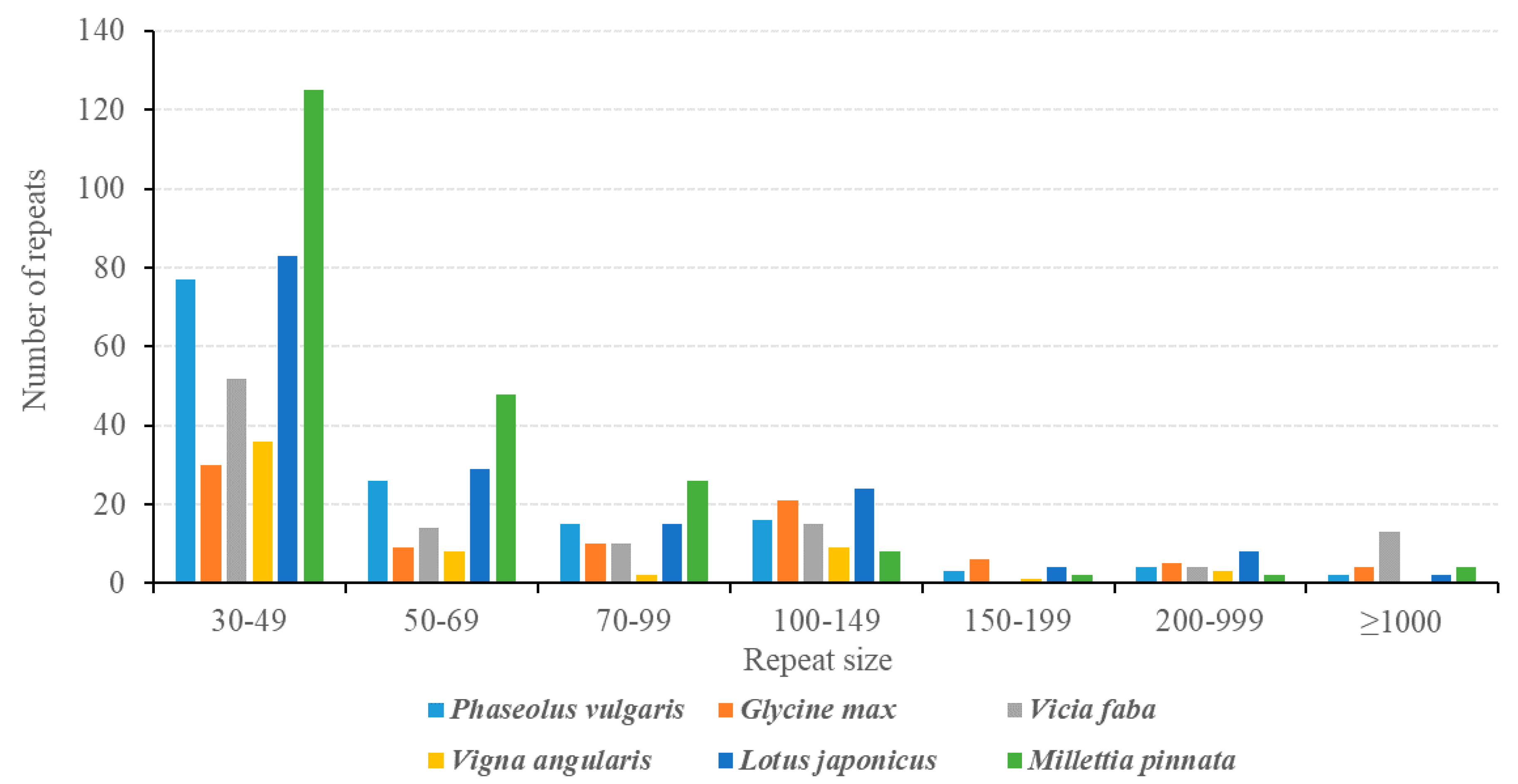
3.5. Analysis of Repeat Sequences in the P. vulgaris Mitogenome
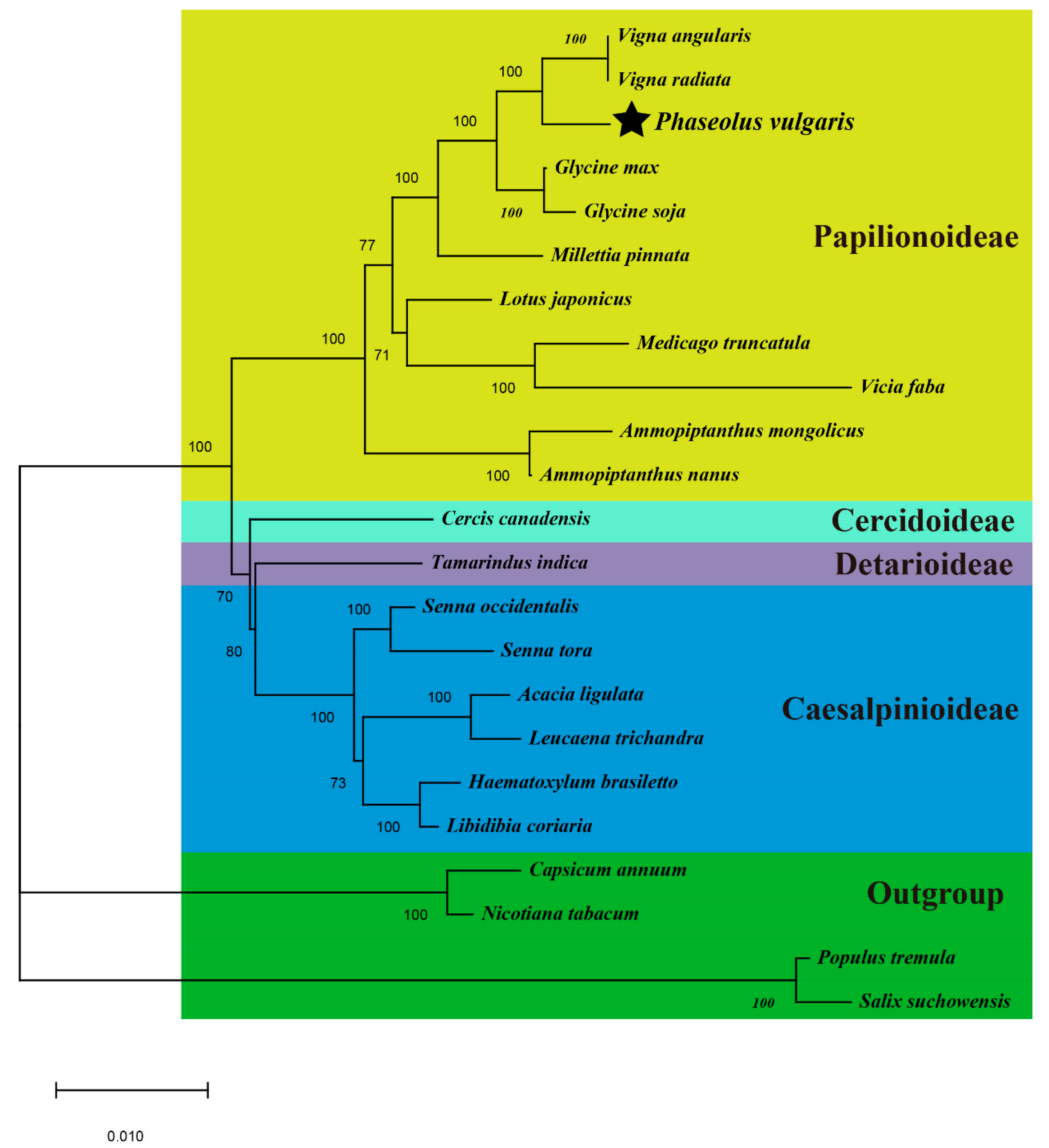
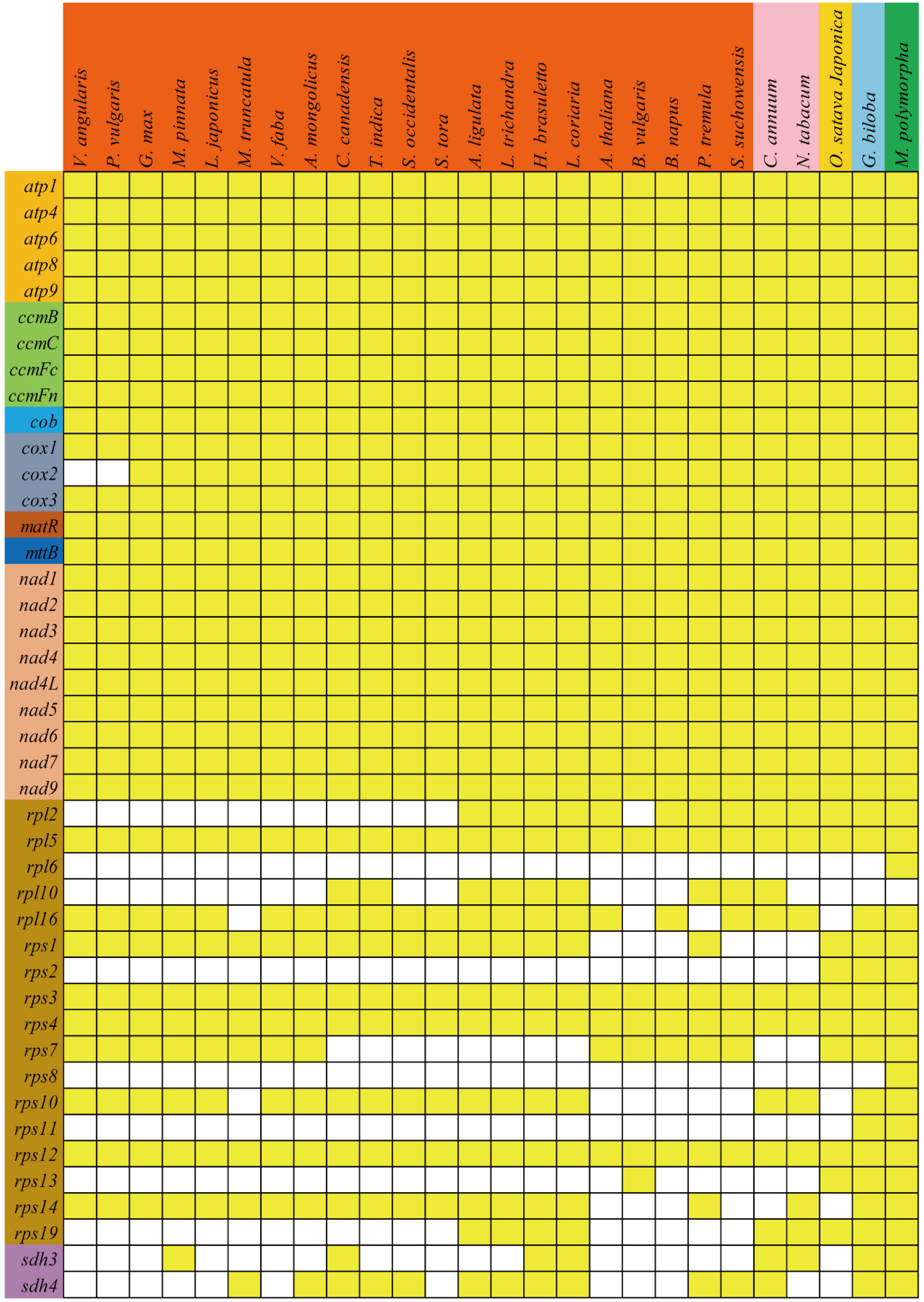
3.6. Phylogenetic Analyses and Multiple Losses of PCGs during Evolution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Greiner, S.; Bock, R. Tuning a menage a trois: Co-evolution and co-adaptation of nuclear and organellar genomes in plants. Bioessays 2013, 35, 354–365. [Google Scholar] [CrossRef]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef]
- Hsu, C.L.; Mullin, B.C. Physical characterization of mitochondrial DNA from cotton. Plant Mol. Biol. 1989, 13, 467–468. [Google Scholar] [CrossRef]
- Kubo, T.; Mikami, T. Organization and variation of angiosperm mitochondrial genome. Physiol. Plant. 2007, 129, 6–13. [Google Scholar] [CrossRef]
- Palmer, J.D.; Herbon, L.A. Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J. Mol. Evol. 1988, 28, 87–97. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial Evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [Green Version]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial Genome Evolution and the Origin of Eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef]
- Alverson, A.J.; Wei, X.X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the Evolution of Mitochondrial Genome Size from Complete Sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Alverson, A.J.; Zhuo, S.; Rice, D.W.; Sloan, D.B.; Palmer, J.D. The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS ONE 2011, 6, e16404. [Google Scholar] [CrossRef] [Green Version]
- Handa, H. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): Comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 2003, 31, 5907–5916. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, R.M.; Chang, K.L.; Chou, C.C. Computational analysis of RNA editing sites in plant mitochondrial genomes reveals similar information content and a sporadic distribution of editing sites. Mol. Biol. Evol. 2007, 24, 1971–1981. [Google Scholar] [CrossRef] [Green Version]
- Andre, C.; Levy, A.; Walbot, V. Small repeated sequences and the structure of plant mitochondrial genomes. Trends Genet. 1992, 8, 128–132. [Google Scholar] [CrossRef]
