In Vitro Effects of Ligand Bias on Primate Mu Opioid Receptor Downstream Signaling


Abstract
:1. Introduction
2. Results
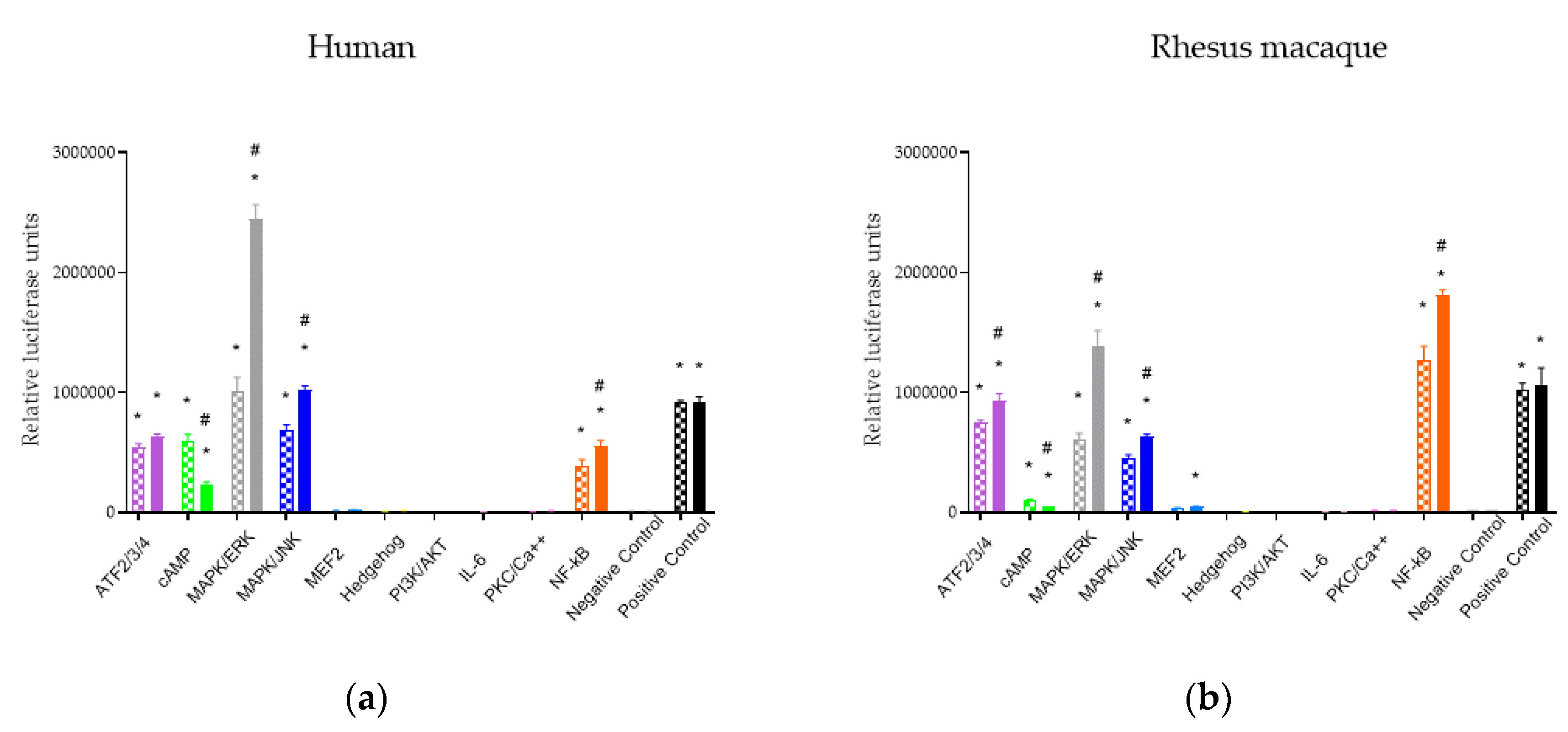
2.1. Downstream Signaling Pathway Responses to MOR Activation
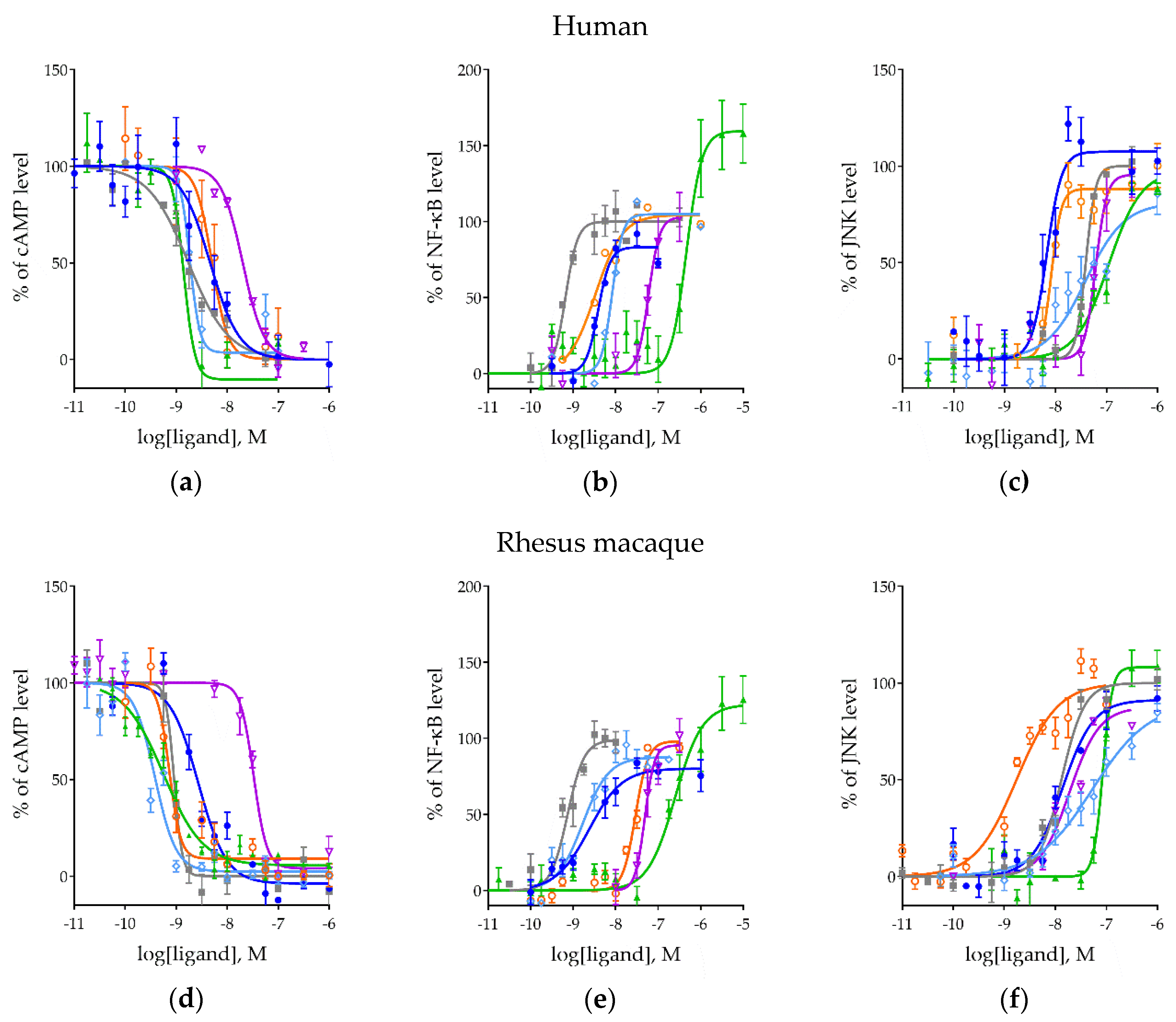
2.2. Effects of MOR Activation on NF-ĸB, MAPK/JNK, and cAMP Pathways Across Ligands
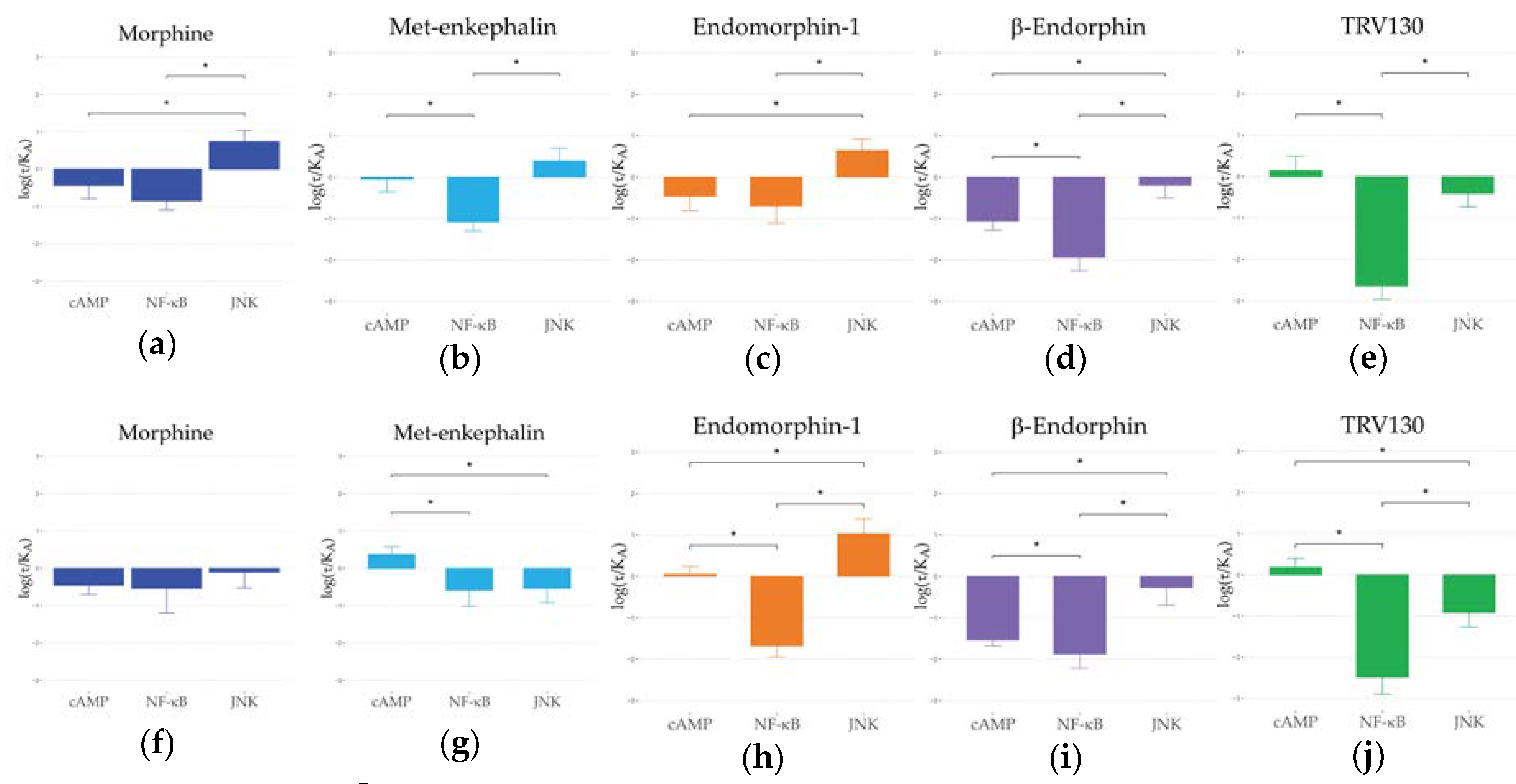
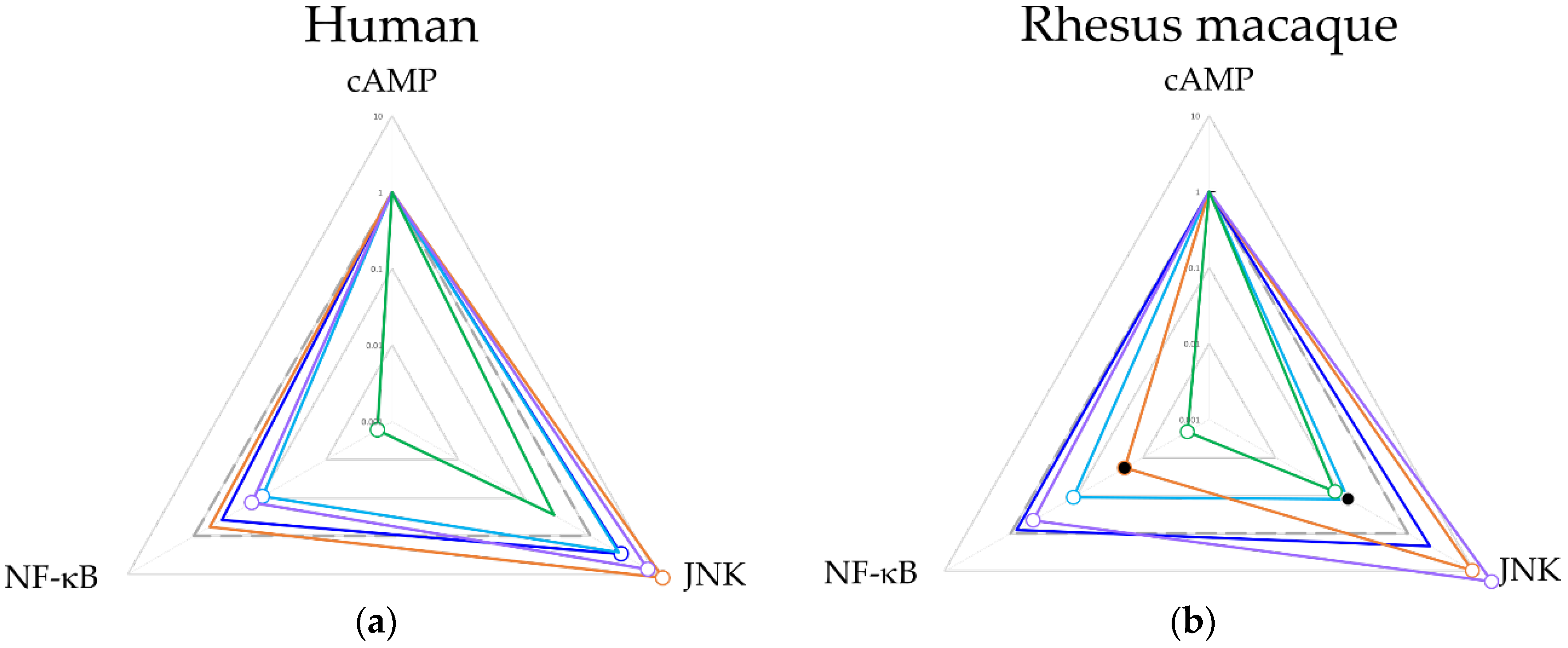
2.3. Ligand Bias and Species Differences Exist for MORs
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cell Lines
4.3. GPCR Signaling Reporter Array
4.4. Drugs
4.5. Concentration Response Determinations
4.6. Data Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MOR | µ opioid receptor |
| NHP | nonhuman primate |
| GPCR | G-protein coupled receptor |
| cAMP | cyclic adenosine 5′-monophosphate |
| NF-ĸB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| MAPK | mitogen-activated protein kinase |
| JNK | Jun N-terminal kinase |
