Pathogenic Pathways and Therapeutic Approaches Targeting Inflammation in Diabetic Nephropathy

 , , ,
, , ,  and
and
Abstract
:1. Introduction
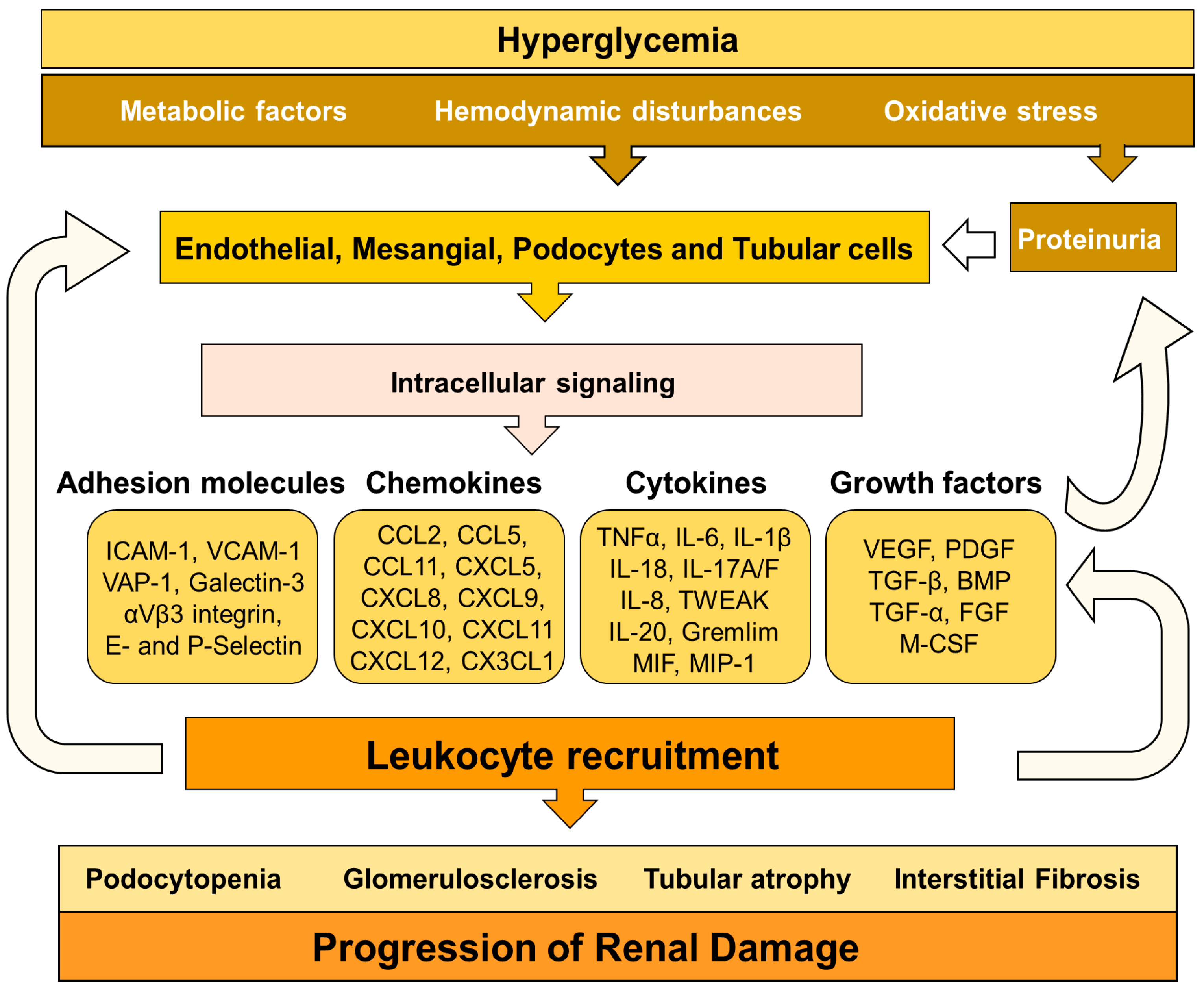
2. Mechanisms Triggering DN-Associated Inflammation
3. Immune Cells and DN
3.1. Macrophages
3.2. Dendritic Cells
3.3. T Lymphocytes
3.4. B Lymphocytes
3.5. Mast Cells
3.6. Neutrophils
4. Inflammatory Mediators in DN
4.1. Cell Adhesion Molecules
4.2. Chemokines

{kind=link}
{kind=link}
{kind=link}
| Target | Diabetic Model | Strategy | Category | Conclusion | Ref. |
|---|---|---|---|---|---|
| CAM | SD rats (65 mg/kg) | Anti-ICAM-1 antibody | ICAM-1 antagonist | Prevents glomerular mononuclear cell infiltration. | [84] |
| Yorkshire pigs + STZ (50 mg/kg) | Anti-αVβ3 antibody | αVβ3 antagonist | Attenuates proteinuria and renal histological changes. | [96] | |
| ZSF1 rats | MK-0429 | αVβ3 inhibitor | Reduces proteinuria and renal fibrosis | [97] | |
| Chemokines | SD rats + STZ (60 mg/kg) | AMD3100 | CXCR4 inhibitor | Increases albuminuria and accelerated tubular cell death. | [106] |
| db/db mice | NOX-A12 | CXCL12 inhibitor | Decreases glomerulosclerosis and albuminuria. | [119] | |
| db/db mice | Recombinant CXCL10 | Mimetic CXCL10 | Reduces mesangial matrix expansion, albuminuria, and glomerular hypertrophy. | [120] | |
| iNOS-Tg mice | Propagermanium | CCR2 antagonist | Decreases mesangial matrix expansion and macrophage infiltration. | [121] | |
| db/db mice | RS504393 | CCR2 antagonist | Ameliorates inflammation, oxidative stress, and fibrosis. | [123] | |
| db/db mice | CCX140-B | CCR2 antagonist | Reduces albuminuria, glomerular hypertrophy and increases podocyte number. | [124] | |
| Cytokines | Wistar rats+ STZ (40 mg/kg) | Infliximab | TNF-α inhibitor | Decreases albuminuria. | [129] |
| KK-A(y) mice | Etanercept | TNF-α inhibitor | Improves albuminuria, macrophage infiltrate and CAM expression. | [130] | |
| SD rats + STZ (65 mg/kg) | Tocilizumab | IL-6 inhibitor | Decreases albuminuria, oxidative stress, inflammation. | [131] | |
| BKS db/db mice | Anti-IL-1β antibody | IL-1β inhibitor | Improves kidney injury markers and attenuates decline of eGFR. | [132] | |
| db/db, and Akita mice; STZ (150 mg/kg) | Recombinant IL-17A | Mimetic IL-17A | Prevents fibrosis, podocytes loss, tubular atrophy, and albuminuria. | [133] | |
| BTBR ob/ob mice | Anti-IL-17A antibody | IL-17A inhibitor | Ameliorates renal function, macrophage infiltration and podocyte loss. | [134] | |
| Wistar rats + STZ (70 mg kg) | Anti-IL-20 antibody | IL-20 inhibitor | Reduces glomerular area and improves renal functions. | [135] | |
| db/db mice | ISO-1 | MIF inhibitor | Decreases albuminuria, fibrosis and inflammation. | [136] | |
| Wistar rats + STZ (50 mg kg) | p425 | MIF antagonist | Decreases UACR, serum BUN and creatinine. | [137] |
4.3. Cytokines
4.3.1. TNF-α
4.3.2. IL-6
4.3.3. IL-1β
4.3.4. IL-18
4.3.5. IL17A
4.4. Other Cytokines/Proinflammatory Proteins in DN
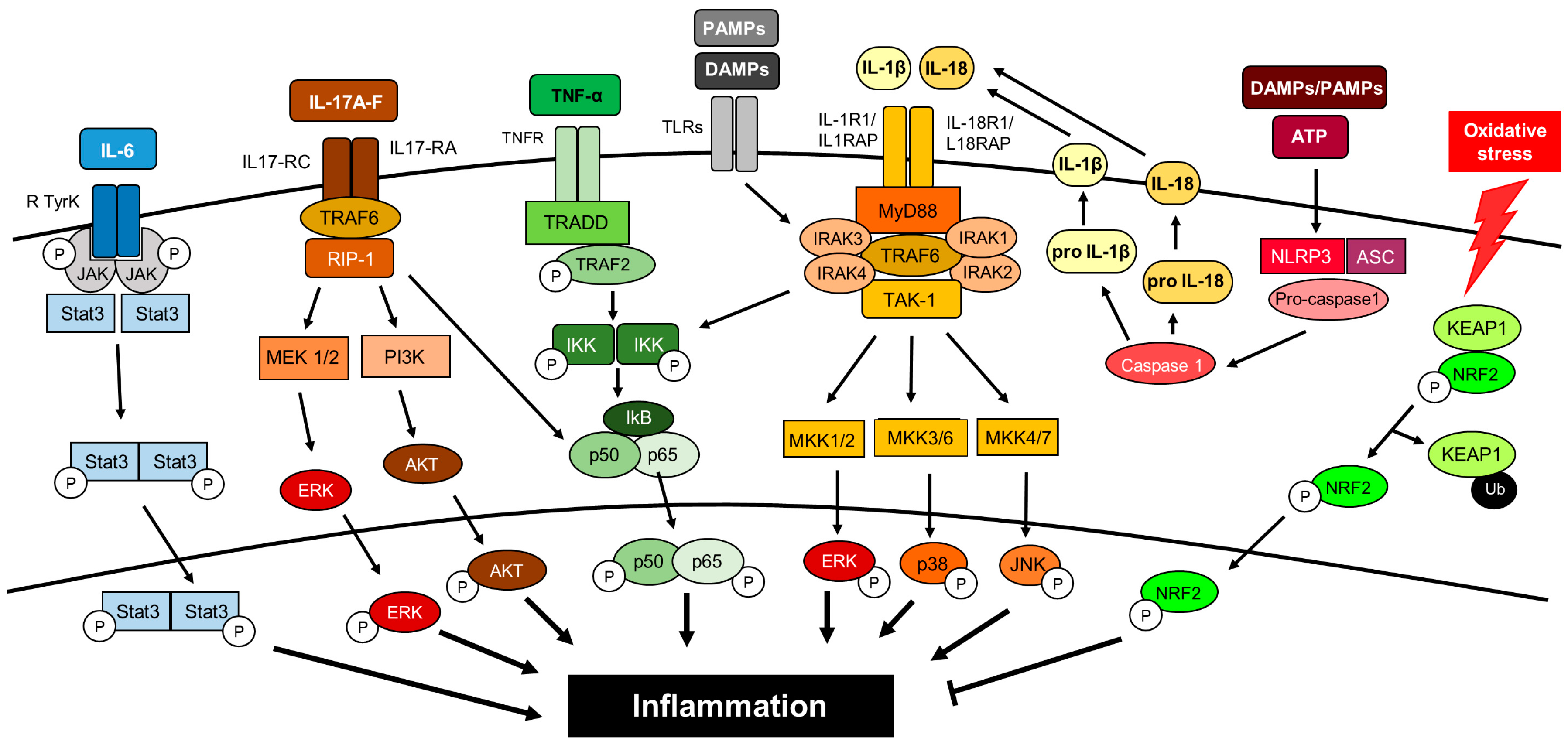
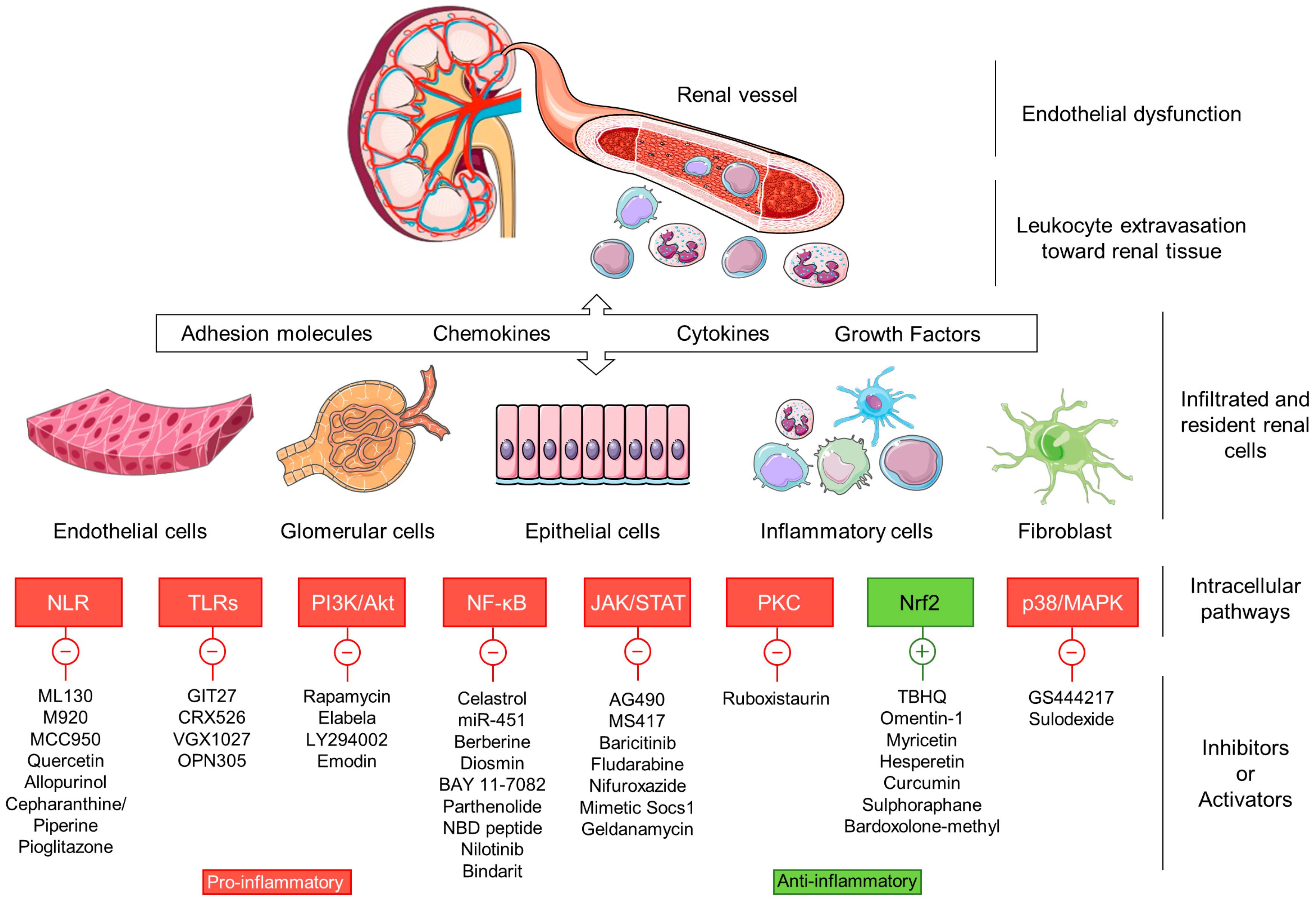
5. Inflammatory Intracellular Signaling Pathways
5.1. Nod-Like Receptors
5.2. Toll-Like Receptors
5.3. PI3K/AKT/mTOR
5.4. Nuclear Factor-κB
5.5. JAK/STAT
5.6. Protein Kinase C
5.7. Nrf2
5.8. p38/MAPK
6. Novel Antidiabetic Drugs with Anti-Inflammatory Actions in DN
7. Perspectives and Conclusions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP binding cassette transporter A1 |
| ACEi | Angiotensin-converting-enzyme inhibitors |
| AGEs | Advanced glycation end-products |
| Akt | Protein kinase B |
| ApoE | Apolipoprotein E |
| AP-1 | Activator protein-1 |
| ARBs | Angiotensin Receptor Blockers |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| BMP-7 | Bone morphogenetic protein 7 |
| BTBR | Black and tan brachyuric |
| BUN | Blood urea nitrogen |
| CASP1 | Caspase-1 |
| CCL11 | Chemokine-CC motif ligand 11 |
| CCL2 | Chemokine-CC motif ligand 2 |
| CCL5 | Chemokine-CC motif ligand 5 |
| CCR2 | C-C chemokine receptor 2 |
| CCR5 | C-C chemokine receptor 5 |
| CD206/MRC1 | Mannose receptor |
| CD4+ | Cluster of differentiation 4+ |
| CD74 | Cluster of differentiation 74 |
| CD8+ | Cluster of differentiation 8+ |
| CKD | Chronic kidney disease |
| Col IV | Collagen IV |
| CVD | Cardiovascular Disease |
| CX3CL1 | Fractalkine |
| CX3CR1 | Fractalkine receptor |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 |
| CXCL10 | Chemokine (C-X-C motif) ligand 10 |
| CXCL12 | Chemokine (C-X-C motif) ligand 12 |
| CXCL16 | Chemokine (C-X-C motif) ligand 16 |
| CXCL5 | Chemokine (C-X-C motif) ligand 5 |
| CXCL8 | Chemokine (C-X-C motif) ligand 8 |
| CXCL9 | Chemokine (C-X-C motif) ligand 9 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| db/db | Diabetic/diabetic |
| DCs | Dendritic cells |
| DN | Diabetic nephropathy |
| DNA | Deoxyribonucleic acid |
| DPP4i | Dipeptidyl peptidase-4 inhibitors |
| eGFR | Estimated glomerular filtration rate |
| EMT | Epithelial to mesenchymal transition |
| EndMT | Endothelial to mesenchymal transition |
| ERK 1/2 | Extracellular Signal-Regulated Kinase 1/2 |
| ESRD | End-stage renal disease |
| FGF1 | Acidic fibroblast growth factor |
| GBM | Glomerular basement membrane |
| GLP-1 | Glucagon-Like Peptide 1 |
| GLP-1 RA | GLP-1 receptor agonists |
| GSK-3β | Glycogen synthase kinase 3β |
| HbA1c | Hemoglobin A1c |
| HK-2 | Human kidney 2 cells |
| HO-1 | Heme Oxygenase 1 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IFN-γ | Interferon-γ |
| IgG | Immunoglobulin G |
| IgM | ImmunoglobulinM |
| IKKα/β/γ | IκB kinase α/β/γ |
| IL-10 | Interleukin-10 |
| IL-17A/F | Interleukin-17A/F |
| IL-18 | Interleukin-18 |
| IL-1β | Interleukin-1β |
| IL-2 | Interleukin-2 |
| IL-20 | Interleukin-20 |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| IP-10 | Interferon-inducible protein 10 |
| JAK | Janus Kinase/ Just Another Kinase |
| KIR | Kinase inhibitory region |
| KRIS | Risk inflammatory signature |
| LDL | Low Density Lipoprotein |
| Ly6C | Lymphocyte antigen 6C |
| mAb | Monoclonal Antibody |
| MAP3K5 | Mitogen-activated protein kinase 5 |
| MAPK | Mitogen-Activated Protein Kinase |
| MDA | Malondialdehyde |
| MEK | Mitogen-activated protein kinase kinase |
| MIF | Macrophage migration inhibitory factor |
| MIP-1 | Macrophage Inflammatory Protein-1 |
| MMP | Matrix metalloproteinase |
| mTOR | Mammalian target of rapamycin |
| NADPH | Nicotinamide adenine dinucleotide phosphate hydrogen |
| NBD | Nucleotide-binding domain |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLR | Nucleotide-binding oligomerization domain (NOD)-like receptors |
| NLRP | Pyrin domain-containing protein |
| NOD1 | Nucleotide-binding oligomerization domain-containing protein 1 |
| NOD2 | Nucleotide-binding oligomerization domain-containing protein 2 |
| NOX-4 | NADPH oxidase 4 |
| Nrf2 | Nuclear factor (erythroid-derived 2)-like 2 |
| Ob | obese |
| P38/MAPK | p38 mitogen-activated protein kinase |
| p53 | Cellular tumor antigen p53 |
| PDGF | Platelet derived growth factor |
| PI3K | Phosphatidylinositol 3-kinase |
| PKC | Protein kinase C |
| PPAR-γ | Peroxisome proliferator-activated receptor-γ |
| PTEN | Phosphatase and tensin homolog |
| PTX3 | Pentraxin 3 |
| RAAS | Renin–-angiotensin–-aldosterone system |
| ROS | Reactive Oxygen Species |
| SGLT2 | Sodium-glucose cotransporter-2 |
| SGLT2i | SGLT2 inhibitors |
| sICAM-1 | Soluble intercellular adhesion molecule-1 |
| Smad | Mothers against decapentaplegic homolog |
| SOCS | Suppressors of cytokine signaling |
| SREBP-1 | Sterol regulatory element binding transcription factor 1 |
| STAT | Signal Transducer and Activator of Transcription |
| sTWEAK | Soluble TNF-related weak inducer of apoptosis |
| STZ | Streptozotocin |
| T1DM | Type 1 and type 2 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TBHQ | Tert-butylhydroquinone |
| TGF-β | Transforming growth factor-β |
| Th1 | T helper cell type 1 |
| Th17 | T helper cell type 17 |
| Th2 | T helper cell type 2 |
| TLRs | Toll-like receptors |
| TNF-α | Tumor necrosis factor-α |
| TNFR1 | Tumor necrosis factor receptor 1 |
| TNFR2 | Tumor necrosis factor receptor 2 |
| Tregs | Regulatory T cells |
| TYK2 | Tyrosine kinase 2 |
| UACR | Urine albumin to creatinine ratio |
| VAP-1 | Vascular adhesion protein-1 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| ZSF1 | Zucker Diabetic Fatty 1 |
| α-SMA | α-smooth muscle actin |
References
- Bell, S.; Fletcher, E.H.; Brady, I.; Looker, H.C.; Levin, D.; Joss, N.; Traynor, J.P.; Metcalfe, W.; Conway, B.; Livingstone, S.; et al. End-stage renal disease and survival in people with diabetes: A national database linkage study. QJM 2015, 108, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Porrini, E.; Ruggenenti, P.; Mogensen, C.E.; Barlovic, D.P.; Praga, M.; Cruzado, J.M.; Hojs, R.; Abbate, M.; de Vries, A.P.J. ERA-EDTA diabesity working group Non-proteinuric pathways in loss of renal function in patients with type 2 diabetes. Lancet Diabetes Endocrinol. 2015, 3, 382–391. [Google Scholar] [CrossRef]
- Anders, H.-J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361. [Google Scholar] [CrossRef] [PubMed]
- National Kidney Foundation. KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 Update. Am. J. Kidney Dis. 2012, 60, 850–886. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.F.; Emanuele, N.; Zhang, J.H.; Brophy, M.; Conner, T.A.; Duckworth, W.; Leehey, D.J.; McCullough, P.A.; O’Connor, T.; Palevsky, P.M.; et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N. Engl. J. Med. 2013, 369, 1892–1903. [Google Scholar] [CrossRef] [Green Version]
- Goldszmid, R.S.; Trinchieri, G. The price of immunity. Nat. Immunol. 2012, 13, 932–938. [Google Scholar] [CrossRef]
- Schena, F.P.; Gesualdo, L. Pathogenetic mechanisms of diabetic nephropathy. J. Am. Soc. Nephrol. 2005, 16, S30–S33. [Google Scholar] [CrossRef]
- Wada, J.; Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 2016, 12, 13. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Md Dom, Z.I.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.-J.; et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Mora-Fernández, C. The role of inflammatory cytokines in diabetic nephropathy. J. Am. Soc. Nephrol. 2008, 19, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Pichler, R.; Afkarian, M.; Dieter, B.P.; Tuttle, K.R. Immunity and inflammation in diabetic kidney disease: Translating mechanisms to biomarkers and treatment targets. Am. J. Physiol. Ren. Physiol. 2017, 312, F716–F731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Prim. 2015, 1, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Berthier, C.C.; Zhang, H.; Schin, M.; Henger, A.; Nelson, R.G.; Yee, B.; Boucherot, A.; Neusser, M.A.; Cohen, C.D.; Carter-Su, C.; et al. Enhanced expression of janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009, 58, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkey, J.M.; Haidacher, S.J.; LeJeune, W.S.; Zhang, X.; Tieu, B.C.; Choudhary, S.; Brasier, A.R.; Denner, L.A.; Tilton, R.G. Diabetes-induced activation of canonical and noncanonical nuclear factor-κB pathways in renal cortex. Diabetes 2006, 55, 1252–1259. [Google Scholar] [CrossRef] [Green Version]
- Rabelink, T.J.; De Zeeuw, D. The glycocalyx-Linking albuminuria with renal and cardiovascular disease. Nat. Rev. Nephrol. 2015, 11, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Agere, S.A.; Kim, E.Y.; Akhtar, N.; Ahmed, S. Syndecans in chronic inflammatory and autoimmune diseases: Pathological insights and therapeutic opportunities. J. Cell. Physiol. 2018, 233, 6346–6358. [Google Scholar] [CrossRef]
- Moreno, J.A.; Moreno, S.; Rubio-Navarro, A.; Gómez-Guerrero, C.; Ortiz, A.; Egido, J. Role of chemokines in proteinuric kidney disorders. Expert Rev. Mol. Med. 2014, 16, e3. [Google Scholar] [CrossRef]
- Langer, H.F.; Chavakis, T. Leukocyte-endothelial interactions in inflammation. J. Cell. Mol. Med. 2009, 13, 1211–1220. [Google Scholar] [CrossRef]