- Chang, S.; Wang, Y.; Lu, J.; Gai, J.; Li, J.; Chu, P.; Guan, R.; Zhao, T. Correction: The Mitochondrial Genome of Soybean Reveals Complex Genome Structures and Gene Evolution at Intercellular and Phylogenetic Levels. PLoS ONE 2013, 8, e56502. [Google Scholar] [CrossRef]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, C.; Wang, X.; Xu, Y.; Wei, S.; Shi, Y.; Dai, X.; Yin, T.; Ye, N. The complete mitochondrial genome of Medicago truncatula. Mitochondrial DNA Part B 2016, 1, 122–123. [Google Scholar] [CrossRef] [Green Version]
- Negruk, V. Mitochondrial genome sequence of the legume Vicia faba. Front. Plant Sci. 2013, 4, 128. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.-S.; Schwarz, E.N.; Ruhlman, T.A.; Khiyami, M.A.; Sabir, J.S.; Hajarah, N.H.; Sabir, M.J.; Rabah, S.O.; Jansen, R.K. Fluctuations in Fabaceae mitochondrial genome size and content are both ancient and recent. BMC Plant Biol. 2019, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Wynn, E.L.; Christensen, A.C. Repeats of Unusual Size in Plant Mitochondrial Genomes: Identification, Incidence and Evolution. G3 Genes Genomes Genet. 2019, 9, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Bergthorsson, U.; Adams, K.L.; Thomason, B.; Palmer, J.D. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 2003, 424, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Li, S.; Bi, C.; Hao, Z.; Sun, C.; Ye, N. Complete chloroplast genome sequence of a major economic species, Ziziphus jujuba (Rhamnaceae). Curr. Genet. 2017, 63, 117–129. [Google Scholar] [CrossRef]
- Sperisen, C.; Büchler, U.; Gugerli, F.; Mátyás, G.; Geburek, T.; Vendramin, G. Tandem repeats in plant mitochondrial genomes: Application to the analysis of population differentiation in the conifer Norway spruce. Mol. Ecol. 2001, 10, 257–263. [Google Scholar] [CrossRef]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and Recombination of the Bacterial-Sized Multichromosomal Mitochondrial Genome of Cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef] [Green Version]
- Backert, S.; Nielsen, B.L.; Börner, T. The mystery of the rings: Structure and replication of mitochondrial genomes from higher plants. Trends Plant Sci. 1997, 2, 477–483. [Google Scholar] [CrossRef]