| ERK | extracellular-signal-regulated kinase |
| ATF | activating transcription factor |
| DAMGO | [D-Ala2-MePhe4-Gly-ol]-enkephalin |
| PMA | phorbol 12-myristate 13-acetate |
| TNF- α | tumor necrosis factor α |
| GFP | green fluorescent protein |
References
- Kenakin, T. Inverse, protean, and ligand-selective agonism: Matters of receptor conformation. FASEB J. 2001, 15, 598–611. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. Mu-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Walker, J.K.; Bohn, L.M. Morphine side effects in β-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [Green Version]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Burke, D.S. Forecasting the opioid epidemic. Science 2016, 354, 529. [Google Scholar] [CrossRef] [Green Version]
- Violin, J.D.; Crombie, A.L.; Soergel, D.G.; Lark, M.W. Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharmacol. Sci. 2014, 35, 308–316. [Google Scholar] [CrossRef]
- Siuda, E.R.; Carr, R., 3rd; Rominger, D.H.; Violin, J.D. Biased mu-opioid receptor ligands: A promising new generation of pain therapeutics. Curr. Opin. Pharmacol. 2017, 32, 77–84. [Google Scholar] [CrossRef]
- Singla, N.; Minkowitz, H.S.; Soergel, D.G.; Burt, D.A.; Subach, R.A.; Salamea, M.Y.; Fossler, M.J.; Skobieranda, F. A randomized, Phase IIb study investigating oliceridine (TRV130), a novel micro-receptor G-protein pathway selective (mu-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J. Pain Res. 2017, 10, 2413–2424. [Google Scholar] [CrossRef] [Green Version]
- Singla, N.K.; Skobieranda, F.; Soergel, D.G.; Salamea, M.; Burt, D.A.; Demitrack, M.A.; Viscusi, E.R. APOLLO-2: A Randomized, Placebo and Active-Controlled Phase III Study Investigating Oliceridine (TRV130), a G Protein-Biased Ligand at the mu-Opioid Receptor, for Management of Moderate to Severe Acute Pain Following Abdominoplasty. Pain Pract. 