The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
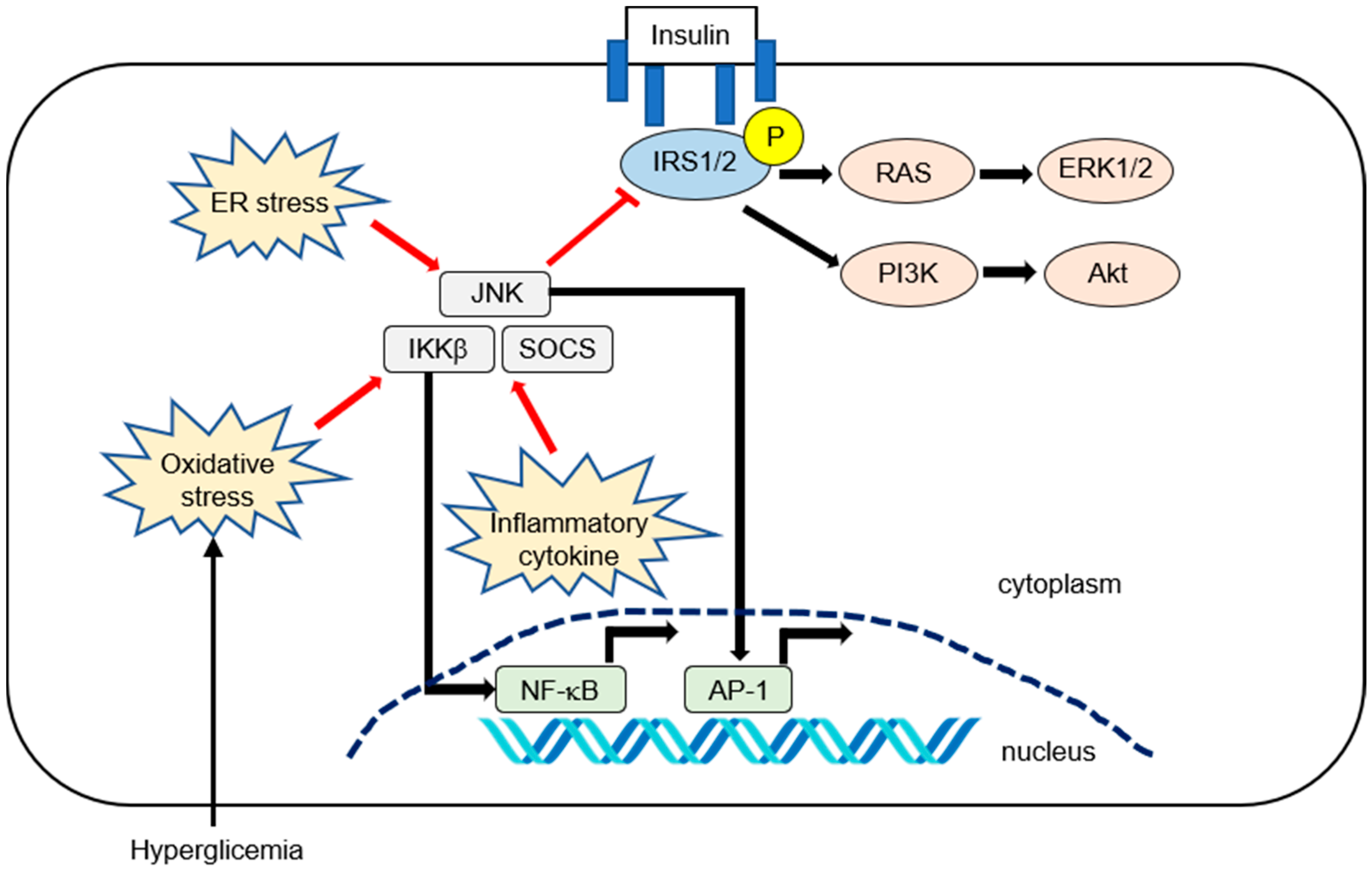
2. Molecular Mechanism of Inflammation in IR
3. Genetic Factors Affecting IR and NAFLD
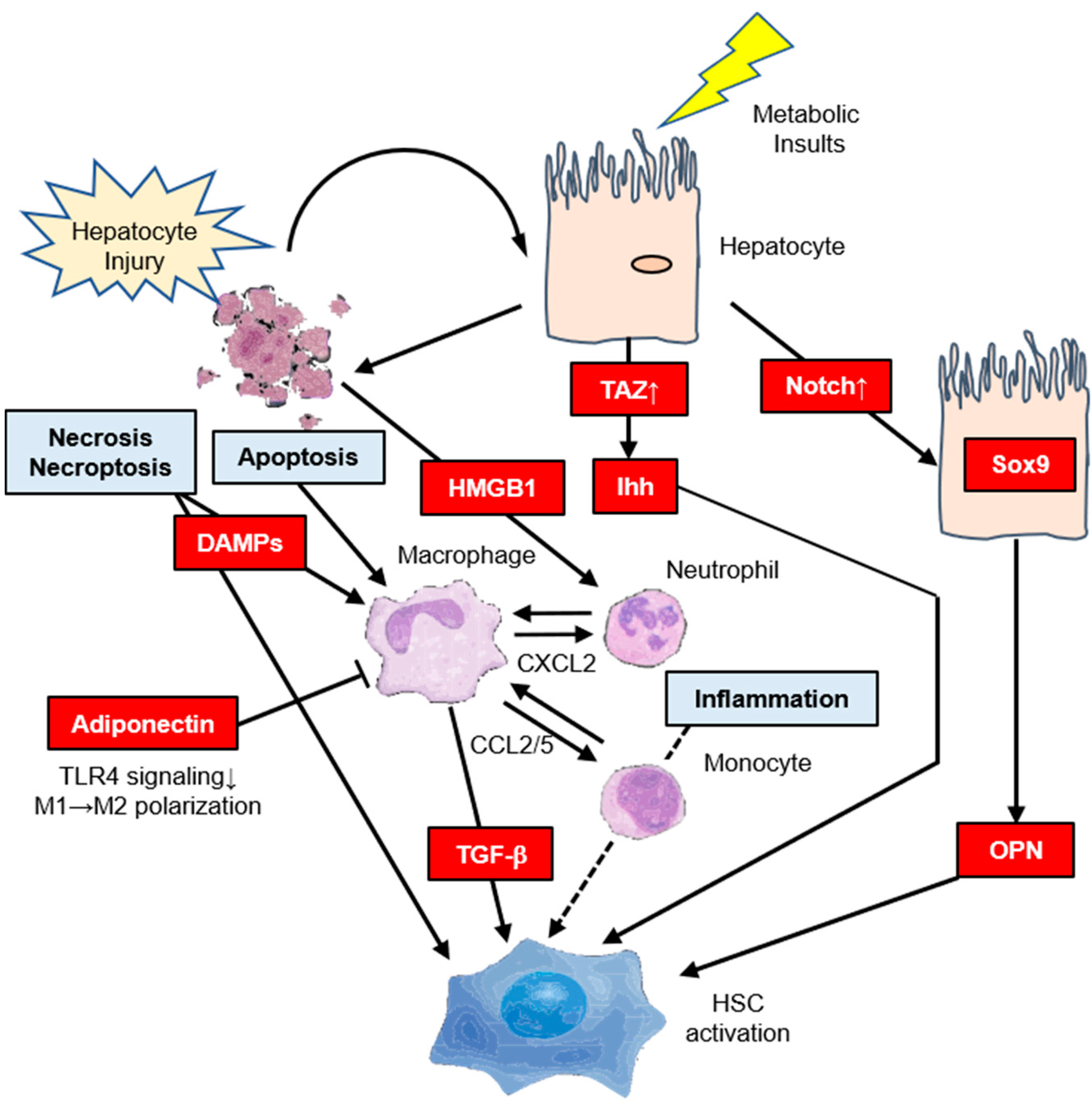
4. Molecular Mechanism of Hepatic IR Affects Hepatic Fibrosis
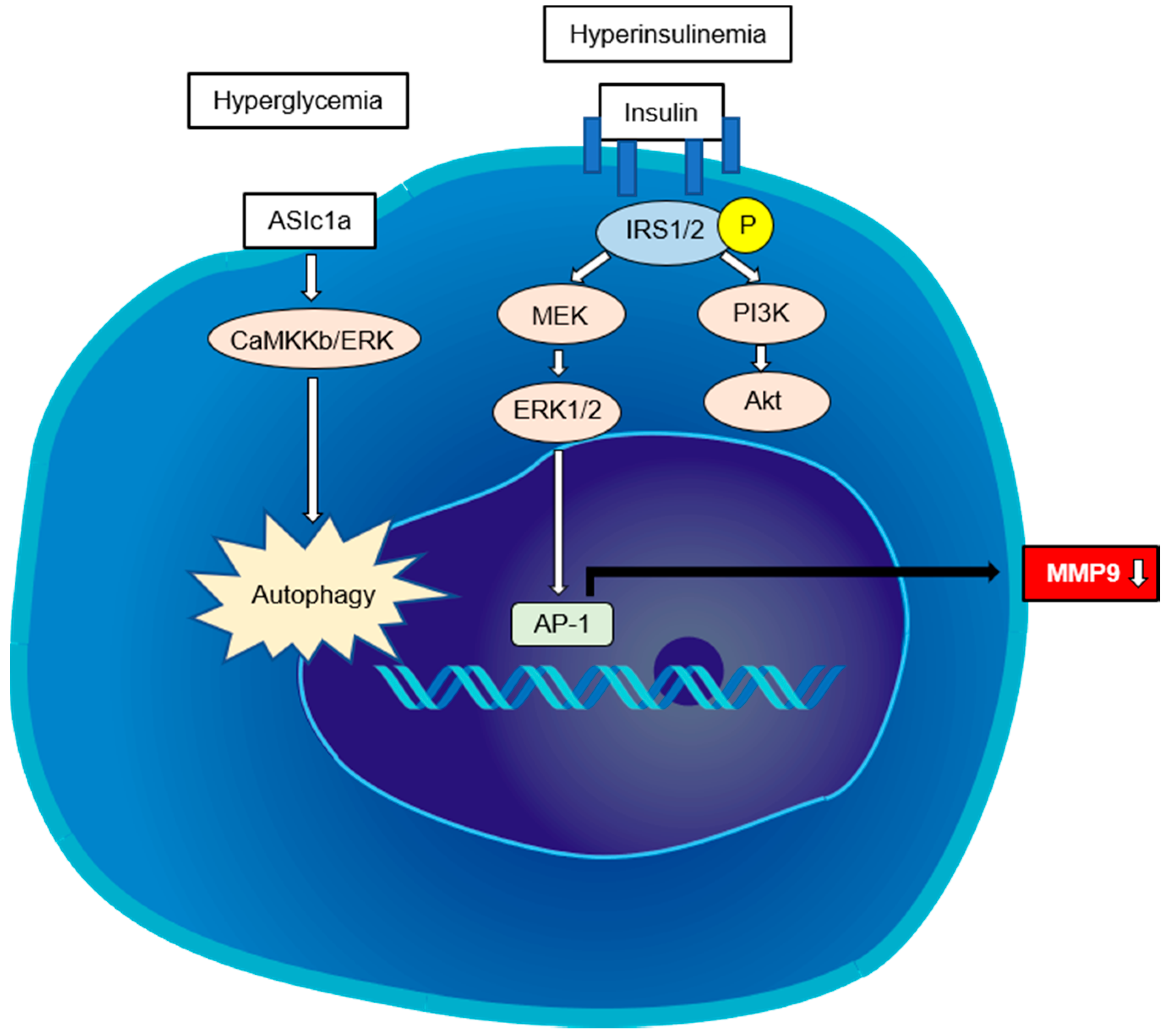
4.1. Indirect Pathway
4.2. Direct Pathway
5. Relationship Between T2DM and Hepatic Fibrosis in Patients with NAFLD
5.1. Liver Biopsy
5.2. Serum Biomarkers
5.3. Vibration-Controlled Transient Elastography
5.4. Magnetic Resonance Elastography
6. The Role of IR in Hepatic Fibrosis in NAFLD Patients
7. Hepatocellular Carcinoma
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| ALD | Alcoholic liver disease |
| AST | Aspartate transaminase |
| ATX | Autotaxin |
| AUROC | Area under the receiver operating characteristic |
| BMI | Body mass index |
| CCR | C-C chemokine receptors |
| CI | Confidence interval |
| ECM | Extracellular matrix |
| ER | Endoplasmic reticulum |
| ERK | RAS extracellular signal-regulated kinase |
| GCK | Glucokinase regulator |
| HCC | Hepatocellular carcinoma |
| HOMA | Homeostasis model assessment parameter |
| HR | Hazard ratio |
| HSC | Hepatic stellate cell |
| HSD17B13 | Hydroxysteroid 17β-dehydrogenase |
| IGF | Insulin-like growth factor |
| IGN | Intestinal gluconeogenesis |
| IKK | Inhibitor of nuclear factor-κB kinase |
| IL | Interleukin |
| IR | Insulin resistance |
| IRS | Insulin receptor substrate |
| JNK | c-Jun N-terminal kinase |
| JSG-NAFLD | Japan Study Group of NAFLD |
| LOXL2 | Lysyl oxidase-like 2 |
| LSM | Liver stiffness measurement |
| Mac-2bp | Mac-2 binding protein |
| MBOAT7 | Membrane bound O-acyltransferase domain- containing 7 |
| MMP | Matrix metalloproteinase |
| MRE | Magnetic resonance elastography |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| NF-κB | Nuclear factor-kappa B |
| NFS | NAFLD fibrosis score |
| OPN | Osteopontin |
| OR | Odds ratio |
| PDK | Phosphoinositide-dependent kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| PNPLA3 | Patatin-like phospholipase domain containing 3 |
| SOCS | Suppressor of cytokine signaling |
| TAZ | Transcriptional coactivator with PDZ- binding motif |
| TEAD | TAZ/TEA domain |
| TGF-β | Transforming growth factor-β |
| T2DM | Type2 diabetes mellitus |
| TM6SF2 | Transmembrane 6 superfamily member 2 |
| TNF | Tumor necrosis factor |
| VCTE | Vibration-controlled transient elastography |
| WFA+-M2bp | Wisteria floribunda agglutinin-positive Mac-2-binding protein |
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Wong, V.W.; Chan, W.K.; Chitturi, S.; Chawla, Y.; Dan, Y.Y.; Duseja, A.; Fan, J.; Goh, K.L.; Hamaguchi, M.; Hashimoto, E.; et al. Asia-Pacific Working Party on Non-alcoholic Fatty Liver Disease guidelines 2017-Part 1: Definition, risk factors and assessment. J. Gastroenterol. Hepatol. 2018, 33, 70–85. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander, S.B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs. nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Thuluvath, P.J.; Kantsevoy, S.; Thuluvath, A.J.; Savva, Y. Is cryptogenic cirrhosis different from NASH cirrhosis? J. Hepatol. 2018, 68, 519–525. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quinones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457. [Google Scholar] [CrossRef]