- Luis-Rodríguez, D.; Martínez-Castelao, A.; Górriz, J.L.; De-Álvaro, F.; Navarro-González, J.F. Pathophysiological role and therapeutic implications of inflammation in diabetic nephropathy. World J. Diabetes 2012, 3, 7–18. [Google Scholar] [CrossRef]
- Cachofeiro, V.; Goicochea, M.; De Vinuesa, S.G.; Oubĩa, P.; Lahera, V.; Lũo, J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int. 2008, 74, S4–S9. [Google Scholar] [CrossRef] [Green Version]
- Sagoo, M.K.; Gnudi, L. Diabetic nephropathy: Is there a role for oxidative stress? Free Radic. Biol. Med. 2018, 116, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Gomez-Guerrero, C.; Mas, S.; Sanz, A.B.; Lorenzo, O.; Ruiz-Ortega, M.; Opazo, L.; Mezzano, S.; Egido, J. Targeting inflammation in diabetic nephropathy: A tale of hope. Expert Opin. Investig. Drugs 2018, 27, 917–930. [Google Scholar] [CrossRef]
- Elmarakby, A.A.; Sullivan, J.C. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc. Ther. 2012, 30, 49–59. [Google Scholar] [CrossRef]
- Kuhad, A.; Chopra, K. Attenuation of diabetic nephropathy by tocotrienol: Involvement of NFkB signaling pathway. Life Sci. 2009, 84, 296–301. [Google Scholar] [CrossRef]
- Ebenezer, P.J.; Mariappan, N.; Elks, C.M.; Haque, M.; Francis, J. Diet-induced renal changes in zucker rats are ameliorated by the superoxide dismutase mimetic TEMPOL. Obesity 2009, 17, 1994–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef] [Green Version]
- Onozato, M.L.; Tojo, A.; Goto, A.; Fujita, T. Radical scavenging effect of gliclazide in diabetic rats fed with a high cholesterol diet. Kidney Int. 2004, 65, 951–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, L.; Yu, Y.; Wang, Y.; Niu, N. The protective effects of taurine against early renal injury in STZ-induced diabetic rats, correlated with inhibition of renal LOX-1-mediated ICAM-1 expression. Ren. Fail. 2008, 30, 763–771. [Google Scholar] [CrossRef]
- Arnalich, F.; Hernanz, A.; Lopez-Maderuelo, D.; Pena, J.M.; Camacho, J.; Madero, R.; Vazquez, J.J.; Montiel, C. Enhanced acute-phase response and oxidative stress in older adults with type II diabetes. Horm. Metab. Res. 2000, 32, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Cipollone, F.; Chiarelli, F.; Iezzi, A.; Fazia, M.L.; Cuccurullo, C.; Pini, B.; De Cesare, D.; Torello, M.; Tumini, S.; Cuccurullo, F.; et al. Relationship between reduced BCL-2 expression in circulating mononuclear cells and early nephropathy in type 1 diabetes. Int. J. Immunopathol. Pharmacol. 2005, 18, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarelli, F.; Cipollone, F.; Mohn, A.; Marini, M.; Iezzi, A.; Fazia, M.; Tumini, S.; De Cesare, D.; Pomilio, M.; Pierdomenico, S.D.; et al. Circulating monocyte chemoattractant protein-1 and early development of nephropathy in type 1 diabetes. Diabetes Care 2002, 25, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, H.; Boucherot, A.; Yasuda, Y.; Henger, A.; Brunner, B.; Eichinger, F.; Nitsche, A.; Kiss, E.; Bleich, M.; Gröne, H.J.; et al. Modular activation of nuclear factor-κB transcriptional programs in human diabetic nephropathy. Diabetes 2006, 55, 2993–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-García, P.M. Inflammation in diabetic kidney disease. World J. Diabetes 2014, 5, 431. [Google Scholar] [CrossRef]
- Meyer, M.; Schreck, R.; Baeuerle, P.A. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993, 12, 2005–2015. [Google Scholar] [CrossRef]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.; Ping, F.; Mu, W.; Hill, P.; Atkins, R.C.; Chadban, S.J. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology 2006, 11, 226–231. [Google Scholar] [CrossRef]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Rollin, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006, 69, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ma, F.Y.; Ozols, E.; Rollins, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia 2007, 50, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Okada, S.; Shikata, K.; Matsuda, M.; Ogawa, D.; Usui, H.; Kido, Y.; Nagase, R.; Wada, J.; Shikata, Y.; Makino, H. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes 2003, 52, 2586–2593. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Gao, T.; Cooper, T.K.; Brian Reeves, W.; Awad, A.S. Macrophages directly mediate diabetic renal injury. Am. J. Physiol. Renal Physiol. 2013, 305, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Ikezumi, Y.; Hurst, L.A.; Masaki, T.; Atkins, R.C.; Nikolic-Paterson, D.J. Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int. 2003, 63, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawluczyk, I.Z.; Harris, K.P. Macrophages Promote Prosclerotic Responses in Cultured Rat Mesangial Cells: A Mechanism for the Initiation of Glomerulosclerosis. J. Am. Soc. Nephrol. 1997, 8, 1525–1536. [Google Scholar] [PubMed]
- Cui, X.; Kushiyama, A.; Yoneda, M.; Nakatsu, Y.; Guo, Y.; Zhang, J.; Ono, H.; Kanna, M.; Sakoda, H.; Ono, H.; et al. Macrophage foam cell formation is augmented in serum from patients with diabetic angiopathy. Diabetes Res. Clin. Pract. 2010, 87, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Sano, J.; Shirakura, S.; Oda, S.; Hara, T.; Ishihara, T. Foam cells generated by a combination of hyperglycemia and hyperlipemia in rats. Pathol. Int. 2004, 54, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fu, S.; Fan, X.; Lotze, M.T.; Zeh, H.J.; Tang, D. Nuclear DAMP complex-mediated RAGE-dependent macrophage cell death. Biochem. Biophys. Res. Commun. 2015, 458, 650–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, M.; Kerry, R.; Aviram, M.; Hayek, T. High glucose concentration increases macrophage cholesterol biosynthesis in diabetes through activation of the sterol regulatory element binding protein 1 (SREBP1): Inhibitory effect of insulin. J. Cardiovasc. Pharmacol. 2008, 52, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zheng, F. Immune Cells and Inflammation in Diabetic Nephropathy. J. Diabetes Res. 2016, 1841690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yao, B.; Wang, Y.; Fan, X.; Wang, S.; Niu, A.; Yang, H.; Fogo, A.; Zhang, M.-Z.; Harris, R.C. Macrophage Cyclooxygenase-2 Protects Against Development of Diabetic Nephropathy. Diabetes 2017, 66, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef] [Green Version]
- Gobert, A.P.; Verriere, T.; Asim, M.; Barry, D.P.; Piazuelo, M.B.; Sablet, T.; Delgado, A.G.; Bravo, L.E.; Correa, P.; Peek, R.M., Jr.; et al. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to Helicobacter pylori. J. Immunol. 2014, 193, 3013–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, N.M.; Ferenbach, D.A.; Isenberg, J.S.; Thomson, A.W.; Hughes, J. Dendritic cells and macrophages in the kidney: A spectrum of good and evil. Nat. Publ. Gr. 2014, 10, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Weisheit, C.K.; Engel, D.R.; Kurts, C. Dendritic cells and macrophages: Sentinels in the kidney. Clin. J. Am. Soc. Nephrol. 2015, 10, 1841–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu Manh, T.-P.; Bertho, N.; Hosmalin, A.; Schwartz-Cornil, I.; Dalod, M. Investigating Evolutionary Conservation of Dendritic Cell Subset Identity and Functions. Front. Immunol. 2015, 6, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Wang, C.; Wen, X.; Chen, Y.; Mao, R.; Cui, D.; Li, L.; Liu, J.; Chen, Y.; Cheng, J.; et al. Mesenchymal stem cells alleviate rat diabetic nephropathy by suppressing CD103+ DCs-mediated CD8+ T cell responses. J. Cell. Mol. Med. 2020, 24, 5817–5831. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Chen, T.; Wang, C.; Zhang, Z.; Wang, X.M.; Li, Q.; Lee, V.W.S.; Wang, Y.M.; Zheng, G.; Alexander, S.I.; et al. Flt3 inhibition alleviates chronic kidney disease by suppressing CD103+ dendritic cell-mediated T cell activation. Nephrol. Dial. Transplant. 2018, 34, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-Y.; Jeong, K.-H.; Lee, T.-W.; Ihm, C.-G.; Lim, S.J.; Lee, S.-H. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am. J. Nephrol. 2012, 35, 164–174. [Google Scholar] [CrossRef]
- Peng, X.; Xiao, Z.; Zhang, J.; Li, Y.; Dong, Y.; Du, J. IL-17A produced by both γδ T and Th17 cells promotes renal fibrosis via RANTES-mediated leukocyte infiltration after renal obstruction. J. Pathol. 2015, 235, 79–89. [Google Scholar] [CrossRef]
- Lim, A.K.H.; Ma, F.Y.; Nikolic-Paterson, D.J.; Kitching, A.R.; Thomas, M.C.; Tesch, G.H. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia 2010, 53, 1772–1782. [Google Scholar] [CrossRef] [Green Version]
- Herrera, M.; Soderberg, M.; Sabirsh, A.; Valastro, B.; Molne, J.; Santamaria, B.; Valverde, A.M.; Guionaud, S.; Heasman, S.; Bigley, A.; et al. Inhibition of T-cell activation by the CTLA4-Fc Abatacept is sufficient to ameliorate proteinuric kidney disease. Am. J. Physiol. Renal Physiol. 2017, 312, F748–F759. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-M.; Lee, S.-H.; Lee, A.; Kim, D.-J.; Kim, Y.-G.; Kim, S.-Y.; Jeong, K.-H.; Lee, T.-W.; Ihm, C.-G.; Lim, S.-J.; et al. Targeting T helper 17 by mycophenolate mofetil attenuates diabetic nephropathy progression. Transl. Res. 2015, 166, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Eller, K.; Kirsch, A.; Wolf, A.M.; Sopper, S.; Tagwerker, A.; Stanzl, U.; Wolf, D.; Patsch, W.; Rosenkranz, A.R.; Eller, P. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 2011, 60, 2954–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Virella, M.F.; Carter, R.E.; Baker, N.L.; Lachin, J.; Virella, G. High levels of oxidized LDL in circulating immune complexes are associated with increased odds of developing abnormal albuminuria in Type 1 diabetes. Nephrol. Dial. Transplant. 2012, 27, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Virella, M.F.; Hunt, K.J.; Baker, N.L.; Virella, G.; VADT Group of Investigators. High levels of AGE-LDL, and of IgG antibodies reacting with MDA-lysine epitopes expressed by oxLDL and MDA-LDL in circulating immune complexes predict macroalbuminuria in patients with type 2 diabetes. J. Diabetes Complications 2016, 30, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Bergtold, A.; Gavhane, A.; D’Agati, V.; Madaio, M.; Clynes, R. FcR-bearing myeloid cells are responsible for triggering murine lupus nephritis. J. Immunol. 2006, 177, 7287–7295. [Google Scholar] [CrossRef] [Green Version]
- Ding, A.; Wright, S.D.; Nathan, C. Activation of mouse peritoneal macrophages by monoclonal antibodies to MAC-1 (complement receptor type 3). J. Exp. Med. 1987, 165, 733–749. [Google Scholar] [CrossRef] [Green Version]
- Mise, K.; Hoshino, J.; Ueno, T.; Sumida, K.; Hiramatsu, R.; Hasegawa, E.; Yamanouchi, M.; Hayami, N.; Suwabe, T.; Sawa, N.; et al. Clinical implications of linear immunofluorescent staining for immunoglobulin G in patients with diabetic nephropathy. Diabetes Res. Clin. Pract. 2014, 106, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Parra, V.; Mallavia, B.; Lopez-Franco, O.; Ortiz-Muñoz, G.; Oguiza, A.; Recio, C.; Blanco, J.; Nimmerjahn, F.; Egido, J.; Gomez-Guerrero, C. Fcγ receptor deficiency attenuates diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Okoń, K.; Stachura, J. Increased mast cell density in renal interstitium is correlated with relative interstitial volume, serum creatinine and urea especially in diabetic nephropathy but also in primary glomerulonephritis. Pol. J. Pathol. 2007, 58, 193–197. [Google Scholar]
- Zheng, J.M.; Yao, G.H.; Cheng, Z.; Wang, R.; Liu, Z.H. Pathogenic role of mast cells in the development of diabetic nephropathy: A study of patients at different stages of the disease. Diabetologia 2012, 55, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.-D.; Luo, J.-H.; Zhao, Z.-Y.; Liao, Y.-J.; Li, Y. Tranilast prevents renal interstitial fibrosis by blocking mast cell infiltration in a rat model of diabetic kidney disease. Mol. Med. Rep. 2018, 17, 7356–7364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, B.J.; Takai, S.; Seth, D.M.; Satou, R.; Harrison-Bernard, L.M. Chymase inhibition retards albuminuria in type 2 diabetes. Physiol. Rep. 2019, 7, e14302. [Google Scholar] [CrossRef]