- Ogihara, Y.; Yamazaki, Y.; Murai, K.; Kanno, A.; Terachi, T.; Shiina, T.; Miyashita, N.; Nasuda, S.; Nakamura, C.; Mori, N. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005, 33, 6235–6250. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Paterson, A.H.; Wang, X.; Xu, Y.; Wu, D.; Qu, Y.; Jiang, A.; Ye, Q.; Ye, N. Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. BioMed. Res. Int. 2016, 2016, 5040598. [Google Scholar] [CrossRef] [Green Version]
- Christenhusz, M.J.; Byng, J.W. The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Azani, N.; Babineau, M.; Bailey, C.D.; Banks, H.; Barbosa, A.R.; Pinto, R.B.; Boatwright, J.S.; Borges, L.M.; Brown, G.K.; Bruneau, A.; et al. A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny: The Legume Phylogeny Working Group (LPWG). Taxon 2017, 66, 44–77. [Google Scholar] [CrossRef] [Green Version]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707. [Google Scholar] [CrossRef] [Green Version]
- Fonsêca, A.; Ferreira, J.; dos Santos, T.R.B.; Mosiolek, M.; Bellucci, E.; Kami, J.; Gepts, P.; Geffroy, V.; Schweizer, D.; dos Santos, K.G. Cytogenetic map of common bean (Phaseolus vulgaris L.). Chrom. Res. 2010, 18, 487–502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Fang, Y.; Wang, X.; Deng, X.; Zhang, X.; Hu, S.; Yu, J. The Complete Chloroplast and Mitochondrial Genome Sequences of Boea hygrometrica: Insights into the Evolution of Plant Organellar Genomes. PLoS ONE 2012, 7, e30531. [Google Scholar] [CrossRef] [Green Version]
- Iorizzo, M.; Senalik, D.; Szklarczyk, M.; Grzebelus, D.; Spooner, D.; Simon, P. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 2012, 12, 61. [Google Scholar] [CrossRef] [Green Version]
- Ye, N.; Wang, X.; Li, J.; Bi, C.; Xu, Y.; Wu, D.; Ye, Q. Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. PeerJ 2017, 5, e3148. [Google Scholar] [CrossRef] [Green Version]
- Nederbragt, A.J. On the middle ground between open source and commercial software-the case of the Newbler program. Genome Biol. 2014, 15, 113. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, X.; Hu, S.; Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods 2011, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Zhao, C.; Chen, F.; Liu, Y.; Zhang, S.; Wu, H.; Zhang, L.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 2018, 19, 614. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating Synonymous and Nonsynonymous Substitution Rates Under Realistic Evolutionary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit Incorporating Gamma-Series Methods and Sliding Window Strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Cao, D.; Li, S.; Su, A.; Geng, J.; Grover, C.E.; Hu, S.; Hua, J. The Complete Mitochondrial Genome of Gossypium hirsutum and Evolutionary Analysis of Higher Plant Mitochondrial Genomes. PLoS ONE 2013, 8, e69476. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Du, H.; Liu, Y.; Ni, L.; Wang, Z.; Liang, C.; Tian, Z. Update soybean Zhonghuang 13 genome to a golden reference. Sci. China Life Sci. 2019, 62, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-H.; Wu, C.-I.; Luo, C.-C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 1985, 2, 150–174. [Google Scholar] [PubMed] [Green Version]
- Betrán, E.; Bai, Y.; Motiwale, M. Fast Protein Evolution and Germ Line Expression of a Drosophila Parental Gene and Its Young Retroposed Paralog. Mol. Biol. Evol. 2006, 23, 2191–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbiza, L.; Dopazo, J.; Dopazo, H. Positive Selection, Relaxation, and Acceleration in the Evolution of the Human and Chimp Genome. PLoS Comp. Biol. 2006, 2, e38. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.H.; Giegé, P.; Gelhaye, E.; Rayapuram, N.; Ahuja, U.; Thöny-Meyer, L.; Grienenberger, J.-M.; Bonnard, G. AtCCMH, an essential component of the c-type cytochrome maturation pathway in Arabidopsis mitochondria, interacts with apocytochrome c. Proc. Natl. Acad. Sci. USA 2005, 102, 16113–16118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre-Nitschke, S.E.; Nazoa, P.; Gualberto, J.M.; Grienenberger, J.M.; Bonnard, G. Wheat mitochondria ccmB encodes the membrane domain of a putative ABC transporter involved in cytochrome c biogenesis. Biochim. Biophys. Acta 2001, 1519, 199–208. [Google Scholar] [CrossRef]
- Sanders, C.; Turkarslan, S.; Lee, D.-W.; Daldal, F. Cytochrome c biogenesis: The Ccm system. Trends Microbiol. 2010, 18, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Robles, P.; Quesada, V. Emerging Roles of Mitochondrial Ribosomal Proteins in Plant Development. Int. J. Mol. Sci. 2017, 18, 2595. [Google Scholar] [CrossRef] [Green Version]
- Mauro, V.P.; Edelman, G.M. The Ribosome Filter Redux. Cell Cycle 2007, 6, 2246–2251. [Google Scholar] [CrossRef] [Green Version]
- Schippers, J.H.M.; Mueller-Roeber, B. Ribosomal composition and control of leaf development. Plant Sci. 2010, 179, 307–315. [Google Scholar] [CrossRef]