- Cui, J.; Chen, C.H.; Lo, M.T.; Schork, N.; Bettencourt, R.; Gonzalez, M.P.; Bhatt, A.; Hooker, J.; Shaffer, K.; Nelson, K.E.; et al. Shared genetic effects between hepatic steatosis and fibrosis: A prospective twin study. Hepatology 2016, 64, 1547–1558. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Overi, D.; Carpino, G.; Franchitto, A.; Onori, P.; Gaudio, E. Hepatocyte injury and hepatic stem cell niche in the progression of non-alcoholic steatohepatitis. Cells 2020, 9, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Tordjman, J.; Clement, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Vily-Petit, J.; Soty-Roca, M.; Silva, M.; Raffin, M.; Gautier-Stein, A.; Rajas, F.; Mithieux, G. Intestinal gluconeogenesis prevents obesity-linked liver steatosis and non-alcoholic fatty liver disease. Gut 2020. [Google Scholar] [CrossRef]
- Soty, M.; Gautier-Stein, A.; Rajas, F.; Mithieux, G. Gut-Brain Glucose Signaling in Energy Homeostasis. Cell Metab. 2017, 25, 1231–1242. [Google Scholar] [CrossRef] [Green Version]
- Rhee, E.J. Nonalcoholic fatty liver disease and diabetes: An epidemiological perspective. Endocrinol. Metab. 2019, 34, 226–233. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in NASH. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Eckstein, S.S.; Weigert, C.; Lehmann, R. Divergent Roles of IRS (Insulin Receptor Substrate) 1 and 2 in Liver and Skeletal Muscle. Curr. Med. Chem. 2017, 24, 1827–1852. [Google Scholar] [CrossRef]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef]
- Freeman, A.M.; Pennings, N. Insulin Resistance; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Rametta, R.; Mozzi, E.; Dongiovanni, P.; Motta, B.M.; Milano, M.; Roviaro, G.; Fargion, S.; Valenti, L. Increased insulin receptor substrate 2 expression is associated with steatohepatitis and altered lipid metabolism in obese subjects. Int. J. Obes. 2013, 37, 986–992. [Google Scholar] [CrossRef] [Green Version]
- Honma, M.; Sawada, S.; Ueno, Y.; Murakami, K.; Yamada, T.; Gao, J.; Kodama, S.; Izumi, T.; Takahashi, K.; Tsukita, S.; et al. Selective insulin resistance with differential expressions of IRS-1 and IRS-2 in human NAFLD livers. Int. J. Obes. 2018, 42, 1544–1555. [Google Scholar] [CrossRef] [Green Version]
- Enooku, K.; Kondo, M.; Fujiwara, N.; Sasako, T.; Shibahara, J.; Kado, A.; Okushin, K.; Fujinaga, H.; Tsutsumi, T.; Nakagomi, R. Hepatic IRS1 and ss-catenin expression is associated with histological progression and overt diabetes emergence in NAFLD patients. J. Gastroenterol. 2018, 53, 1261–1275. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Cipolla, U.; Cassader, M.; Pinach, S.; Saba, F.; De Michieli, F.; Paschetta, E.; Bongiovanni, D.; Framarin, L.; Leone, N.; et al. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J. Lipid Res. 2017, 58, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef]