- Takahashi, T.; Hato, F.; Yamane, T.; Inaba, M.; Okuno, Y.; Nishizawa, Y.; Kitagawa, S. Increased spontaneous adherence of neutrophils from type 2 diabetic patients with overt proteinuria: Possible role of the progression of diabetic nephropathy. Diabetes Care 2000, 23, 417–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, A.; Tomino, Y.; Yokoyama, K.; Koide, H. Production of hydrogen peroxide by neutrophilic polymorphonuclear leukocytes in patients with diabetic nephropathy. J. Clin. Lab. Anal. 1993, 7, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, M.; Harada, K.; Kitoh, Y.; Nagasaka, S.I.; Saito, T.; Fujimura, A. The functions of circulatory polymorphonuclear leukocytes in diabetic patients with and without diabetic triopathy. Life Sci. 2000, 66, 1861–1870. [Google Scholar] [CrossRef]
- Kawamoto, R.; Ninomiya, D.; Kikuchi, A.; Akase, T.; Kasai, Y.; Kusunoki, T.; Ohtsuka, N.; Kumagi, T. Association of neutrophil-to-lymphocyte ratio with early renal dysfunction and albuminuria among diabetic patients. Int. Urol. Nephrol. 2019, 51, 483–490. [Google Scholar] [CrossRef]
- Shikata, K.; Makino, H. Microinflammation in the pathogenesis of diabetic nephropathy. J. Diabetes Investig. 2013, 4, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Sucosky, P.; Balachandran, K.; Elhammali, A.; Jo, H.; Yoganathan, A.P. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-β1-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Sumagin, R.; Sarelius, I.H. TNF-α activation of arterioles and venules alters distribution and levels of ICAM-1 and affects leukocyte-endothelial cell interactions. Am. J. Physiol. Hear. Circ. Physiol. 2006, 291, H2116–H2125. [Google Scholar] [CrossRef]
- Coimbra, T.M.; Janssen, U.; Gröne, H.J.; Ostendorf, T.; Kunter, U.; Schmidt, H.; Brabant, G.; Floege, J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000, 57, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Karimi, Z.; Kahe, F.; Jamil, A.; Marszalek, J.; Ghanbari, A.; Afarideh, M.; Khajeh, E.; Noshad, S.; Esteghamati, A.; Chi, G. Intercellular adhesion molecule-1 in diabetic patients with and without microalbuminuria. Diabetes Metab. Syndr. Clin. Res. Rev. 2018, 12, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Glynn, R.J.; Rifai, N.; Manson, J.E.; Ridker, P.M.; Nathan, D.M.; Schaumberg, D.A. Inflammation and progressive nephropathy in type 1 diabetes in the diabetes control and complications trial. Diabetes Care 2008, 31, 2338–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; He, D.; Fang, L.; Yan, X. Association between E469K polymorphism in the ICAM1 gene and the risk of diabetic nephropathy: A meta-analysis. Lipids Health Dis. 2018, 17, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, H.; Shikata, K.; Hirata, K.; Akiyama, K.; Matsuda, M.; Kushiro, M.; Shikata, Y.; Miyatake, N.; Miyasaka, M.; Makino, H. Increased expression of intercellular adhesion molecule-1 (ICAM-1) in diabetic rat glomeruli: Glomerular hyperfiltration is a potential mechanism of ICAM-1 upregulation. Diabetes 1997, 46, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Guerra, A.F.; Vargas-Robles, H.; Lozano Nuevo, J.J.; Escalante-Acosta, B.A. Correlation between circulating adhesion molecule levels and albuminuria in type-2 diabetic hypertensive patients. Kidney Blood Press. Res. 2009, 32, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Stehouwer, C.D.A.; Gall, M.-A.; Twisk, J.W.R.; Knudsen, E.; Emeis, J.J.; Parving, H.-H. Increased Urinary Albumin Excretion, Endothelial Dysfunction, and Chronic Low-Grade Inflammation in Type 2 Diabetes. Diabetes 2002, 51, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmi, M.; Jalkanen, S. Vascular adhesion protein-1: A cell surface amine oxidase in translation. Antioxidants Redox Signal. 2019, 30, 314–332. [Google Scholar] [CrossRef]
- Li, H.Y.; Wei, J.N.; Lin, M.S.; Smith, D.J.; Vainio, J.; Lin, C.H.; Chiang, F.T.; Shih, S.R.; Huang, C.H.; Wu, M.Y.; et al. Serum vascular adhesion protein-1 is increased in acute and chronic hyperglycemia. Clin. Chim. Acta 2009, 404, 149–153. [Google Scholar] [CrossRef]
- Li, H.Y.; Lin, H.A.; Nien, F.J.; Wu, V.C.; Jiang, Y.D.; Chang, T.J.; Kao, H.L.; Lin, M.S.; Wei, J.N.; Lin, C.H.; et al. Serum Vascular Adhesion Protein-1 Predicts End-Stage Renal Disease in Patients with Type 2 Diabetes. PLoS ONE 2016, 11, e0147981. [Google Scholar] [CrossRef]
- Li, H.Y.; Jiang, Y.D.; Chang, T.J.; Wei, J.N.; Lin, M.S.; Lin, C.H.; Chiang, F.T.; Shih, S.R.; Hung, C.S.; Hua, C.H.; et al. Serum vascular adhesion protein-1 predicts 10-year cardiovascular and cancer mortality in individuals with type 2 diabetes. Diabetes 2011, 60, 993–999. [Google Scholar] [CrossRef] [Green Version]
- De Zeeuw, D.; Renfurm, R.W.; Bakris, G.; Rossing, P.; Perkovic, V.; Hou, F.F.; Nangaku, M.; Sharma, K.; Heerspink, H.J.L.; Garcia-Hernandez, A.; et al. Efficacy of a novel inhibitor of vascular adhesion protein-1 in reducing albuminuria in patients with diabetic kidney disease (ALBUM): A randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2018, 6, 925–933. [Google Scholar] [CrossRef]
- Pugliese, G.; Iacobini, C.; Pesce, C.M.; Menini, S. Galectin-3: An emerging all-out player in metabolic disorders and their complications. Glycobiology 2015, 25, 136–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drechsler, C.; Delgado, G.; Wanner, C.; Blouin, K.; Pilz, S.; Tomaschitz, A.; Kleber, M.E.; Dressel, A.; Willmes, C.; Krane, V.; et al. Galectin-3, renal function, and clinical outcomes: Results from the luric and 4D studies. J. Am. Soc. Nephrol. 2015, 26, 2213–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, A.; Zent, R. Integrins in kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Gingras, D.; Bendayan, M. Alterations of vitronectin and its receptor αv integrin in the rat renal glomerular wall during diabetes. Am. J. Kidney Dis. 2001, 38, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Maile, L.A.; Busby, W.H.; Gollahon, K.A.; Flowers, W.; Garbacik, N.; Garbacik, S.; Stewart, K.; Nichols, T.; Bellinger, D.; Patel, A.; et al. Blocking ligand occupancy of the αVβ3 integrin inhibits the development of nephropathy in diabetic pigs. Endocrinology 2014, 155, 4665–4675. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhang, J.; Haimbach, R.; Zhu, W.; Mayer-Ezell, R.; Garcia-Calvo, M.; Smith, E.; Price, O.; Kan, Y.; Zycband, E.; et al. An integrin antagonist (MK-0429) decreases proteinuria and renal fibrosis in the ZSF1 rat diabetic nephropathy model. Pharmacol. Res. Perspect. 2017, 5, e00354. [Google Scholar] [CrossRef] [Green Version]
- Hirata, K.; Shikata, K.; Matsuda, M.; Akiyama, K.; Sugimoto, H.; Kushiro, M.; Makino, H. Increased expression of selectins in kidneys of patients with diabetic nephropathy. Diabetologia 1998, 41, 185–192. [Google Scholar] [CrossRef]
- Narumi, S.; Onozato, M.L.; Tojo, A.; Sakamoto, S.; Tamatani, T. Tissue-specific induction of E-selectin in glomeruli is augmented following diabetes mellitus. Nephron 2001, 89, 161–171. [Google Scholar] [CrossRef]
- Soedamah-Muthu, S.S.; Chaturvedi, N.; Schalkwijk, C.G.; Stehouwer, C.D.A.; Ebeling, P.; Fuller, J.H.; EURODIAB Prospective Complications Study Group. Soluble vascular cell adhesion molecule-1 and soluble E-selectin are associated with micro- and macrovascular complications in Type 1 diabetic patients. J. Diabetes Complications 2006, 20, 188–195. [Google Scholar] [CrossRef]
- Wang, F.; Xing, T.; Wang, N.; Liu, L. Clinical significance of plasma CD146 and P-selectin in patients with type 2 diabetic nephropathy. Cytokine 2012, 57, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Utsumi, K.; Yasuda, F.; Watanabe, Y.; Higo, S.; Hirama, A.; Fujita, E.; Ueda, K.; Mii, A.; Kaneko, T.; Mishina, M.; et al. Effects of olmesartan and imidapril on the plasma adiponectin, P-selectin, and MDA-LDL levels of diabetic nephropathy patients. Clin. Chim. Acta 2012, 413, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.C.K.; Lan, H.Y. Chemokines in renal injury. J. Am. Soc. Nephrol. 2011, 22, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzano, S.; Aros, C.; Droguett, A.; Burgos, M.E.; Ardiles, L.; Flores, C.; Schneider, H.; Ruiz-Ortega, M.; Egido, J. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol. Dial. Transplant. 2004, 19, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Ruster, C.; Wolf, G. The role of chemokines and chemokine receptors in diabetic nephropathy. Front. Biosci. 2008, 13, 944–955. [Google Scholar] [CrossRef] [Green Version]
- Siddiqi, F.S.; Chen, L.-H.; Advani, S.L.; Thai, K.; Batchu, S.N.; Alghamdi, T.A.; White, K.E.; Sood, M.M.; Gibson, I.W.; Connelly, K.A.; et al. CXCR4 promotes renal tubular cell survival in male diabetic rats: Implications for ligand inactivation in the human kidney. Endocrinology 2015, 156, 1121–1132. [Google Scholar] [CrossRef]
- Verhave, J.C.; Bouchard, J.; Goupil, R.; Pichette, V.; Brachemi, S.; Madore, F.; Troyanov, S. Clinical value of inflammatory urinary biomarkers in overt diabetic nephropathy: A prospective study. Diabetes Res. Clin. Pract. 2013, 101, 333–340. [Google Scholar] [CrossRef]
- Higurashi, M.; Ohya, Y.; Joh, K.; Muraguchi, M.; Nishimura, M.; Terawaki, H.; Yagui, K.; Hashimoto, N.; Saito, Y.; Yamada, K. Increased urinary levels of CXCL5, CXCL8 and CXCL9 in patients with Type 2 diabetic nephropathy. J. Diabetes Complications 2009, 23, 178–184. [Google Scholar] [CrossRef]
- Wang, G.; Lai, F.M.-M.; Chow, K.-M.; Kwan, B.C.-H.; Pang, W.-F.; Luk, C.C.-W.; Leung, C.-B.; Li, P.K.-T.; Szeto, C.-C. Urinary mRNA levels of ELR-negative CXC chemokine ligand and extracellular matrix in diabetic nephropathy. Diabetes Metab. Res. Rev. 2015, 31, 699–706. [Google Scholar] [CrossRef]
- Uematsu, M.; Nakamura, T.; Yoshizaki, T.; Watanabe, Y.; Deyama, J.; Watanabe, K.; Kobayashi, T.; Fujioka, D.; Saito, Y.; Nakamura, K.; et al. High levels of stromal cell-derived factor-1α predict short-term progression of renal dysfunction in patients with coronary artery disease. Clin. Exp. Nephrol. 2019, 23, 920–927. [Google Scholar] [CrossRef]
- Ihm, C.G.; Park, J.K.; Hong, S.P.; Lee, T.W.; Cho, B.S.; Kim, M.J.; Cha, D.R.; Ha, H. A high glucose concentration stimulates the expression of monocyte chemotactic peptide 1 in human mesangial cells. Nephron 1998, 79, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Cheng, K.; Ming, Y. CX3CL1/CX3CR1 Axis, as the Therapeutic Potential in Renal Diseases: Friend or Foe? Curr. Gene Ther. 2018, 17, 442–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, Y.; Ikee, R.; Hemmi, N.; Hyodo, N.; Saigusa, T.; Namikoshi, T.; Yamada, M.; Suzuki, S.; Miura, S. Fractalkine and its receptor, CX3CR1, upregulation in streptozotocin-induced diabetic kidneys. Nephron. Exp. Nephrol. 2004, 97, e17–e25. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Song, K.H.; Ha, H. Fractalkine increases mesangial cell proliferation through reactive oxygen species and mitogen-activated protein kinases. Transplant. Proc. 2012, 44, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Song, K.H.; Park, J.; Park, J.H.; Natarajan, R.; Ha, H. Fractalkine and its receptor mediate extracellular matrix accumulation in diabetic nephropathy in mice. Diabetologia 2013, 56, 1661–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.-X.; Zhao, Y.-F.; Qiao, H.-X.; Zhang, Y.-P.; Li, X.-J.; Ren, W.-D.; Yu, P. CXCR3 knockdown protects against high glucose-induced podocyte apoptosis and inflammatory cytokine production at the onset of diabetic nephropathy. Int. J. Clin. Exp. Pathol. 2017, 10, 8829–8838. [Google Scholar]
- Wang, Y.; Wei, Q.; Liu, Q.; Li, Z.; Zhou, L.; Zou, F.; Yuan, Y.; Sun, Z. Crosstalk between monocytes and renal mesangial cells via interaction of metalloproteinases and fractalkine in diabetic nephropathy. Mol. Med. Rep. 2013, 8, 1817–1823. [Google Scholar] [CrossRef]