- Cui, P.; Liu, H.; Lin, Q.; Ding, F.; Zhuo, G.; Hu, S.; Liu, D.; Yang, W.; Zhan, K.; Zhang, A.; et al. A complete mitochondrial genome of wheat (Triticum aestivum cv. Chinese Yumai), and fast evolving mitochondrial genes in higher plants. J. Genet. 2009, 88, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Li, N.; Yang, W.; Li, Y.; Wang, C.-M.; Tong, S.-W.; He, J.-X. Analyses of mitochondrial genomes of the genus Ammopiptanthus provide new insights into the evolution of legume plants. Plant Syst. Evol. 2019, 305, 385–399. [Google Scholar] [CrossRef]
- Wernegreen, J.J.; Riley, M.A. Comparison of the evolutionary dynamics of symbiotic and housekeeping loci: A case for the genetic coherence of rhizobial lineages. Mol. Biol. Evol. 1999, 16, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Bock, R.; Khan, M.S. Taming plastids for a green future. Trends Biotechnol. 2004, 22, 311–318. [Google Scholar] [CrossRef]
- Chen, H.; Deng, L.; Jiang, Y.; Lu, P.; Yu, J. RNA Editing Sites Exist in Protein-coding Genes in the Chloroplast Genome of Cycas taitungensis. J. Integr. Plant Biol. 2011, 53, 961–970. [Google Scholar] [CrossRef]
- Raman, G.; Park, S. Analysis of the Complete Chloroplast Genome of a Medicinal Plant, Dianthus superbus var. longicalyncinus, from a Comparative Genomics Perspective. PLoS ONE 2015, 10, e0141329. [Google Scholar] [CrossRef] [Green Version]
- Wakasugi, T.; Hirose, T.; Horihata, M.; Tsudzuki, T.; Kössel, H.; Sugiura, M. Creation of a novel protein-coding region at the RNA level in black pine chloroplasts: The pattern of RNA editing in the gymnosperm chloroplast is different from that in angiosperms. Proc. Natl. Acad. Sci. USA 1996, 93, 8766–8770. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. PREP-Mt: Predictive RNA editor for plant mitochondrial genes. BMC Bioinform. 2005, 6, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, O.; Lättig, K.; Hiesel, R.; Brennicke, A.; Knoop, V. RNA editing in bryophytes and a molecular phylogeny of land plants. EMBO J. 1996, 15, 1403–1411. [Google Scholar] [CrossRef]
- Steinhauser, S.; Beckert, S.; Capesius, I.; Malek, O.; Knoop, V. Plant Mitochondrial RNA Editing. J. Mol. Evol. 1999, 48, 303–312. [Google Scholar] [CrossRef]