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K.; et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. Elife 2019, 8, e49882. [Google Scholar] [CrossRef]
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine 2020, 52, 102658. [Google Scholar] [CrossRef] [Green Version]
- Umano, G.R.; Caprio, S.; Di Sessa, A.; Chalasani, N.; Dykas, D.J.; Pierpont, B.; Bale, A.E.; Santoro, N. The rs626283 variant in the MBOAT7 gene is associated with insulin resistance and fatty liver in Caucasian obese youth. Am. J. Gastroenterol. 2018, 113, 376–383. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Investig. 2003, 83, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar] [CrossRef]
- Huebener, P.; Pradere, J.P.; Hernandez, C.; Gwak, G.Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Schwabe, R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2015, 125, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; De Minicis, S.; Gwak, G.Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Investig. 2009, 119, 1858–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berres, M.L.; Koenen, R.R.; Rueland, A.; Zaldivar, M.M.; Heinrichs, D.; Sahin, H.; Schmitz, P.; Streetz, K.L.; Berg, T.; Gassler, N.; et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Investig. 2010, 120, 4129–4140. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults with Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zheng, Z.; Caviglia, J.M.; Corey, K.E.; Herfel, T.M.; Cai, B.; Masia, R.; Chung, R.T.; Lefkowitch, J.H.; Schwabe, R.F.; et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016, 24, 848–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Kim, K.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, 468. [Google Scholar] [CrossRef] [PubMed]
- Pajvani, U.B.; Shawber, C.J.; Samuel, V.T.; Birkenfeld, A.L.; Shulman, G.I.; Kitajewski, J.; Accili, D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med. 2011, 17, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Rametta, R.; Meroni, M.; Valenti, L. The role of insulin resistance in nonalcoholic steatohepatitis and liver disease development—A potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 229–242. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Baselli, G.A.; Bassani, G.A.; Rametta, R.; Pietrelli, A.; Maggioni, M.; Facciotti, F.; Trunzo, V.; Badiali, S.; et al. Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin. Sci. 2017, 131, 1301–1315. [Google Scholar] [CrossRef] [Green Version]
- Ranjbar, G.; Mikhailidis, D.P.; Sahebkar, A. Effects of newer antidiabetic drugs on nonalcoholic fatty liver and steatohepatitis: Think out of the box! Metabolism 2019, 101, 154001. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017, 16, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar] [PubMed]