- Gómez-Garre, D.; Largo, R.; Tejera, N.; Fortes, J.; Manzarbeitia, F.; Egido, J. Activation of NF-kappaB in tubular epithelial cells of rats with intense proteinuria: Role of angiotensin II and endothelin-1. Hypertension 2001, 37, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Sayyed, S.G.; Hägele, H.; Kulkarni, O.P.; Endlich, K.; Segerer, S.; Eulberg, D.; Klussmann, S.; Anders, H.-J. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 2009, 52, 2445–2454. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Thai, K.; Kepecs, D.M.; Winer, D.; Gilbert, R.E. Reversing CXCL10 Deficiency Ameliorates Kidney Disease in Diabetic Mice. Am. J. Pathol. 2018, 188, 2763–2773. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, H.; Matsubara, T.; Mima, A.; Sumi, E.; Nagai, K.; Takahashi, T.; Abe, H.; Iehara, N.; Fukatsu, A.; Okamoto, H.; et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem. Biophys. Res. Commun. 2007, 360, 772–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.Y.; Chung, C.H.; Khoury, C.C.; Yeo, T.K.; Pyagay, P.E.; Wang, A.; Chen, S. The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-beta, increases podocyte motility and albumin permeability. Am. J. Physiol. Renal Physiol. 2009, 297, F85–F94. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.S.; Lee, M.H.; Song, H.K.; Ko, G.J.; Kwon, O.S.; Lim, T.K.; Kim, S.H.; Han, S.Y.; Han, K.H.; Lee, J.E.; et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int. 2010, 78, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, T.; Miao, Z.; Dairaghi, D.J.; Krasinski, A.; Wang, Y.; Zhao, B.N.; Baumgart, T.; Ertl, L.S.; Pennell, A.; Seitz, L.; et al. CCR2 antagonist CCX140-B provides renal and glycemic benefits in diabetic transgenic human CCR2 knockin mice. Am. J. Physiol. Renal Physiol. 2013, 305, F1288–F1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darisipudi, M.N.; Kulkarni, O.P.; Sayyed, S.G.; Ryu, M.; Migliorini, A.; Sagrinati, C.; Parente, E.; Vater, A.; Eulberg, D.; Klussmann, S.; et al. Dual blockade of the homeostatic chemokine CXCL12 and the proinflammatory chemokine CCL2 has additive protective effects on diabetic kidney disease. Am. J. Pathol. 2011, 179, 116–124. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Bekker, P.; Henkel, E.; Hasslacher, C.; Gouni-Berthold, I.; Mehling, H.; Potarca, A.; Tesar, V.; Heerspink, H.J.L.; Schall, T.J.; et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: A randomised trial. Lancet Diabetes Endocrinol. 2015, 3, 687–696. [Google Scholar] [CrossRef]
- Menne, J.; Eulberg, D.; Beyer, D.; Baumann, M.; Saudek, F.; Valkusz, Z.; Więcek, A.; Haller, H.; Emapticap Study Group. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol. Dial. Transplant. 2017, 32, 307–315. [Google Scholar]
- Gale, J.D.; Gilbert, S.; Blumenthal, S.; Elliott, T.; Pergola, P.E.; Goteti, K.; Scheele, W.; Perros-Huguet, C. Effect of PF-04634817, an Oral CCR2/5 Chemokine Receptor Antagonist, on Albuminuria in Adults with Overt Diabetic Nephropathy. Kidney Int. Rep. 2018, 3, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, Y.; Inokuchi, T.; Yamamoto, A.; Ka, T.; Tsutsumi, Z.; Takahashi, S.; Yamamoto, T. Effect of TNF-alpha inhibition on urinary albumin excretion in experimental diabetic rats. Acta Diabetol. 2007, 44, 215–218. [Google Scholar] [CrossRef]
- Omote, K.; Gohda, T.; Murakoshi, M.; Sasaki, Y.; Kazuno, S.; Fujimura, T.; Ishizaka, M.; Sonoda, Y.; Tomino, Y. Role of the TNF pathway in the progression of diabetic nephropathy in KK-A(y) mice. Am. J. Physiol. Renal Physiol. 2014, 306, F1335–F1347. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahman, A.M.; Al Suleimani, Y.; Shalaby, A.; Ashique, M.; Manoj, P.; Ali, B.H. Effect of tocilizumab, an interleukin-6 inhibitor, on early stage streptozotocin-induced diabetic nephropathy in rats. Naunyn. Schmiedebergs. Arch. Pharmacol. 2019, 392, 1005–1013. [Google Scholar] [CrossRef]
- Lei, Y.; Devarapu, S.K.; Motrapu, M.; Cohen, C.D.; Lindenmeyer, M.T.; Moll, S.; Kumar, S.V.; Anders, H.-J. Interleukin-1β Inhibition for Chronic Kidney Disease in Obese Mice With Type 2 Diabetes. Front. Immunol. 2019, 10, 1223. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.; Jayakumar, C.; Chen, F.; Fulton, D.; Stepp, D.; Gansevoort, R.T.; Ramesh, G. Low-Dose IL-17 Therapy Prevents and Reverses Diabetic Nephropathy, Metabolic Syndrome, and Associated Organ Fibrosis. J. Am. Soc. Nephrol. 2016, 27, 745–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoz, C.; Matus, Y.S.; Orejudo, M.; Carpio, J.D.; Droguett, A.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Interleukin-17A blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int. 2019, 95, 1418–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.H.; Li, H.H.; Sung, J.M.; Chen, W.Y.; Hou, Y.C.; Weng, Y.H.; Lai, W.T.; Wu, C.H.; Chang, M.S. Interleukin-20 targets podocytes and is upregulated in experimental murine diabetic nephropathy. Exp. Mol. Med. 2017, 49, e310. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wei, M.; Wang, M.; Chen, L.; Liu, H.; Ren, Y.; Shi, K.; Jiang, H. Inhibition of Macrophage Migration Inhibitory Factor Reduces Diabetic Nephropathy in Type II Diabetes Mice. Inflammation 2014, 37, 2020–2029. [Google Scholar] [CrossRef] [PubMed]
- Khalilpour, J.; Roshan-Milani, S.; Gharalari, F.H.; Fard, A.A. Macrophage migration inhibitory factor antagonist (p425) ameliorates kidney histopathological and functional changes in diabetic rats. J. Bras. Nefrol. 2019, 41, 315–322. [Google Scholar] [CrossRef]
- Navarro, J.F.; Mora-Fernández, C. The role of TNF-α in diabetic nephropathy: Pathogenic and therapeutic implications. Cytokine Growth Factor Rev. 2006, 17, 441–450. [Google Scholar] [CrossRef]
- Navarro, J.F.; Milena, F.J.; Mora, C.; León, C.; Claverie, F.; Flores, C.; García, J. Tumor necrosis factor-alpha gene expression in diabetic nephropathy: Relationship with urinary albumin excretion and effect of angiotensin-converting enzyme inhibition. Kidney Int. Suppl. 2005, 68, S98–S102. [Google Scholar] [CrossRef] [Green Version]
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef]
- Navarro, J.F.; Mora, C.; Rivero, A.; Gallego, E.; Chahin, J.; Macia, M.; Mendez, M.L.; Garcia, J. Urinary protein excretion and serum tumor necrosis factor in diabetic patients with advanced renal failure: Effects of pentoxifylline administration. Am. J. Kidney Dis. 1999, 33, 458–463. [Google Scholar] [CrossRef]
- Moriwaki, Y.; Yamamoto, T.; Shibutani, Y.; Aoki, E.; Tsutsumi, Z.; Takahashi, S.; Okamura, H.; Koga, M.; Fukuchi, M.; Hada, T. Elevated levels of interleukin-18 and tumor necrosis factor-α in serum of patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy. Metabolism 2003, 52, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Gohda, T.; Tomino, Y. Novel biomarkers for the progression of diabetic nephropathy: Soluble TNF receptors. Curr. Diab. Rep. 2013, 13, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Gohda, T.; Maruyama, S.; Kamei, N.; Yamaguchi, S.; Shibata, T.; Murakoshi, M.; Horikoshi, S.; Tomino, Y.; Ohsawa, I.; Gotoh, H.; et al. Circulating TNF Receptors 1 and 2 Predict Mortality in Patients with End-stage Renal Disease Undergoing Dialysis. Sci. Rep. 2017, 7, 43520. [Google Scholar] [CrossRef]
- DiPetrillo, K.; Gesek, F.A. Pentoxifylline ameliorates renal tumor necrosis factor expression, sodium retention, and renal hypertrophy in diabetic rats. Am. J. Nephrol. 2004, 24, 352–359. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Mora-Fernández, C.; Muros De Fuentes, M.; Chahin, J.; Méndez, M.L.; Gallego, E.; Macía, M.; Del Castillo, N.; Rivero, A.; Getino, M.A.; et al. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: The PREDIAN trial. J. Am. Soc. Nephrol. 2015, 26, 220–229. [Google Scholar] [CrossRef] [Green Version]
- DiPetrillo, K.; Coutermarsh, B.; Gesek, F.A. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am. J. Physiol. Ren. Physiol. 2003, 284, F113–F121. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Wu, M.; Zang, C.; Li, Y.; Xu, Z. Association Between IL-6 Polymorphisms and Diabetic Nephropathy Risk: A Meta-analysis. Am. J. Med. Sci. 2019, 358, 363–373. [Google Scholar] [CrossRef]
- Cui, Z.-H.; Lu, X.-T.; Xiao, K.-L.; Chen, Y.; Li, H.-Q. Association of Interleukin-6-174G/C Polymorphism with the Risk of Diabetic Nephropathy in Type 2 Diabetes: A Meta-analysis. Curr. Med. Sci. 2019, 39, 250–258. [Google Scholar] [CrossRef]
- Wolkow, P.P.; Niewczas, M.A.; Perkins, B.; Ficociello, L.H.; Lipinski, B.; Warram, J.H.; Krolewski, A.S. Association of urinary inflammatory markers and renal decline in microalbuminuric type 1 diabetics. J. Am. Soc. Nephrol. 2008, 19, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef]
- Al-Mutairi, N.; Shabaan, D. Effects of tumor necrosis factor α inhibitors extend beyond psoriasis: Insulin sensitivity in psoriasis patients with type 2 diabetes mellitus. Cutis 2016, 97, 235–241. [Google Scholar] [PubMed]
- Otsuka, Y.; Kiyohara, C.; Kashiwado, Y.; Sawabe, T.; Nagano, S.; Kimoto, Y.; Ayano, M.; Mitoma, H.; Akahoshi, M.; Arinobu, Y.; et al. Effects of tumor necrosis factor inhibitors and tocilizumab on the glycosylated hemoglobin levels in patients with rheumatoid arthritis; an observational study. PLoS ONE 2018, 13, e0196368. [Google Scholar] [CrossRef] [PubMed]
- Buraczynska, M.; Ksiazek, K.; Wacinski, P.; Zaluska, W. Interleukin-1β Gene (IL1B) Polymorphism and Risk of Developing Diabetic Nephropathy. Immunol. Investig. 2019, 48, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Dalmas, E.; Böni-Schnetzler, M.; Donath, M.Y. The IL-1 Pathway in Type 2 Diabetes and Cardiovascular Complications. Trends Endocrinol. Metab. 2015, 26, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Stahel, M.; Becker, M.; Graf, N.; Michels, S. Systemic interleukin 1β inhibition in proliferative diabetic retinopathy: A Prospective Open-Label Study Using Canakinumab. Retina 2016, 36, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Lemos, D.R.; McMurdo, M.; Karaca, G.; Wilflingseder, J.; Leaf, I.A.; Gupta, N.; Miyoshi, T.; Susa, K.; Johnson, B.G.; Soliman, K.; et al. Interleukin-1β Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 1690–1705. [Google Scholar] [CrossRef] [Green Version]
- Vila, E.; Salaices, M. Cytokines and vascular reactivity in resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, 1016–1021. [Google Scholar] [CrossRef] [Green Version]
- Vallejo, S.; Palacios, E.; Romacho, T.; Villalobos, L.; Peiró, C.; Sánchez-Ferrer, C.F. The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2014, 13, 158. [Google Scholar] [CrossRef] [Green Version]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.C.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Cavelti-Weder, C.; Babians-Brunner, A.; Keller, C.; Stahel, M.A.; Kurz-Levin, M.; Zayed, H.; Solinger, A.M.; Mandrup-Poulsen, T.; Dinarello, C.A.; Donath, M.Y. Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care 2012, 35, 1654–1662. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, K.L.; Chonchol, M.; Ikizler, T.A.; Farmer-Bailey, H.; Salas, N.; Chaudhry, R.; Wang, W.; Smits, G.; Tengesdal, I.; Dinarello, C.A.; et al. IL-1 inhibition and vascular function in CKD. J. Am. Soc. Nephrol. 2017, 28, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.C.; Adhikari, S.; Grishman, E.K.; Sumpter, K.M. A phase I study of anti-inflammatory therapy with rilonacept in adolescents and adults with type 1 diabetes mellitus. Pediatr. Diabetes 2018, 19, 788–793. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Interleukin-18 and diabetic nephropathy: A review. J. Cell. Physiol. 2019, 234, 5674–5682. [Google Scholar] [CrossRef]
- Park, C.C.; Morel, J.C.M.; Amin, M.A.; Connors, M.A.; Harlow, L.A.; Koch, A.E. Evidence of IL-18 as a Novel Angiogenic Mediator. J. Immunol. 2001, 167, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Mohammadi, M.T.; Rezaee, R.; Sahebkar, A. Crocin improves renal function by declining Nox-4, IL-18, and p53 expression levels in an experimental model of diabetic nephropathy. J. Cell. Biochem. 2018, 119, 6080–6093. [Google Scholar] [CrossRef]