- Shu-Miaw, C.; Arthur, C.C.S.; Wang, D.; Yu-Wei, W.; Shu-Mei, L.; The-Yuan, C. The Mitochondrial Genome of the Gymnosperm Cycas taitungensis Contains a Novel Family of Short Interspersed Elements, Bpu Sequences, and Abundant RNA Editing Sites. Mol. Biol. Evol. 2008, 25, 603–615. [Google Scholar]
- Hiesel, R.; Combettes, B.; Brennicke, A. Evidence for RNA editing in mitochondria of all major groups of land plants except the Bryophyta. Proc. Natl. Acad. Sci. USA 1994, 91, 629–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goremykin, V.V.; Salamini, F.; Velasco, R.; Viola, R. Mitochondrial DNA of Vitis vinifera and the Issue of Rampant Horizontal Gene Transfer. Mol. Biol. Evol. 2009, 26, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.B.; Alverson, A.J.; Štorchová, H.; Palmer, J.D.; Taylor, D.R. Extensive loss of translational genes in the structurally dynamic mitochondrial genome of the angiosperm Silene latifolia. BMC Evol. Biol. 2010, 10, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, A.O.; Rice, D.W.; Young, G.J.; Alverson, A.J.; Palmer, J.D. The “fossilized” mitochondrial genome of Liriodendron tulipifera: Ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.L.; Kohchi, T.; Yamato, K.T.; Jenkins, J.; Shu, S. Insights into Land Plant Evolution Garnered from the Marchantia polymorpha Genome. Cell 2017, 171, 287–304. [Google Scholar] [CrossRef]
- Guo, W.; Felix, G.; Fan, W.; Young, G.J.; Volker, K.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia Mitogenomes Reveal Extreme Contrasts in Gymnosperm Mitochondrial Evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef]
- Clifton, S.W.; Minx, P.; Fauron, C.M.-R.; Gibson, M.; Allen, J.O.; Sun, H.; Thompson, M.; Barbazuk, W.B.; Kanuganti, S.; Tayloe, C. Sequence and Comparative Analysis of the Maize NB Mitochondrial Genome. Plant Physiol. 2004, 136, 3486–3503. [Google Scholar] [CrossRef] [Green Version]
- Unseld, M.; Marienfeld, J.R.; Brandt, P.; Brennicke, A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 1997, 15, 57–61. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Watase, Y.; Nagase, M.; Makita, N.; Yagura, S.; Hirai, A.; Sugiura, M. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: Comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genom. 2005, 272, 603–615. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | A % | C % | G % | T % | GC % | Size (bp) | Proportion in Genome (%) |