- Svegliati-Baroni, G.; Ridolfi, F.; Di Sario, A.; Casini, A.; Marucci, L.; Gaggiotti, G.; Orlandoni, P.; Macarri, G.; Perego, L.; Benedetti, A.; et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: Differential effects on signal transduction pathways. Hepatology 1999, 29, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Takamura, T.; Kurita, S.; Matsuzawa, N.; Kita, Y.; Uno, M.; Akahori, H.; Misu, H.; Sakurai, M.; Zen, Y.; et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology 2007, 132, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Villar-Lorenzo, A.; Rada, P.; Rey, E.; Maranon, P.; Arroba, A.I.; Santamaria, B.; Saiz, J.; Ruperez, F.J.; Barbas, C.; Garcia-Monzon, C.; et al. Insulin receptor substrate 2 (IRS2) deficiency delays liver fibrosis associated with cholestatic injury. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Zhang, Q.; Sun, W.; Xin, Y.; Zhang, Z.; Tan, Y.; Zhou, S.; Zhang, C.; Cai, L.; Lu, X.; et al. Zinc treatment prevents type 1 diabetes-induced hepatic oxidative damage, endoplasmic reticulum stress, and cell death, and even prevents possible steatohepatitis in the OVE26 mouse model: Important role of metallothionein. Toxicol. Lett. 2015, 233, 114–124. [Google Scholar] [CrossRef]
- Kiss, K.; Regos, E.; Rada, K.; Firneisz, G.; Baghy, K.; Kovalszky, I. Chronic Hyperglycaemia Induced Alterations of Hepatic Stellate Cells Differ from the Effect of TGFB1, and Point toward Metabolic Stress. Pathol. Oncol. Res. 2018, 26, 291–299. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Y.; Zuo, L.; Wang, Y.; Huang, Y. ASIC1a promotes high glucose and PDGF-induced hepatic stellate cell activation by inducing autophagy through CaMKKbeta/ERK signaling pathway. Toxicol. Lett. 2019, 300, 1–9. [Google Scholar] [CrossRef]
- Fujii, H.; Enomoto, M.; Fukushima, W.; Tamori, A.; Sakaguchi, H.; Kawada, N. Applicability of BARD score to Japanese patients with NAFLD. Gut 2009, 58, 1566–1567. [Google Scholar] [CrossRef]
- Nakahara, T.; Hyogo, H.; Yoneda, M.; Sumida, Y.; Eguchi, Y.; Fujii, H.; Ono, M.; Kawaguchi, T.; Imajo, K.; Aikata, H.; et al. Type 2 diabetes mellitus is associated with the fibrosis severity in patients with nonalcoholic fatty liver disease in a large retrospective cohort of Japanese patients. J. Gastroenterol. 2014, 49, 1477–1484. [Google Scholar] [CrossRef] [Green Version]
- Kwok, R.; Choi, K.C.; Wong, G.L.; Zhang, Y.; Chan, H.L.; Luk, A.O.; Shu, S.S.; Chan, A.W.; Yeung, M.W.; Chan, J.C.; et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: A prospective cohort study. Gut 2016, 65, 1359–1368. [Google Scholar] [CrossRef]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Mosca, A.; Comparcola, D.; Romito, I.; Mantovani, A.; Nobili, V.; Byrne, C.D.; Alisi, A.; Targher, G. Plasma N-terminal propeptide of type III procollagen accurately predicts liver fibrosis severity in children with non-alcoholic fatty liver disease. Liver Int. 2019, 39, 2317–2329. [Google Scholar] [CrossRef] [PubMed]
- Daniels, S.J.; Leeming, D.J.; Eslam, M.; Hashem, A.M.; Nielsen, M.J.; Krag, A.; Karsdal, M.A.; Grove, J.I.; Neil Guha, I.; Kawaguchi, T.; et al. ADAPT: An Algorithm Incorporating PRO-C3 Accurately Identifies Patients With NAFLD and Advanced Fibrosis. Hepatology 2019, 69, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castera, L.; Friedrich-Rust, M.; Loomba, R. Noninvasive Assessment of Liver Disease in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1264–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wai, C.T.; Greenson, J.K.; Fontana, R.J.; Kalbfleisch, J.D.; Marrero, J.A.; Conjeevaram, H.S.; Lok, A.S. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003, 38, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, R.K.; Lissen, E.; Clumeck, N.; Sola, R.; Correa, M.C.; Montaner, J.; S Sulkowski, M.; Torriani, F.J.; Dieterich, D.T.; Thomas, D.L.; et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006, 43, 1317–1325. [Google Scholar] [CrossRef]