- Miyauchi, K.; Takiyama, Y.; Honjyo, J.; Tateno, M.; Haneda, M. Upregulated IL-18 expression in type 2 diabetic subjects with nephropathy: TGF-beta1 enhanced IL-18 expression in human renal proximal tubular epithelial cells. Diabetes Res. Clin. Pract. 2009, 83, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Collins, M.; Jennings, C.; van der Merwe, L.; Söderström, I.; Olsson, T.; Levitt, N.S.; Lambert, E.V.; Goedecke, J.H. The association of interleukin-18 genotype and serum levels with metabolic risk factors for cardiovascular disease. Eur. J. Endocrinol. 2007, 157, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T.; Ogihara, N.; Kamura, Y.; Satomura, A.; Fuke, Y.; Shimizu, C.; Wada, Y.; Matsumoto, K. Interleukin-18 contributes more closely to the progression of diabetic nephropathy than other diabetic complications. Acta Diabetol. 2012, 49, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Shikata, K.; Hiramatsu, M.; Nakatou, T.; Kitamura, T.; Wada, J.; Itoshima, T.; Makino, H. Serum interleukin-18 levels are associated with nephropathy and atherosclerosis in Japanese patients with type 2 diabetes. Diabetes Care 2005, 28, 2890–2895. [Google Scholar] [CrossRef] [Green Version]
- Sueud, T.; Hadi, N.R.; Abdulameer, R.; Jamil, D.A.; Al-Aubaidy, H.A. Assessing urinary levels of IL-18, NGAL and albumin creatinine ratio in patients with diabetic nephropathy. Diabetes Metab. Syndr. 2019, 13, 564–568. [Google Scholar] [CrossRef]
- McKie, E.A.; Reid, J.L.; Mistry, P.C.; DeWall, S.L.; Abberley, L.; Ambery, P.D.; Gil-Extremera, B. A Study to Investigate the Efficacy and Safety of an Anti-Interleukin-18 Monoclonal Antibody in the Treatment of Type 2 Diabetes Mellitus. PLoS ONE 2016, 11, e0150018. [Google Scholar] [CrossRef]
- Mistry, P.; Reid, J.; Pouliquen, I.; McHugh, S.; Abberley, L.; DeWall, S.; Taylor, A.; Tong, X.; Del Cura, M.R.; McKie, E. Safety, tolerability, pharmacokinetics, and pharmacodynamics of single-dose anti-interleukin-18 mAb GSK1070806 in healthy and obese subjects. Int. J. Clin. Pharmacol. Ther. 2014, 52, 867–879. [Google Scholar] [CrossRef]
- Cortvrindt, C.; Speeckaert, R.; Moerman, A.; Delanghe, J.R.; Speeckaert, M.M. The role of interleukin-17A in the pathogenesis of kidney diseases. Pathology 2017, 49, 247–258. [Google Scholar] [CrossRef]
- Biswas, P.S. IL-17 in Renal Immunity and Autoimmunity. J. Immunol. 2018, 201, 3153–3159. [Google Scholar] [CrossRef] [Green Version]
- Robert, M.; Miossec, P. Effects of Interleukin 17 on the cardiovascular system. Autoimmun. Rev. 2017, 16, 984–991. [Google Scholar] [CrossRef]
- Ryba-Stanisławowska, M.; Skrzypkowska, M.; Myśliwiec, M.; Myśliwska, J. Loss of the balance between CD4(+)Foxp3(+) regulatory T cells and CD4(+)IL17A(+) Th17 cells in patients with type 1 diabetes. Hum. Immunol. 2013, 74, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-R.; Mueller, E.E.; Bradley, L.M. Islet antigen-specific Th17 cells can induce TNF-α-dependent autoimmune diabetes. J. Immunol. 2014, 192, 1425–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baharlou, R.; Ahmadi-Vasmehjani, A.; Davami, M.H.; Faraji, F.; Atashzar, M.R.; Karimipour, F.; Sadeghi, A.; Asadi, M.-A.; Khoubyari, M. Elevated Levels of T-helper 17-associated Cytokines in Diabetes Type I Patients: Indicators for Following the Course of Disease. Immunol. Investig. 2016, 45, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, A.K.; Panagiotopoulos, C.; Biggs, C.M.; Staiger, S.; Del Bel, K.L.; Hirschfeld, A.F.; Priatel, J.J.; Turvey, S.E.; Tan, R. Pre-diagnostic genotyping identifies T1D subjects with impaired Treg IL-2 signaling and an elevated proportion of FOXP3+IL-17+ cells. Genes Immun. 2017, 18, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan, D.L.; Danesh, F.R. Paradoxical role of IL-17 in progression of diabetic nephropathy. J. Am. Soc. Nephrol. 2016, 27, 657–658. [Google Scholar] [CrossRef] [Green Version]
- Orejudo, M.; Rodrigues-Diez, R.R.; Rodrigues-Diez, R.; Garcia-Redondo, A.; Santos-Sánchez, L.; Rández-Garbayo, J.; Cannata-Ortiz, P.; Ramos, A.M.; Ortiz, A.; Selgas, R.; et al. Interleukin 17A Participates in Renal Inflammation Associated to Experimental and Human Hypertension. Front. Pharmacol. 2019, 10, 1015. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Sun, Y.; Wang, X.; Niu, K. Elevated Serum Level of Interleukin 17 in a Population With Prehypertension. J. Clin. Hypertens. (Greenwich) 2015, 17, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti–Interleukin-17 Monoclonal Antibody Ixekizumab in Chronic Plaque Psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Mease, P.; van der Heijde, D.; Landewé, R.; Mpofu, S.; Rahman, P.; Tahir, H.; Singhal, A.; Boettcher, E.; Navarra, S.; Meiser, K.; et al. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: Primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann. Rheum. Dis. 2018, 77, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Vasanthakumar, R.; Mohan, V.; Anand, G.; Deepa, M.; Babu, S.; Aravindhan, V. Serum IL-9, IL-17, and TGF-β levels in subjects with diabetic kidney disease (CURES-134). Cytokine 2015, 72, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Hetta, H.F.; Elkady, A.; Morsy, K.H.; Mohamed, I.S.; Ibrahim, M.A. Serum Level of IL17a among Cirrhotic Hepatitis C Virus Infected Patients with Incidence of Diabetes Mellitus. Egypt. J. Immunol. 2017, 24, 79–88. [Google Scholar] [PubMed]
- Liu, S.-Y.; Chen, J.; Li, Y.-F. Clinical significance of serum interleukin-8 and soluble tumor necrosis factor-like weak inducer of apoptosis levels in patients with diabetic nephropathy. J. Diabetes Investig. 2018, 9, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, X.; Pan, Z.; Liu, S.; Han, H.; Zhao, C.; Tang, X. The role of IL-8/CXCR2 signaling in microcystin-LR triggered endothelial cell activation and increased vascular permeability. Chemosphere 2018, 194, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Yahya, M.J.; Ismail, P.; Nordin, N.; Md Akim, A.; Md. Yusuf, W.S.; Adam, N.L.; Yusoff, M.J. Association of CCL2, CCR5, ELMO1, and IL8 Polymorphism with Diabetic Nephropathy in Malaysian Type 2 Diabetic Patients. Int. J. Chronic Dis. 2019, 2019, 2053015. [Google Scholar]
- Shen, Z.; Chen, Q.; Ying, H.; Ma, Z.; Bi, X.; Li, X.; Wang, M.; Jin, C.; Lai, D.; Zhao, Y.; et al. Identification of differentially expressed genes in the endothelial precursor cells of patients with type 2 diabetes mellitus by bioinformatics analysis. Exp. Ther. Med. 2019, 19, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Sharif, M.N.; Campanholle, G.; Nagiec, E.E.; Wang, J.; Syed, J.; O’Neil, S.P.; Zhan, Y.; Brenneman, K.; Homer, B.; Neubert, H.; et al. Soluble Fn14 Is Detected and Elevated in Mouse and Human Kidney Disease. PLoS ONE 2016, 11, e0155368. [Google Scholar] [CrossRef]
- Umapathy, D.; Dornadula, S.; Krishnamoorthy, E.; Mariappanadar, V.; Viswanathan, V.; Ramkumar, K.M. YKL-40: A biomarker for early nephropathy in type 2 diabetic patients and its association with inflammatory cytokines. Immunobiology 2018, 223, 718–727. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Carrero, J.J.; Martín-Ventura, J.L.; Sonmez, A.; Saglam, M.; Celik, T.; Yaman, H.; Yenicesu, M.; Eyileten, T.; Moreno, J.A.; et al. Combined therapy with renin-angiotensin system and calcium channel blockers in type 2 diabetic hypertensive patients with proteinuria: Effects on soluble TWEAK, PTX3, and flow-mediated dilation. Clin. J. Am. Soc. Nephrol. 2010, 5, 1174–1181. [Google Scholar] [CrossRef]
- Mayer, C.; Bergholdt, R.; Cucak, H.; Rolin, B.C.; Sams, A.; Rosendahl, A. Neutralizing anti-il20 antibody treatment significantly modulates low grade inflammation without affecting HbA1c in Type 2 diabetic db/db mice. PLoS ONE 2015, 10, e0131306. [Google Scholar] [CrossRef] [Green Version]
- Blumberg, H.; Conklin, D.; Xu, W.F.; Grossmann, A.; Brender, T.; Carollo, S.; Eagan, M.; Foster, D.; Haldeman, B.A.; Hammond, A.; et al. Interleukin 20: Discovery, receptor identification, and role in epidermal function. Cell 2001, 104, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Mezzano, S.; Droguett, A.; Lavoz, C.; Krall, P.; Egido, J.; Ruiz-Ortega, M. Gremlin and renal diseases: Ready to jump the fence to clinical utility? Nephrol. Dial. Transplant. 2018, 33, 735–741. [Google Scholar] [CrossRef]
- Roxburgh, S.A.; Kattla, J.J.; Curran, S.P.; O’Meara, Y.M.; Pollock, C.A.; Goldschmeding, R.; Godson, C.; Martin, F.; Brazil, D.P. Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes 2009, 58, 1641–1650. [Google Scholar] [CrossRef] [Green Version]
- Marchant, V.; Droguett, A.; Valderrama, G.; Eugenia Burgos, M.; Carpio, D.; Kerr, B.; Ruiz-Ortega, M.; Egido, J.; Mezzano, S. Tubular overexpression of gremlin in transgenic mice aggravates renal damage in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2015, 309, F559–F568. [Google Scholar] [CrossRef] [Green Version]
- Dolan, V.; Murphy, M.; Sadlier, D.; Lappin, D.; Doran, P.; Godson, C.; Martin, F.; O’Meara, Y.; Schmid, H.; Henger, A.; et al. Expression of gremlin, a bone morphogenetic protein antagonist, in human diabetic nephropathy. Am. J. Kidney Dis. 2005, 45, 1034–1039. [Google Scholar] [CrossRef]
- Marquez-Exposito, L.; Lavoz, C.; Rodrigues-Diez, R.R.; Rayego-Mateos, S.; Orejudo, M.; Cantero-Navarro, E.; Ortiz, A.; Egido, J.; Selgas, R.; Mezzano, S.; et al. Gremlin regulates tubular epithelial to mesenchymal transition via VEGFR2: Potential role in renal fibrosis. Front. Pharmacol. 2018, 9, 1195. [Google Scholar] [CrossRef]
- Lavoz, C.; Alique, M.; Rodrigues-Diez, R.; Pato, J.; Keri, G.; Mezzano, S.; Egido, J.; Ruiz-Ortega, M. Gremlin regulates renal inflammation via the vascular endothelial growth factor receptor 2 pathway. J. Pathol. 2015, 236, 407–420. [Google Scholar] [CrossRef]
- Lavoz, C.; Rodrigues-Diez, R.R.; Plaza, A.; Carpio, D.; Egido, J.; Ruiz-Ortega, M.; Mezzano, S. VEGFR2 Blockade Improves Renal Damage in an Experimental Model of Type 2 Diabetic Nephropathy. J. Clin. Med. 2020, 9, 302. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Zamora, Y.I.; Rodriguez-Sosa, M. The Role of MIF in Type 1 and Type 2 Diabetes Mellitus. J. Diabetes Res. 2014, 2014, 804519. [Google Scholar] [CrossRef] [Green Version]
- Herder, C.; Kolb, H.; Koenig, W.; Haastert, B.; Müller-Scholze, S.; Rathmann, W.; Holle, R.; Thorand, B.; Wichmann, H.E. Association of systemic concentrations of macrophage migration inhibitory factor with impaired glucose tolerance and type 2 diabetes: Results from the Cooperative Health Research in the Region of Augsburg, Survey 4 (KORA S4). Diabetes Care 2006, 29, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Sanz, A.B.; Ihalmo, P.; Lassila, M.; Holthofer, H.; Mezzano, S.; Aros, C.; Groop, P.H.; Saleem, M.A.; Mathieson, P.W.; et al. The MIF receptor CD74 in diabetic podocyte injury. J. Am. Soc. Nephrol. 2009, 20, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Perlman, A.S.; Chevalier, J.M.; Wilkinson, P.; Liu, H.; Parker, T.; Levine, D.M.; Sloan, B.J.; Gong, A.; Sherman, R.; Farrell, F.X. Serum inflammatory and immune mediators are elevated in early stage diabetic nephropathy. Ann. Clin. Lab. Sci. 2015, 45, 256–263. [Google Scholar]
- Liu, J.; Zhao, Z.; Willcox, M.D.P.; Xu, B.; Shi, B. Multiplex bead analysis of urinary cytokines of type 2 diabetic patients with normo- and microalbuminuria. J. Immunoass. Immunochem. 2010, 31, 279–289. [Google Scholar] [CrossRef]
- Kim, Y.K.; Shin, J.S.; Nahm, M.H. NOD-like receptors in infection, immunity, and diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Du, P.; Fan, B.; Han, H.; Zhen, J.; Shang, J.; Wang, X.; Li, X.; Shi, W.; Tang, W.; Bao, C.; et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int. 2013, 84, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Gou, F.; Long, Y.; Li, Y.; Feng, H.; Zhang, Q.; Gao, C.; Chen, G.; Xu, Y. High Glucose and Lipopolysaccharide Activate NOD1- RICK-NF-κB Inflammatory Signaling in Mesangial Cells. Exp. Clin. Endocrinol. Diabetes 2016, 124, 512–517. [Google Scholar] [CrossRef]
- Shang, J.; Zhang, Y.; Jiang, Y.; Li, Z.; Duan, Y.; Wang, L.; Xiao, J.; Zhao, Z. NOD2 promotes endothelial-to-mesenchymal transition of glomerular endothelial cells via MEK/ERK signaling pathway in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2017, 484, 435–441. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Q.; Wang, X.; Duan, Y.; Wang, Z.; Wei, X.; Zhang, Y.; Wang, H.; Wang, R.; Yi, F. Identification of NOD2 as a novel target of RNA-binding protein HuR: Evidence from NADPH oxidase-mediated HuR signaling in diabetic nephropathy. Free Radic. Biol. Med. 2015, 79, 217–227. [Google Scholar] [CrossRef]