|---|---|---|---|---|---|---|---|
| Whole genome | 27.37 | 22.40 | 22.71 | 27.52 | 45.11 | 395,516 | 100 |
| Protein-coding genes a | 26.34 | 21.29 | 21.62 | 30.75 | 42.91 | 28,725 | 7.26 |
| cis-spliced introns a | 24.49 | 25.56 | 24.79 | 25.16 | 50.34 | 32,584 | 8.24 |
| tRNA genes a | 23.07 | 24.34 | 27.49 | 25.09 | 51.84 | 1335 | 0.34 |
| rRNA genes a | 26.14 | 22.62 | 29.04 | 22.20 | 51.66 | 5252 | 1.33 |
| Non-coding regions | 27.78 | 22.18 | 22.48 | 27.56 | 44.65 | 327,620 | 82.83 |
| Group of Genes | Gene Name |
|---|---|
| Complex I (NADH dehydrogenase) | nad1 *, nad2 *, nad3, nad4 *, nad4L, nad5 *, nad6, nad7 *, nad9 |
| Complex II (succinate dehydrogenase) | - |
| Complex III (ubiquinol cytochrome c reductase) | cob |
| Complex IV (cytochrome c oxidase) | cox1, cox3 |
| Complex V (ATP synthase) | atp1, atp4, atp6, atp8, atp9 |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFC *, ccmFN |
| Ribosomal proteins (SSU) | rps1, rps3 *, rps4, rps10 *, rps12, rps14 |
| Ribosomal proteins (LSU) | rpl5, rpl16 |
| Maturases | matR |
| Transport membrane protein | mttB |
| Ribosomal RNAs | rrn5, rrnS, rrnL |
| Transfer RNAs | trnC-GCA (2 copies), trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnfM-CAU (3 copies), trnH-GUG, trnI-CAU, trnK-UUU, trnM-CAU, trnN-GUU, trnP-UGG, trnQ-UUG, trnW-CCA, trnY-GUA |
| Gene Name | Length | Start Codon | Stop Codon | Direction |
|---|---|---|---|---|
| atp1 | 1527 | ATG | TGA | F |
| atp4 | 588 | ATG | TAA | F |
| atp6 | 726 | ATG | TAG | F |
| atp8 | 483 | ATG | TAA | R |
| atp9 | 225 | ATG | TAA | R |
| ccmB | 621 | ATG | TGA | F |
| ccmC | 741 | ATG | TGA | R |
| ccmFC | 1329 | ATG | CGA | F |
| ccmFN | 1740 | ATG | TGA | R |
| cob | 1176 | ATG | TAG | F |
| cox1 | 1584 | ATG | TAA | F |
| cox3 | 798 | ATG | TGA | F |
| matR | 2010 | ATG | TGA | F |
| mttB | 723 | ACG | TGA | F |
| nad1 | 978 | ACG | TAA | F |
| nad2 | 1467 | ATG | TAA | R |
| nad3 | 357 | ATG | TAA | R |
| nad4 | 1488 | ATG | TGA | R |
| nad4L | 303 | ACG | TAA | F |
| nad5 | 2019 | ATG | TAA | F/R |
| nad6 | 618 | ATG | TAA | F |
| nad7 | 1185 | ATG | TAG | R |
| nad9 | 573 | ATG | TAA | F |
| rpl5 | 558 | ATG | TAA | F |
| rpl16 | 516 | ATG | TAA | F |
| rps1 | 618 | ATG | TAA | R |
| rps3 | 1689 | ATG | TAG | F |
| rps4 | 1041 | ATG | TAA | F |
| rps10 | 363 | ACG | TGA | F |