- Yoneda, M.; Fujii, H.; Sumida, Y.; Hyogo, H.; Itoh, Y.; Ono, M.; Eguchi, Y.; Suzuki, Y.; Aoki, N.; Kanemasa, K.; et al. Platelet count for predicting fibrosis in nonalcoholic fatty liver disease. J. Gastroenterol. 2011, 46, 1300–1306. [Google Scholar] [CrossRef]
- Shirab, K.; Bekki, Y.; Gantumur, D.; Araki, K.; Ishii, N.; Kuno, A.; Narimatsu, H.; Mizokami, M. Mac-2 binding protein glycan isomer (M2BPGi) is a new serum biomarker for assessing liver fibrosis: More than a biomarker of liver fibrosis. J. Gastroenterol. 2018, 53, 819–826. [Google Scholar] [CrossRef]
- Abe, M.; Miyake, T.; Kuno, A.; Imai, Y.; Sawai, Y.; Hino, K.; Hara, Y.; Hige, S.; Sakamoto, M.; Yamada, G.; et al. Association between Wisteria floribunda agglutinin-positive Mac-2 binding protein and the fibrosis stage of non-alcoholic fatty liver disease. J. Gastroenterol. 2015, 50, 776–784. [Google Scholar] [CrossRef]
- Nishikawa, H.; Enomoto, H.; Iwata, Y.; Kishino, K.; Shimono, Y.; Hasegawa, K.; Nakano, C.; Takata, R.; Yoh, K.; Nishimura, T.; et al. Clinical significance of serum Wisteria floribunda agglutinin positive Mac-2-binding protein level in non-alcoholic steatohepatitis. Hepatol. Res. 2016, 46, 1194–1202. [Google Scholar] [CrossRef]
- Kamada, Y.; Fujii, H.; Fujii, H.; Sawai, Y.; Doi, Y.; Uozumi, N.; Mizutani, K.; Akita, M.; Sato, M.; Kida, S.; et al. Serum Mac-2 binding protein levels as a novel diagnostic biomarker for prediction of disease severity and nonalcoholic steatohepatitis. Proteom. Clin. Appl. 2013, 7, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Ono, M.; Hyogo, H.; Fujii, H.; Sumida, Y.; Mori, K.; Tanaka, S.; Yamada, M.; Akita, M.; Mizutani, K.; et al. A novel noninvasive diagnostic method for nonalcoholic steatohepatitis using two glycobiomarkers. Hepatology 2015, 62, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Ono, M.; Hyogo, H.; Fujii, H.; Sumida, Y.; Yamada, M.; Mori, K.; Tanaka, S.; Maekawa, T.; Ebisutani, Y.; et al. Use of Mac-2 binding protein as a biomarker for nonalcoholic fatty liver disease diagnosis. Hepatol. Commun. 2017, 1, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Imajo, K.; Kobayashi, T.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Yoneda, M.; Kobayashi, N.; Saito, S.; Nakajima, A. Autotaxin is a valuable biomarker for the prediction of liver fibrosis in patients with non-alcoholic fatty liver disease. Hepatol. Res. 2019, 49, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Kobayashi, M.; Kumada, H.; Enooku, K.; Koike, K.; Kurano, M.; Sato, M.; Nojiri, T.; Kobayashi, T.; Ohkawa, R.; et al. Performance of autotaxin as a serum marker for liver fibrosis. Ann. Clin. Biochem. 2018, 55, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, N.; Umemura, T.; Kimura, T.; Tanaka, N.; Sugiura, A.; Yamazaki, T.; Joshita, S.; Komatsu, M.; Usami, Y.; Sano, K.; et al. Serum autotaxin levels are correlated with hepatic fibrosis and ballooning in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2018, 24, 1239–1249. [Google Scholar] [CrossRef]