- Wen, H.; Ting, J.P.-Y.; O’Neill, L.A.J. A role for the NLRP3 inflammasome in metabolic diseases--did Warburg miss inflammation? Nat. Immunol. 2012, 13, 352–357. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, X.; Yang, S.; Chen, B.; Shi, J. Salidroside alleviates high glucose-induced oxidative stress and extracellular matrix accumulation in rat glomerular mesangial cells by the TXNIP-NLRP3 inflammasome pathway. Chem. Biol. Interact. 2017, 278, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, N.; Zhao, D. Function of NLRP3 in the pathogenesis and development of diabetic nephropathy. Med. Sci. Monit. 2017, 23, 3878–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Qiu, D.; Luo, F.; Wei, J.; Wu, M.; Wu, H.; Du, C.; Du, Y.; Ren, Y.; Chen, N.; et al. Knockdown of NLRP3 alleviates high glucose or TGFB1-induced EMT in human renal tubular cells. J. Mol. Endocrinol. 2018, 61, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, C.K.; Paine, S.K.; Mahanta, J.; Borphukan, S.; Borah, P.K. Expression of inflammasome complex mRNA and its targeted microRNA in type 2 diabetes mellitus: A possible predictor of the severity of diabetic nephropathy. J. Diabetes 2019, 11, 90–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pan, Y.; Zhang, Q.-Y.; Wang, F.-M.; Kong, L.-D. Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PLoS ONE 2012, 7, e38285. [Google Scholar] [CrossRef] [Green Version]
- Samra, Y.A.; Said, H.S.; Elsherbiny, N.M.; Liou, G.I.; El-Shishtawy, M.M.; Eissa, L.A. Cepharanthine and Piperine ameliorate diabetic nephropathy in rats: Role of NF-κB and NLRP3 inflammasome. Life Sci. 2016, 157, 187–199. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, B.; Wang, L.; Yang, M.; Xia, Z.; Wei, W.; Zhang, F.; Yuan, X. Pioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitus. PLoS ONE 2017, 12, e0181248. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhu, X.; Li, L.; Ma, T.; Shi, M.; Yang, Y.; Fan, Q. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 1297–1309. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Kohli, S.; Nazir, S.; Ghosh, S.; Ranjan, S.; Wang, H.; Madhusudhan, T.; et al. Caspase-1, but not caspase-3, promotes diabetic nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2270–2275. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Tang, S.C.W. Toll-like receptors: Sensing and reacting to diabetic injury in the kidney. Nephrol. Dial. Transplant. 2014, 29, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Yiu, W.H.; Li, R.X.; Wu, H.J.; Wong, D.W.L.; Chan, L.Y.Y.; Leung, J.C.K.; Lai, K.N.; Tang, S.C.W. The TLR4 antagonist CRX-526 protects against advanced diabetic nephropathy. Kidney Int. 2013, 83, 887–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, J.J.; Hyun, Y.Y.; Lee, M.H.; Kim, J.E.; Nam, D.H.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Han, J.Y.; et al. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinology 2013, 154, 2144–2155. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Liu, G.; Guo, J.; Su, Z.Q. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Maffei, A.; Lembo, G.; Carnevale, D. PI3Kinases in Diabetes Mellitus and Its Related Complications. Int. J. Mol. Sci. 2018, 19, 4098. [Google Scholar] [CrossRef] [Green Version]
- Lieberthal, W.; Levine, J.S. The role of the mammalian target of rapamycin (mTOR) in renal disease. J. Am. Soc. Nephrol. 2009, 20, 2493–2502. [Google Scholar] [CrossRef]
- Lee, C.-H.; Inoki, K.; Guan, K.-L. mTOR pathway as a target in tissue hypertrophy. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 443–467. [Google Scholar] [CrossRef]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Rüegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, J.; Qin, L.; Shou, Z.; Zhao, J.; Wang, H.; Chen, Y.; Chen, J. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am. J. Nephrol. 2007, 27, 495–502. [Google Scholar] [CrossRef]
- Song, Y.; Liu, W.; Tang, K.; Zang, J.; Li, D.; Gao, H. Mangiferin Alleviates Renal Interstitial Fibrosis in Streptozotocin-Induced Diabetic Mice through Regulating the PTEN/PI3K/Akt Signaling Pathway. J. Diabetes Res. 2020, 9481720. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Luo, M.; Xu, F.; Lu, Y.; Zhou, X.; Cui, W.; Miao, L. Elabela protects against podocyte injury in mice with streptozocin-induced diabetes by associating with the PI3K/Akt/mTOR pathway. Peptides 2019, 114, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jin, M.; Zhao, X.; Zhao, T.; Lin, W.; He, Z.; Fan, M.; Jin, W.; Zhou, J.; Jin, L.; et al. FGF1ΔHBS ameliorates chronic kidney disease via PI3K/AKT mediated suppression of oxidative stress and inflammation. Cell Death Dis. 2019, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Sadi, G.; Şahin, G.; Bostancı, A. Modulation of Renal Insulin Signaling Pathway and Antioxidant Enzymes with Streptozotocin-Induced Diabetes: Effects of Resveratrol. Medicina (Kaunas) 2018, 55, 3. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhao, X.; Zhu, H.; Wang, J.; Ma, J.; Gu, M. Apigenin Protects Against Renal Tubular Epithelial Cell Injury and Oxidative Stress by High Glucose via Regulation of NF-E2-Related Factor 2 (Nrf2) Pathway. Med. Sci. Monit. 2019, 25, 5280–5288. [Google Scholar] [CrossRef]
- Jing, D.; Bai, H.; Yin, S. Renoprotective effects of emodin against diabetic nephropathy in rat models are mediated via PI3K/Akt/GSK-3β and Bax/caspase-3 signaling pathways. Exp. Ther. Med. 2017, 14, 5163–5169. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Thaiss, F.; Guo, L. NFκB and Kidney Injury. Front. Immunol. 2019, 10, 815. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-κβ: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Lee, M.H.; Nam, D.H.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Hyun, Y.Y.; Han, S.Y.; et al. Celastrol, an NF-κB inhibitor, improves insulin resistance and attenuates renal injury in db/db mice. PLoS ONE 2013, 8, e62068. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Peng, R.; Peng, H.; Liu, H.; Wen, L.; Wu, T.; Yi, H.; Li, A.; Zhang, Z. miR-451 suppresses the NF-kappaB-mediated proinflammatory molecules expression through inhibiting LMP7 in diabetic nephropathy. Mol. Cell. Endocrinol. 2016, 433, 75–86. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, X.; Liu, P.; Shen, X.; Lan, T.; Li, W.; Jiang, Q.; Xie, X.; Huang, H. Effects of berberine on matrix accumulation and NF-kappa B signal pathway in alloxan-induced diabetic mice with renal injury. Eur. J. Pharmacol. 2010, 638, 150–155. [Google Scholar] [CrossRef]
- Ahmed, S.; Mundhe, N.; Borgohain, M.; Chowdhury, L.; Kwatra, M.; Bolshette, N.; Ahmed, A.; Lahkar, M. Diosmin Modulates the NF-kB Signal Transduction Pathways and Downregulation of Various Oxidative Stress Markers in Alloxan-Induced Diabetic Nephropathy. Inflammation 2016, 39, 1783–1797. [Google Scholar] [CrossRef]
- Kolati, S.R.; Kasala, E.R.; Bodduluru, L.N.; Mahareddy, J.R.; Uppulapu, S.K.; Gogoi, R.; Barua, C.C.; Lahkar, M. BAY 11-7082 ameliorates diabetic nephropathy by attenuating hyperglycemia-mediated oxidative stress and renal inflammation via NF-κB pathway. Environ. Toxicol. Pharmacol. 2015, 39, 690–699. [Google Scholar] [CrossRef]
- López-Franco, O.; Suzuki, Y.; Sanjuán, G.; Blanco, J.; Hernández-Vargas, P.; Yo, Y.; Kopp, J.; Egido, J.; Gómez-Guerrero, C. Nuclear factor-κB inhibitors as potential novel anti-inflammatory agents for the treatment of immune glomerulonephritis. Am. J. Pathol. 2002, 161, 1497–1505. [Google Scholar] [CrossRef]
- Oguiza, A.; Recio, C.; Lazaro, I.; Mallavia, B.; Blanco, J.; Egido, J.; Gomez-Guerrero, C. Peptide-based inhibition of IκB kinase/nuclear factor-κB pathway protects against diabetes-associated nephropathy and atherosclerosis in a mouse model of type 1 diabetes. Diabetologia 2015, 58, 1656–1667. [Google Scholar] [CrossRef] [Green Version]
- Elsherbiny, N.M.; El-Sherbiny, M.; Said, E. Amelioration of experimentally induced diabetic nephropathy and renal damage by nilotinib. J. Physiol. Biochem. 2015, 71, 635–648. [Google Scholar] [CrossRef]
- Lazaro, I.; Oguiza, A.; Recio, C.; Mallavia, B.; Madrigal-Matute, J.; Blanco, J.; Egido, J.; Martin-Ventura, J.L.; Gomez-Guerrero, C. Targeting HSP90 ameliorates nephropathy and atherosclerosis through suppression of NF-κB and STAT signaling pathways in diabetic mice. Diabetes 2015, 64, 3600–3613. [Google Scholar] [CrossRef] [Green Version]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [Green Version]
- Stark, G.R.; Darnell, J.E. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Cheon, H.J.; Yang, J.; Stark, G.R. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J. Interf. Cytokine Res. 2011, 31, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Sanz, L.; Bernal, S.; Recio, C.; Lazaro, I.; Oguiza, A.; Melgar, A.; Jimenez-Castilla, L.; Egido, J.; Gomez-Guerrero, C. SOCS1-targeted therapy ameliorates renal and vascular oxidative stress in diabetes via STAT1 and PI3K inhibition. Lab. Investig. 2018, 98, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lv, C.; Wu, C.; Chen, F.; Shao, Y.; Wang, Q. Suppressor of cytokine signaling (SOCS) 2 attenuates renal lesions in rats with diabetic nephropathy. Acta Histochem. 2014, 116, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrero, M.B.; Banes-berceli, A.K.; Stern, D.M.; Eaton, D.C.; Mario, B.; Douglas, C. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am. J. Physiol.-Renal Physiol. 2006, 290, 762–768. [Google Scholar] [CrossRef]
- Brosius, F.C.; Tuttle, K.R.; Kretzler, M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia 2016, 59, 1624–1627. [Google Scholar] [CrossRef]
- Ortiz-Muñoz, G.; Lopez-Parra, V.; Lopez-Franco, O.; Fernandez-Vizarra, P.; Mallavia, B.; Flores, C.; Sanz, A.; Blanco, J.; Mezzano, S.; Ortiz, A.; et al. Suppressors of cytokine signaling abrogate diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.-Y.; Wang, S.-J.; Li, X.-Q.; Shen, Y.-L.; Lu, J.-R.; Tian, X.-H.; Rahman, K.; Zhang, L.-J.; Nian, H.; Zhang, H. CXCL6 Promotes Renal Interstitial Fibrosis in Diabetic Nephropathy by Activating JAK/STAT3 Signaling Pathway. Front. Pharmacol. 2019, 10, 224. [Google Scholar] [CrossRef]
- Hashimoto, R.; Kakigi, R.; Miyamoto, Y.; Nakamura, K.; Itoh, S.; Daida, H.; Okada, T.; Katoh, Y. JAK-STAT-dependent regulation of scavenger receptors in LPS-activated murine macrophages. Eur. J. Pharmacol. 2020, 871, 172940. [Google Scholar] [CrossRef]
- Banes, A.K.; Shaw, S.; Jenkins, J.; Redd, H.; Amiri, F.; Pollock, D.M.; Marrero, M.B. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am. J. Physiol. Renal Physiol. 2004, 286, F653–F659. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Nair, V.; Saha, J.; Atkins, K.B.; Hodgin, J.B.; Saunders, T.L.; Myers, M.G.; Werner, T.; Kretzler, M.; Brosius, F.C. Podocyte-specific JAK2 overexpression worsens diabetic kidney disease in mice. Kidney Int. 2017, 92, 909–921. [Google Scholar] [CrossRef]