| rps12 | 378 | ATG | TGA | R |
| rps14 | 303 | ATG | TAG | F |
| Motif Type | Number of Repeats | Total | Proportion (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | |||
| Monomer | - | - | - | - | - | 91 | 32 | 10 | 4 | 1 | 1 | 139 | 44.27 |
| Dimer | - | 120 | 16 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 140 | 44.59 |
| Trimer | - | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 1.59 |
| Tetramer | 21 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 22 | 7.01 |
| Pentamer | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0.96 |
| Hexamer | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 1.59 |
| Total | 29 | 126 | 16 | 4 | 0 | 91 | 32 | 10 | 4 | 1 | 1 | 314 | 100 |
| No | Size (bp) | Start | End | Repeat (bp) × Copy Number | Location |
|---|---|---|---|---|---|
| 1 | 57 | 75,208 | 75,322 | (TTGGATCAAAACGATGTTCAACAACCTTTGCCGCGTCTGTTTCTTGGAGGAAAATAG) × 2 | IGS (trnD, atp6) |
| 2 | 27 | 92,189 | 92,243 | (AGAGCAGGTCGGTCTAGGTAGTTGAAA) × 2 | IGS (trnC, trnN) |
| 3 | 38 | 108,846 | 108,922 | (AAAAATATACATAACATATCCCAAACTCTATAGAGATA) × 2 | IGS (rrn5, ccmFN) |
| 4 | 13 | 231,727 | 231,753 | (TCTTAAGTAAAGT) × 2 | IGS (nad2-exon1, trnH) |
| 5 | 18 | 252,332 | 252,368 | (CATAGTCGCGAGCTGTTT) × 2 | rrnL |
| 6 | 15 | 315,763 | 315,793 | (GTATAGTATAGTAGG) × 2 | IGS (nad1-exon1, nad6) |
| 7 | 33 | 356,560 | 356,626 | (CCTTGCCCCCTGCAGAGCCTCAAGCCCCTGAGC) × 2 | IGS (atp1, nad4L) |
| IGS: intergenic pacers. | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and Analysis of the Mitochondrial Genome of Common Bean (Phaseolus vulgaris) by Comparative Genomic Approaches. Int. J. Mol. Sci. 2020, 21, 3778. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113778
Bi C, Lu N, Xu Y, He C, Lu Z. Characterization and Analysis of the Mitochondrial Genome of Common Bean (Phaseolus vulgaris) by Comparative Genomic Approaches. International Journal of Molecular Sciences. 2020; 21(11):3778. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113778
Chicago/Turabian StyleBi, Changwei, Na Lu, Yiqing Xu, Chunpeng He, and Zuhong Lu. 2020. "Characterization and Analysis of the Mitochondrial Genome of Common Bean (Phaseolus vulgaris) by Comparative Genomic Approaches" International Journal of Molecular Sciences 21, no. 11: 3778. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113778