- Okanoue, T.; Ebise, H.; Kai, T.; Mizuno, M.; Shima, T.; Ichihara, J.; Aoki, M. A simple scoring system using type IV collagen 7S and aspartate aminotransferase for diagnosing nonalcoholic steatohepatitis and related fibrosis. J. Gastroenterol. 2018, 53, 129–139. [Google Scholar] [CrossRef]
- Singh, A.; Gosai, F.; Siddiqui, M.T.; Gupta, M.; Lopez, R.; Lawitz, E.; Poordad, F.; Carey, W.; McCullough, A.; Alkhouri, N. Accuracy of Noninvasive Fibrosis Scores to Detect Advanced Fibrosis in Patients With Type-2 Diabetes With Biopsy-proven Nonalcoholic Fatty Liver Disease. J. Clin. Gastroenterol. 2020. [Google Scholar] [CrossRef]
- Alkayyali, T.; Qutranji, L.; Kaya, E.; Bakir, A.; Yilmaz, Y. Clinical utility of noninvasive scores in assessing advanced hepatic fibrosis in patients with type 2 diabetes mellitus: A study in biopsy-proven non-alcoholic fatty liver disease. Acta Diabetol. 2020. [Google Scholar] [CrossRef]
- Patel, K.; Sebastiani, G. Limitations of non-invasive tests for assessment of liver fibrosis. JHEP Rep. 2020, 2, 100067. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R. Role of imaging-based biomarkers in NAFLD: Recent advances in clinical application and future research directions. J. Hepatol. 2018, 68, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Tapper, E.B.; Loomba, R. Noninvasive imaging biomarker assessment of liver fibrosis by elastography in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 274–282. [Google Scholar] [CrossRef]
- Friedrich-Rust, M.; Poynard, T.; Castera, L. Critical comparison of elastography methods to assess chronic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 402–411. [Google Scholar] [CrossRef]
- Eddowes, P.J.; Sasso, M.; Allison, M.; Tsochatzis, E.; Anstee, Q.M.; Sheridan, D.; Guha, I.N.; Cobbold, J.F.; Deeks, J.J.; Paradis, V.; et al. Accuracy of FibroScan Controlled Attenuation Parameter and Liver Stiffness Measurement in Assessing Steatosis and Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1717–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeda, S.; Takahashi, H.; Imajo, K.; Seko, Y.; Ogawa, Y.; Moriguchi, M.; Yoneda, M.; Anzai, K.; Aishima, S.; Kage, M. Accuracy of liver stiffness measurement and controlled attenuation parameter using FibroScan((R)) M/XL probes to diagnose liver fibrosis and steatosis in patients with nonalcoholic fatty liver disease: A multicenter prospective study. J. Gastroenterol. 