- Dieter, B.P.; Meek, R.L.; Anderberg, R.J.; Cooney, S.K.; Bergin, J.L.; Zhang, H.; Nair, V.; Kretzler, M.; Brosius, F.C.; Tuttle, K.R. Serum amyloid A and Janus kinase 2 in a mouse model of diabetic kidney disease. PLoS ONE 2019, 14, e0211555. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Huang, L.; Luo, W.; Yu, W.; Hu, X.; Guan, X.; Cai, Y.; Zou, C.; Yin, H.; Xu, Z.; et al. Inhibition of STAT3 in tubular epithelial cells prevents kidney fibrosis and nephropathy in STZ-induced diabetic mice. Cell Death Dis. 2019, 10, 848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, E.; Zaitone, S.A.; Eldosoky, M.; Elsherbiny, N.M. Nifuroxazide, a STAT3 inhibitor, mitigates inflammatory burden and protects against diabetes-induced nephropathy in rats. Chem. Biol. Interact. 2018, 281, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhong, Y.; Li, X.; Chen, H.; Jim, B.; Zhou, M.-M.; Chuang, P.Y.; He, J.C. Role of transcription factor acetylation in diabetic kidney disease. Diabetes 2014, 63, 2440–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C.; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 2018, 33, 1950–1959. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Muñoz, G.; Martin-Ventura, J.L.; Hernandez-Vargas, P.; Mallavia, B.; Lopez-Parra, V.; Lopez-Franco, O.; Muñoz-Garcia, B.; Fernandez-Vizarra, P.; Ortega, L.; Egido, J.; et al. Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, Inflammation, and Autoimmunity. Front. Immunol. 2012, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Recio, C.; Oguiza, A.; Lazaro, I.; Mallavia, B.; Egido, J.; Gomez-Guerrero, C. Suppressor of cytokine signaling 1-derived peptide inhibits janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1953–1960. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.; Bogdanov, P.; Gómez-Guerrero, C.; Sampedro, J.; Solà-Adell, C.; Espejo, C.; García-Ramírez, M.; Prieto, I.; Egido, J.; Simó, R. SOCS1-derived peptide administered by eye drops prevents retinal neuroinflammation and vascular leakage in experimental diabetes. Int. J. Mol. Sci. 2019, 20, 3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Du, C.; Zhang, Y.; Ren, Y.; Hao, J.; Zhao, S.; Yao, F.; Duan, H. Suppressor of cytokine signaling-1 ameliorates expression of MCP-1 in diabetic nephropathy. Am. J. Nephrol. 2010, 31, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Lazaro, I.; Oguiza, A.; Lopez-Sanz, L.; Bernal, S.; Blanco, J.; Egido, J.; Gomez-Guerrero, C. Suppressor of Cytokine Signaling-1 Peptidomimetic Limits Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieper, G.M. Riaz-ul-Haq Activation of nuclear factor-κb in cultured endothelial cells by increased glucose concentration: Prevention by calphostin C. J. Cardiovasc. Pharmacol. 1997, 30, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Ohshiro, Y.; Ma, R.C.; Yasuda, Y.; Hiraoka-Yamamoto, J.; Clermont, A.C.; Isshiki, K.; Yagi, K.; Arikawa, E.; Kern, T.S.; King, G.L. Reduction of diabetes-induced oxidative stress, fibrotic cytokine expression, and renal dysfunction in protein kinase Cβ-null mice. Diabetes 2006, 55, 3112–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mima, A.; Kitada, M.; Geraldes, P.; Li, Q.; Matsumoto, M.; Mizutani, K.; Qi, W.; Li, C.; Leitges, M.; Rask-Madsen, C.; et al. Glomerular VEGF resistance induced by PKCδ/SHP-1 activation and contribution to diabetic nephropathy. FASEB J. 2012, 26, 2963–2974. [Google Scholar] [CrossRef] [Green Version]
- Al-Onazi, A.S.; Al-Rasheed, N.M.; Attia, H.A.; Al-Rasheed, N.M.; Ahmed, R.M.; Al-Amin, M.A.; Poizat, C. Ruboxistaurin attenuates diabetic nephropathy via modulation of TGF-β1/Smad and GRAP pathways. J. Pharm. Pharmacol. 2016, 68, 219–232. [Google Scholar] [CrossRef]
- Wang, Z.B.; Zhang, S.; Li, Y.; Wang, R.M.; Tong, L.C.; Wang, Y.; Liu, W.Y.; Su, D.F.; Tu, Y.; Zhang, L.C.; et al. LY333531, a PKCβ inhibitor, attenuates glomerular endothelial cell apoptosis in the early stage of mouse diabetic nephropathy via down-regulating swiprosin-1. Acta Pharmacol. Sin. 2017, 38, 1009–1023. [Google Scholar] [CrossRef]
- Tuttle, K.R.; McGill, J.B.; Bastyr, E.J.; Poi, K.K.; Shahri, N.; Anderson, P.W. Effect of ruboxistaurin on albuminuria and estimated GFR in people with diabetic peripheral neuropathy: Results from a randomized trial. Am. J. Kidney Dis. 2015, 65, 634–636. [Google Scholar] [CrossRef]
- Guerrero-Hue, M.; Farre-Alins, V.; Palomino-Antolin, A.; Parada, E.; Rubio-Navarro, A.; Egido, J.; Egea, J.; Moreno, J.A. Targeting Nrf2 in Protection Against Renal Disease. Curr. Med. Chem. 2017, 24, 3583–3605. [Google Scholar] [CrossRef]
- Cui, W.; Min, X.; Xu, X.; Du, B.; Luo, P. Role of Nuclear Factor Erythroid 2-Related Factor 2 in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 3797802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagishita, Y.; Fukutomi, T.; Sugawara, A.; Kawamura, H.; Takahashi, T.; Pi, J.; Uruno, A.; Yamamoto, M. Nrf2 protects pancreatic β-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 2014, 63, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, L.; Wang, F.; Shi, Y.; Ren, Y.; Liu, Q.; Cao, Y.; Duan, H. Attenuation of glomerular injury in diabetic mice with tert- butylhydroquinone through nuclear factor erythroid 2-related factor 2-dependent antioxidant gene activation. Am. J. Nephrol. 2011, 33, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Mou, Y.; Xu, X.; Zhao, D.; Lai, Y.; Xu, Y.; Chen, C.; Li, P.; Peng, S.; Tian, J.; et al. Novel Derivative of Bardoxolone Methyl Improves Safety for the Treatment of Diabetic Nephropathy. J. Med. Chem. 2017, 60, 8847–8857. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Bai, Y.; Miao, X.; Luo, P.; Chen, Q.; Tan, Y.; Rane, M.J.; Miao, L.; Cai, L. Prevention of Diabetic Nephropathy by Sulforaphane: Possible Role of Nrf2 Upregulation and Activation. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Kong, L.; Tang, Z.-Z.; Zhang, Y.-M.; Liu, Y.; Wang, T.-Y.; Liu, Y.-W. Hesperetin ameliorates diabetic nephropathy in rats by activating Nrf2/ARE/glyoxalase 1 pathway. Biomed. Pharmacother. 2019, 111, 1166–1175. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Wang, H.-R.; Wang, Y.-I.; Zhai, Z.-H.; Wang, L.-W.; Li, L.; Zhang, C.; Tang, L. Myricetin Attenuated Diabetes-Associated Kidney Injuries and Dysfunction via Regulating Nuclear Factor (Erythroid Derived 2)-Like 2 and Nuclear Factor-κB Signaling. Front. Pharmacol. 2019, 10, 647. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Zhang, H.; Sun, Y.; Guo, R.; Zhong, D.; Xu, R.; Song, M. Omentin-1 protects renal function of mice with type 2 diabetic nephropathy via regulating miR-27a-Nrf2/Keap1 axis. Biomed. Pharmacother. 2018, 107, 440–446. [Google Scholar] [CrossRef]
- Soetikno, V.; Sari, F.R.; Sukumaran, V.; Lakshmanan, A.P.; Harima, M.; Suzuki, K.; Kawachi, H.; Watanabe, K. Curcumin decreases renal triglyceride accumulation through AMPK-SREBP signaling pathway in streptozotocin-induced type 1 diabetic rats. J. Nutr. Biochem. 2013, 24, 796–802. [Google Scholar] [CrossRef]
- Jiménez-Osorio, A.S.; García-Niño, W.R.; González-Reyes, S.; Álvarez-Mejía, A.E.; Guerra-León, S.; Salazar-Segovia, J.; Falcón, I.; Montes de Oca-Solano, H.; Madero, M.; Pedraza-Chaverri, J. The Effect of Dietary Supplementation With Curcumin on Redox Status and Nrf2 Activation in Patients With Nondiabetic or Diabetic Proteinuric Chronic Kidney Disease: A Pilot Study. J. Ren. Nutr. 2016, 26, 237–244. [Google Scholar] [CrossRef]
- Yang, H.; Xu, W.; Zhou, Z.; Liu, J.; Li, X.; Chen, L.; Weng, J.; Yu, Z. Curcumin attenuates urinary excretion of albumin in type II diabetic patients with enhancing nuclear factor erythroid-derived 2-like 2 (Nrf2) system and repressing inflammatory signaling efficacies. Exp. Clin. Endocrinol. Diabetes 2015, 123, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b-4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, M.P.; Bakris, G.L.; Block, G.A.; Chertow, G.M.; Goldsberry, A.; Inker, L.A.; Heerspink, H.J.L.; O’Grady, M.; Pergola, P.E.; Wanner, C.; et al. Bardoxolone Methyl Improves Kidney Function in Patients with Chronic Kidney Disease Stage 4 and Type 2 Diabetes: Post-Hoc Analyses from Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes Study. Am. J. Nephrol. 2018, 47, 40–47. [Google Scholar] [CrossRef]
- Komers, R.; Lindsley, J.N.; Oyama, T.T.; Cohen, D.M.; Anderson, S. Renal p38 MAP kinase activity in experimental diabetes. Lab. Investig. 2007, 87, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, L.; Chow, F.; Nikolic-Paterson, D.J.; Stambe, C.; Dowling, J.; Atkins, R.C.; Tesch, G.H. Abnormal p38 mitogen-activated protein kinase signalling in human and experimental diabetic nephropathy. Diabetologia 2004, 47, 1210–1222. [Google Scholar] [CrossRef]
- Tesch, G.H.; Ma, F.Y.; Han, Y.; Liles, J.T.; Breckenridge, D.G.; Nikolic-Paterson, D.J. ASK1 inhibitor halts progression of diabetic nephropathy in Nos3-deficient mice. Diabetes 2015, 64, 3903–3913. [Google Scholar] [CrossRef] [Green Version]
- Liles, J.T.; Corkey, B.K.; Notte, G.T.; Budas, G.R.; Lansdon, E.B.; Hinojosa-Kirschenbaum, F.; Badal, S.S.; Lee, M.; Schultz, B.E.; Wise, S.; et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J. Clin. Investig. 2018, 128, 4485–4500. [Google Scholar] [CrossRef]
- Cha, J.J.; Kang, Y.S.; Hyun, Y.Y.; Han, S.Y.; Jee, Y.H.; Han, K.H.; Han, J.Y.; Cha, D.R. Sulodexide improves renal function through reduction of vascular endothelial growth factor in type 2 diabetic rats. Life Sci. 2013, 92, 1118–1124. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; Von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim, K.M.; Choi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE −/− mice fed a western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terami, N.; Ogawa, D.; Tachibana, H.; Hatanaka, T.; Wada, J.; Nakatsuka, A.; Eguchi, J.; Horiguchi, C.S.; Nishii, N.; Yamada, H.; et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS ONE 2014, 9, e100777. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [Green Version]
- Davis, H.; Jones Briscoe, V.; Dumbadze, S.; Davis, S.N. Using DPP-4 inhibitors to modulate beta cell function in type 1 diabetes and in the treatment of diabetic kidney disease. Expert Opin. Investig. Drugs 2019, 28, 377–388. [Google Scholar] [CrossRef]
- Liu, W.; Yu, J.; Yan, Q.; Wang, L.; Li, N.; Xiong, W. Meta-analysis of the benefit of sitagliptin treatment in patients with type 2 diabetes complicated with incipient nephropathy. Exp. Ther. Med. 2018, 16, 2545–2553. [Google Scholar] [CrossRef]
- Marques, C.; Gonçalves, A.; Pereira, P.M.R.; Almeida, D.; Martins, B.; Fontes-Ribeiro, C.; Reis, F.; Fernandes, R. The dipeptidyl peptidase 4 inhibitor sitagliptin improves oxidative stress and ameliorates glomerular lesions in a rat model of type 1 diabetes. Life Sci. 2019, 234, 116738. [Google Scholar] [CrossRef]
- Wang, J.; Hu, L.; Chen, Y.; Fu, T.; Jiang, T.; Jiang, A.; You, X. Sitagliptin improves renal function in diabetic nephropathy in male Sprague Dawley rats through upregulating heme oxygenase-1 expression. Endocrine 2019, 63, 70–78. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kume, S.; Chin-Kanasaki, M.; Araki, H.; Araki, S.-I.; Ugi, S.; Sugaya, T.; Uzu, T.; Maegawa, H. Renoprotective effect of DPP-4 inhibitors against free fatty acid-bound albumin-induced renal proximal tubular cell injury. Biochem. Biophys. Res. Commun. 2016, 470, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Li, L.; Song, C.; Jin, H.; Ma, S.; Kang, P. Sitagliptin relieves diabetic nephropathy fibrosis via the MAPK/ERK signaling pathway. Minerva Endocrinol. 2020. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Ninčević, V.; Kolarić, T.O.; Roguljić, H.; Kizivat, T.; Smolić, M.; Ćurčić, I.B. Renal benefits of SGLT 2 inhibitors and GLP-1 receptor agonists: Evidence supporting a paradigm shift in the medical management of type 2 diabetes. Int. J. Mol. Sci. 2019, 20, 5831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieter, B.P.; Alicic, R.Z.; Tuttle, K.R. GLP-1 receptor agonists in diabetic kidney disease: From the patient-side to the bench-side. Am. J. Physiol. Ren. Physiol. 2018, 315, F1519–F1525. [Google Scholar] [CrossRef]
- Moellmann, J.; Klinkhammer, B.M.; Onstein, J.; Stöhr, R.; Jankowski, V.; Jankowski, J.; Lebherz, C.; Tacke, F.; Marx, N.; Boor, P.; et al. Glucagon-Like Peptide 1 and Its Cleavage Products Are Renoprotective in Murine Diabetic Nephropathy. Diabetes 2018, 67, 2410–2419. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.; Zeng, J.; Zhang, T.; Lin, S.; Gao, J.; Jiang, C.; Fan, R.; Yin, D. Hyperglycemia induces NF-κB activation and MCP-1 expression via downregulating GLP-1R expression in rat mesangial cells: Inhibition by metformin. Cell Biol. Int. 2019, 43, 940–953. [Google Scholar] [CrossRef]
- Hendarto, H.; Inoguchi, T.; Maeda, Y.; Ikeda, N.; Zheng, J.; Takei, R.; Yokomizo, H.; Hirata, E.; Sonoda, N.; Takayanagi, R. GLP-1 analog liraglutide protects against oxidative stress and albuminuria in streptozotocin-induced diabetic rats via protein kinase A-mediated inhibition of renal NAD(P)H oxidases. Metabolism 2012, 61, 1422–1434. [Google Scholar] [CrossRef]
- Yin, Q.H.; Zhang, R.; Li, L.; Wang, Y.T.; Liu, J.P.; Zhang, J.; Bai, L.; Cheng, J.Q.; Fu, P.; Liu, F. Exendin-4 ameliorates lipotoxicity-induced glomerular endothelial cell injury by improving ABC transporter A1- mediated cholesterol efflux in diabetic apoE knockout mice. J. Biol. Chem. 2016, 291, 26487–26501. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Xu, S.; Wang, Z.; Liu, H.; Peng, L.; Fang, Q.; Deng, T.; Zhang, W.; Lou, J. Recombinant human GLP-1(rhGLP-1) alleviating renal tubulointestitial injury in diabetic STZ-induced rats. Biochem. Biophys. Res. Commun. 2018, 495, 793–800. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Lakshmanan, M.C.; Rayner, B.; Busch, R.S.; Zimmermann, A.G.; Woodward, D.B.; Botros, F.T. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): A multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol. 2018, 6, 605–617. [Google Scholar] [CrossRef]
- De Boer, I.H. Liraglutide and Renal Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 2198. [Google Scholar] [PubMed] [Green Version]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jodar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Køber, L.V.; Lawson, F.C.; Ping, L.; Wei, X.; Lewis, E.F.; et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Campion, C.G.; Sanchez-Ferras, O.; Batchu, S.N. Potential role of serum and urinary biomarkers in diagnosis and prognosis of diabetic nephropathy. Can. J. Kidney Health Dis. 2017, 4, 2054358117705371. [Google Scholar] [CrossRef] [PubMed]
- Tvarijonaviciute, A.; Castillo, C.; Ceron, J.J.; Martinez-Subiela, S.; Tecles, F.; López-Jornet, P. Leptin and NGF in saliva of patients with diabetes mellitus type 2: A pilot study. J. Oral Pathol. Med. 2017, 46, 853–855. [Google Scholar] [CrossRef]
- Srinivasan, M.; Meadows, M.L.; Maxwell, L. Assessment of Salivary Adipokines Resistin, Visfatin, and Ghrelin as Type 2 Diabetes Mellitus Biomarkers. Biochem. Res. Int. 2018, 7463796. [Google Scholar] [CrossRef] [Green Version]



| Target | Diabetic Model | Strategy | Category | Conclusion | Ref. |
|---|---|---|---|---|---|
| NOD- like receptor | SD rats + STZ (55 mg/kg) | Quercetin or allopurinol | Flavonoid or XO inhibitor | Ameliorates hyperuricemia and reduces IL-1β and IL-18 levels. | [226] |
| SD rats + STZ (50 mg/kg) | Cepharanthine and Piperine | Nutraceuticals | Decreases cytokines NLRP3-dependent. | [227] | |
| ApoE-/- mice + STZ (80 mg/kg) | Pioglitazone | PPARγ agonist | Restores renal function. Downregulates AGEs, RAGEs and cytokines NLRP3-dependent. | [228] | |
| db/db mice | MCC950 | Selective NLRP3 inhibitor | Ameliorates GBM thickness, podocyte injury and renal fibrosis. | [229] | |
| db/db mice | M920 | Pan-caspase inhibitor | Reduces NLRP3-inflammasome (IL-1β, IL-18, NLRP3, caspase-1) and profibrotic markers. | [230] | |
| TLRs | eNOS−/− + STZ mice | CRX-526 | TLR4 antagonist | Ameliorates histological changes, inflammatory and profibrotic markers. | [232] |
| db/db mice | GIT27 | TLR4 inhibitor | Reduces proinflammatory, oxidative stress and lipid metabolism markers. | [233] | |
| PI3K/ AKT | SD + STZ (60 mg/kg) | Rapamycin | mTOR inhibitor | Ameliorates histological changes, inflammation and fibrotic markers. | [239] |
| C57BL/6 + STZ (60 mg/kg) | Mangiferin | Natural phenolic xanthonoid | Reduces inflammation, oxidative stress and profibrotic markers. | [240] | |
| C57BL/6J + STZ (50 mg/kg) | Elabela | Peptide agonist Apelin/APJ receptor | Reduces GBM thickening, podocyte damage, inflammation, apoptosis and fibrosis markers. | [241] | |
| Wistar rats + STZ (60 mg/kg) | Emodin | Derived from root rhubarb | Attenuates inflammation, oxidative stress and apoptosis markers. | [245] | |
| NF-κB | db/db mice | Celastrol | Celastrus regelii root extracts | Improves lipid accumulation, oxidative stress and proinflammatory markers. | [248] |
| db/db mice | miR-451 | LMP7 modulator | Decreases histological lesions, inflammation and fibrosis markers. | [249] | |
| Wistar rats + STZ (65 mg/kg) | Berberine | Flavonoid | Improves systemic and renal cortex inflammatory response. | [250] | |
| SD rats + STZ (50 mg/kg). | BAY 11-7082 | Phospho-IκB inhibitor | Reduces proinflammatory cytokines and oxidative stress markers. | [252] | |
| ApoE−/− mice + STZ (125 mg/kg -2 days) | NBD peptide | Mimetic IKKβ-NEMO complex | Decreases histological lesions, inflammation and fibrosis. | [254] | |
| SD rats + STZ (50 mg/kg) | Nilotinib hydrochloride | BCR-ABL tyrosine kinase inhibitor | Reduces oxidative stress, inflammation and profibrotic marker expression. | [255] | |
| JAK/ STAT | SD rats + STZ (60 mg/kg) | AG-490 (Tyrphostin) | JAK 2 inhibitor | Prevents JAK2 and STATs phosphorylation. | [269] |
| Akita mice + JAK2 Pod-overexpression | Tyrphostin or Baricitinib | JAK 2 inhibitors | Ameliorates histological changes, serum amyloid A3 plasma and kidney tissue. | [228,229] | |
| SD rats + STZ (50 mg/kg) | Nifuroxazide | STAT3 inhibitor | Diminishes inflammatory and profibrotic factors and infiltrating cells. | [273] | |
| Double db/db- SIRT-1−/− mice | MS417 | BET-specific BRD4 inhibitor | Attenuates histological changes and STAT3 activity. | [274] | |
| CD-1 mice + STZ (150 mg/kg) | Gene delivery SOCS1 | SOCS1 Overexpression | Attenuates renal hypertrophy, inflammation and profibrotic markers. | [282] | |
| ApoE−/− + STZ (125 mg/kg) | SOCS1 delivery system | Mimetic SOCS1- KIR region | Reduces atherosclerosis, structural changes and inflammatory markers. | [219,241] | |
| PKC | Wistar rats + STZ (55 mg/kg) | Ruboxistaurin | Selective PKCβ inhibitor | Improves histological changes and attenuates TGFβ/Smad2/3 pathway. | [287] |
| Swiprosin-1−/− C57BL/6 mice + STZ (150 mg/kg) | Ruboxistaurin | Selective PKCβ inhibitor | Ameliorates histological changes, apoptosis and Swiprosin-1 expression. | [288] | |
| Nrf2 | SD rats + IR injury + STZ (65 mg/kg) | TBHQ | Synthetic antioxidant | Ameliorates oxidative stress, inflammation and apoptosis. | [293] |
| db/db and C57BL/6 mice + HFD | γ-glutamyl transpeptidase | Bardoxolone methyl analog | Reduces body weight, histological changes and GBM thickening. | [294] | |
| Wistar rats + STZ (58 mg/kg) | Sulforaphane | Cruciferous vegetables extracts | Diminishes inflammation, oxidative stress markers, DNA damage and cell death. | [295] | |
| SD rats + STZ (60 mg/kg) | Hesperetin | Flavonoid | Ameliorates histological changes, inhibition AGEs/RAGE axis and inflammation. | [296] | |
| C57BL/6 + STZ (50 mg/kg) and Nrf2 knockdown mice | Myricetin | Flavonoid | Mitigates inflammation, oxidative stress and fibrosis markers. | [297] | |
| db/db mice | Omentin-1 | Adipokine | Reduces proinflammatory cytokines and oxidative stress markers. | [298] | |
| SD rats + STZ (55 mg/kg) | Curcumin | Flavonoid | Lipid accumulation, angiogenesis, profibrotic and podocyte damage markers. | [299] | |
| p38/ MAPK | eNOS−/− mice + STZ (55 mg/kg) | GS-444217 | ASK1 inhibitor (Selonsertib) | Ameliorates histological changes, inflammatory and profibrotic markers. | [308] |
| eNOS−/− db/db mice | GS-444217 + ACEi | [309] | |||
| OLETF rats | Sulodexide | Sulfated GAGs | Reduces histological changes, urinary VEGF and profibrotic markers. | [310] |
| Target | Compound | Enrollment | Phase | Status | 1stor 2nd Outcome | Identification |
|---|---|---|---|---|---|---|
| Chymase inhibitor | BAY1142524 Fulacimstat | 152 | 2 | C | UACR | NCT03412006 |
| VAP-1 inhibitor | ASP8232 | 55 | 2 | C | AE—UACR | NCT02218099 |
| Galectin-3 antagonist | GCS-100 | 375 | 2 | U | eGFR | NCT02312050 |
| Anti-Human αVβ3 | VPI-2690B Monoclonal antibody | 165 | 2 | C | Albuminuria—eGFR | NCT02251067 |
| CCL2 inhibitor | AF2838 Bindarit | 100 | 2 | C | UACR | NCT01109212 |
| CCL2 inhibitor | Spiegelmer® NOX-E36 | 76 | 2 | C | UACR | NCT01547897 |
| CCR2 inhibitor | Propagermanium | 45 | 2 | A | UACR—eGFR | NCT03627715 |
| CCR2 inhibitor | CCX140-B | 332 | 2 | C | UACR | NCT01447147 |
| CCR2/5 antagonist | PF-04634817 | 226 | 2 | C | UACR | NCT01712061 |
| CCR2/5 antagonist | BMS-813160 | 319 | 2 | T | UACR | NCT01752985 |
| PDE inhibitor | Pentoxifylline | 196 | 4 | R | eGFR | NCT03664414 |
| PDE inhibitor | Pentoxifylline | 2510 | 4 | A | Death—ESRD—UACR | NCT03625648 |
| Anti-Human IL-1β | Canakinumab | 10066 | 3 | C | MACE—UACR—eGFR | NCT01327846 |
| Anti-Human IL-18 | GSK1070806 Monoclonal antibody | 37 | 2 | C | UACR, HbA1c and metabolic changes | NCT01648153 |
| Anti-Human IL-33 | MEDI3506 Monoclonal Antibody | 168 | 2 | R | UACR | NCT04170543 |
| Anti-Human TGFβ1 | Galunisertib Monoclonal Antibody | 417 | 2 | T | UACR | NCT01113801 |
| Anti-Human TGFα/Epiregulin | LY3016859 Monoclonal antibody | 60 | 2 | C | UACR | NCT01774981 |
| JAK2 inhibitor | Baricitinib | 130 | 2 | C | UACR—eGFR | NCT01703234 |
| PKCβ Inhibitor | Ruboxistaurin | 20 | 3 | C | UACR | NCT00297401 |
| NRF2 inducer | Bardoxolone Methyl | 1323 | 3 | A | eGFR or ESRD | NCT03550443 |
| NRF2 inducer | Bardoxolone Methyl | 2185 | 3 | C | ESRD or death | NCT01351675 |
| NRF2 inducer | Bardoxolone Methyl | 216 | 2 | C | AE—eGFR | NCT02316821 |
| Anti-inflammatory inhibitor | Colchicine | 160 | NA | A | UACR | NCT02035891 |
| Sulfated GAGs | KRX-101 Sulodexide | 1248 | 4 | T | ESRD—UACR | NCT00130312 |
| ASK-1 inhibitor | GS-4997 Selonsertib | 3300 | 3 | A | eGFR—ESRD | NCT04026165 |
| Target | Compound | Enrollment | Phase | Status | 1st or 2nd Outcome | Identification |
|---|---|---|---|---|---|---|
| SGLT2i | Empaglifozin | 70 | 4 | R | Albuminuria—eGFR | NCT04127084 |
| SGLT2i | Empaglifozin | 7064 | 3 | C | MACE and UACR | NCT01131676 |
| SGLT2i + DPP-4i | Empaglifozin + Linagliptin | 66 | 4 | R | eGFR—UACR | NCT03433248 |
| SGLT2i + GLP-1 RA | Empaglifozin + Semaglutide | 80 | 4 | NR | Albuminuria—eGFR | NCT04061200 |
| SGLT2i | Canagliflozin | 4401 | 3 | C | eGFR—ESRD | NCT02065791 |
| SGLT2i | Canagliflozin | 300 | 3 | A | eGFR—UACR | NCT03436693 |
| SGLT2i | Canaglifozin | 1452 | 3 | C | HbA1c change | NCT00968812 |
| SGLT2i | Dapagliflozine | 44 | 4 | C | eGFR—ERPF—UACR | NCT02682563 |
| DPP-4i | Linagliptin | 48 | 4 | C | eGFR—ERPF | NCT02106104 |
| DPP-4i | Linagliptin | 6991 | 4 | C | MACE—eGFR | NCT01897532 |
| DPP-4i | Alogliptin | 5380 | 3 | C | MACE | NCT00968708 |
| DPP-4i | Saxagliptin | 18206 | 4 | C | MACE | NCT01107886 |
| DPP-4i | Sitagliptin | 14671 | 3 | C | MACE—eGFR | NCT00790205 |
| GLP-1 RA | Lixisenatide | 40 | 4 | C | eGFR—ERPF | NCT02276196 |
| GLP-1 RA | Exenatide | 92 | 4 | C | Albuminuria | NCT02690883 |
| GLP-1 RA | Dulaglutide | 577 | 3 | C | HbA1c—eGFR—UACR | NCT01621178 |
| GLP-1 RA | Liraglutide | 9341 | 3 | C | MACE and UACR | NCT01179048 |
| GLP-1 RA | Semaglutide | 3297 | 3 | C | MACE—HbA1c—UACR | NCT01720446 |
| GLP-1 RA | Lixisenatide | 6068 | 3 | C | MACE—UACR | NCT01147250 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayego-Mateos, S.; Morgado-Pascual, J.L.; Opazo-Ríos, L.; Guerrero-Hue, M.; García-Caballero, C.; Vázquez-Carballo, C.; Mas, S.; Sanz, A.B.; Herencia, C.; Mezzano, S.; et al. Pathogenic Pathways and Therapeutic Approaches Targeting Inflammation in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 3798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113798
Rayego-Mateos S, Morgado-Pascual JL, Opazo-Ríos L, Guerrero-Hue M, García-Caballero C, Vázquez-Carballo C, Mas S, Sanz AB, Herencia C, Mezzano S, et al. Pathogenic Pathways and Therapeutic Approaches Targeting Inflammation in Diabetic Nephropathy. International Journal of Molecular Sciences. 2020; 21(11):3798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113798
Chicago/Turabian StyleRayego-Mateos, Sandra, José Luis Morgado-Pascual, Lucas Opazo-Ríos, Melania Guerrero-Hue, Cristina García-Caballero, Cristina Vázquez-Carballo, Sebastián Mas, Ana Belén Sanz, Carmen Herencia, Sergio Mezzano, and et al. 2020. "Pathogenic Pathways and Therapeutic Approaches Targeting Inflammation in Diabetic Nephropathy" International Journal of Molecular Sciences 21, no. 11: 3798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113798