2020, 55, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Roulot, D.; Roudot-Thoraval, F.; NKontchou, G.; Kouacou, N.; Costes, J.L.; Elourimi, G.; Le Clesiau, H.; Ziol, M.; Beaugrand, M. Concomitant screening for liver fibrosis and steatosis in French type 2 diabetic patients using Fibroscan. Liver Int. 2017, 37, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Koehler, E.M.; Plompen, E.P.; Schouten, J.N.; Hansen, B.E.; Darwish Murad, S.; Taimr, P.; Leebeek, F.W.; Hofman, A.; Stricker, B.H.; Castera, L. Presence of diabetes mellitus and steatosis is associated with liver stiffness in a general population: The Rotterdam study. Hepatology 2016, 63, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imajo, K.; Kessoku, T.; Honda, Y.; Tomeno, W.; Ogawa, Y.; Mawatari, H.; Fujita, K.; Yoneda, M.; Taguri, M.; Hyogo, H.; et al. Magnetic Resonance Imaging More Accurately Classifies Steatosis and Fibrosis in Patients With Nonalcoholic Fatty Liver Disease Than Transient Elastography. Gastroenterology 2016, 150, 626–637. [Google Scholar] [CrossRef] [Green Version]
- Park, C.C.; Nguyen, P.; Hernandez, C.; Bettencourt, R.; Ramirez, K.; Fortney, L.; Hooker, J.; Sy, E.; Savides, M.T.; Alquiraish, M.H.; et al. Magnetic Resonance Elastography vs. Transient Elastography in Detection of Fibrosis and Noninvasive Measurement of Steatosis in Patients With Biopsy-Proven Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 598–607. [Google Scholar] [CrossRef] [Green Version]
- Doycheva, I.; Cui, J.; Nguyen, P.; Costa, E.A.; Hooker, J.; Hofflich, H.; Bettencourt, R.; Brouha, S.; Sirlin, C.B.; Loomba, R. Non-invasive screening of diabetics in primary care for NAFLD and advanced fibrosis by MRI and MRE. Aliment. Pharmacol. Ther. 2016, 43, 83–95. [Google Scholar] [CrossRef]
- Kang, K.A.; Jun, D.W.; Kim, M.S.; Kwon, H.J.; Nguyen, M.H. Prevalence of significant hepatic fibrosis using magnetic resonance elastography in a health check-up clinic population. Aliment. Pharmacol. Ther. 2020, 51, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Corcuera-Solano, I.; Lo, G.; Esses, S.; Liao, J.; Besa, C.; Chen, N.; Abraham, G.; Fung, M.; Babb, J.S.; et al. Technical Failure of MR Elastography Examinations of the Liver: Experience from a Large Single-Center Study. Radiology 2017, 284, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Imajo, K.; Takahashi, H.; Ogawa, Y.; Eguchi, Y.; Sumida, Y.; Yoneda, M.; Kawanaka, M.; Saito, S.; Tokushige, K.; et al. Clinical strategy of diagnosing and following patients with nonalcoholic fatty liver disease based on invasive and noninvasive methods. J. Gastroenterol. 2018, 53, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, M.; Imajo, K.; Nakajima, A. Non-Invasive Diagnosis of Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2018, 113, 1409–1411. [Google Scholar] [CrossRef]
- Jung, K.Y.; Cho, S.Y.; Kim, H.J.; Kim, S.B.; Song, I.H. Nonalcoholic steatohepatitis associated with metabolic syndrome: Relationship to insulin resistance and liver histology. J. Clin. Gastroenterol. 2014, 48, 883–888. [Google Scholar] [CrossRef]
- Kessoku, T.; Yoneda, M.; Sumida, Y.; Eguchi, Y.; Fujii, H.; Hyogo, H.; Ono, M.; Kawaguchi, T.; Nakajima, A. Japan Study Group of NAFLD. Insulin resistance correlated with the severity of liver histology in Japanese NAFLD patients: A multicenter retrospective study. J. Clin. Gastroenterol. 2015, 49, 169–170. [Google Scholar] [CrossRef]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of non-alcoholic steatohepatitis and its individual histological features: Insulin resistance, serum uric acid, metabolic syndrome, alanine aminotransferase and serum total cholesterol are a clue to pathogenesis and candidate targets for treatment. Hepatol. Res. 2016, 46, 1074–1087. [Google Scholar]
- Fujii, H.; Imajo, K.; Yoneda, M.; Nakahara, T.; Hyogo, H.; Takahashi, H.; Hara, T.; Tanaka, S.; Sumida, Y.; Eguchi, Y.; et al. HOMA-IR: An independent predictor of advanced liver fibrosis in nondiabetic non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2019, 34, 1390–1395. [Google Scholar] [CrossRef]
- Alkhouri, N.; Poordad, F.; Lawitz, E. Management of nonalcoholic fatty liver disease: Lessons learned from type 2 diabetes. Hepatol. Commun. 2018, 2, 778–785. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M.; Tokushige, K.; Kawanaka, M.; Fujii, H.; Yoneda, M.; Imajo, K.; Takahashi, H.; Eguchi, Y.; Ono, M.; et al. Antidiabetic Therapy in the Treatment of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 1907. [Google Scholar] [CrossRef] [Green Version]
- Golabi, P.; Rhea, L.; Henry, L.; Younossi, Z.M. Hepatocellular carcinoma and non-alcoholic fatty liver disease. Hepatol. Int. 2019, 13, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Tokushige, K.; Hyogo, H.; Nakajima, T.; Ono, M.; Kawaguchi, T.; Honda, K.; Eguchi, Y.; Nozaki, Y.; Kawanaka, M.; Tanaka, S. Hepatocellular carcinoma in Japanese patients with nonalcoholic fatty liver disease and alcoholic liver disease: Multicenter survey. J. Gastroenterol. 2016, 51, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 2019, 17, 95. [Google Scholar] [CrossRef]
- Yang, J.D.; Ahmed, F.; Mara, K.C.; Addissie, B.D.; Allen, A.M.; Gores, G.J.; Roberts, L.R. Diabetes Is Associated With Increased Risk of Hepatocellular Carcinoma in Patients With Cirrhosis From Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 907–916. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Gawrieh, S.; Dakhoul, L.; Miller, E.; Scanga, A.; deLemos, A.; Kettler, C.; Burney, H.; Liu, H.; Abu-Sbeih, H.; Chalasani, N.; et al. Characteristics, aetiologies and trends of hepatocellular carcinoma in patients without cirrhosis: A United States multicentre study. Aliment. Pharmacol. Ther. 2019, 50, 809–821. [Google Scholar] [CrossRef]
- Bengtsson, B.; Stal, P.; Wahlin, S.; Bjorkstrom, N.K.; Hagstrom, H. Characteristics and outcome of hepatocellular carcinoma in patients with NAFLD without cirrhosis. Liver Int. 2019, 39, 1098–1108. [Google Scholar] [CrossRef]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujii, H.; Kawada, N.; Japan Study Group of NAFLD (JSG-NAFLD). The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113863
Fujii H, Kawada N, Japan Study Group of NAFLD (JSG-NAFLD). The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2020; 21(11):3863. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113863
Chicago/Turabian StyleFujii, Hideki, Norifumi Kawada, and Japan Study Group of NAFLD (JSG-NAFLD). 2020. "The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 21, no. 11: 3863. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113863