2019, 19, 715–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Viscusi, E.R.; Skobieranda, F.; Soergel, D.G.; Cook, E.; Burt, D.A.; Singla, N. APOLLO-1: A randomized placebo and active-controlled phase III study investigating oliceridine (TRV130), a G protein-biased ligand at the micro-opioid receptor, for management of moderate-to-severe acute pain following bunionectomy. J. Pain Res. 2019, 12, 927–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viscusi, E.R.; Webster, L.; Kuss, M.; Daniels, S.; Bolognese, J.A.; Zuckerman, S.; Soergel, D.G.; Subach, R.A.; Cook, E.; Skobieranda, F. A randomized, phase 2 study investigating TRV130, a biased ligand of the mu-opioid receptor, for the intravenous treatment of acute pain. Pain 2016, 157, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Collins, F.S. The role of science in addressing the opioid crisis. N. Engl. J. Med. 2017, 377, 391–394. [Google Scholar] [CrossRef]
- Madhusoodanan, J. Inner workings: Safer opioids may be on the horizon, but mitigating addiction is a long shot. Proc. Natl. Acad. Sci. USA 2018, 115, 8229–8231. [Google Scholar] [CrossRef] [Green Version]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmiedel, F.; et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020, 13, eaaz3140. [Google Scholar] [CrossRef]
- Kliewer, A.; Gillis, A.; Hill, R.; Schmidel, F.; Bailey, C.; Kelly, E.; Henderson, G.; Christie, M.J.; Schulz, S. Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 2019, 10, 367. [Google Scholar] [CrossRef]
- Altarifi, A.A.; David, B.; Muchhala, K.H.; Blough, B.E.; Akbarali, H.; Negus, S.S. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 2017, 31, 730–739. [Google Scholar] [CrossRef]
- Zamarripa, C.A.; Edwards, S.R.; Qureshi, H.N.; Yi, J.N.; Blough, B.E.; Freeman, K.B. The G-protein biased mu-opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug Alcohol Depend. 2018, 192, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Schwienteck, K.L.; Faunce, K.E.; Rice, K.C.; Obeng, S.; Zhang, Y.; Blough, B.E.; Grim, T.W.; Negus, S.S.; Banks, M.L. Effectiveness comparisons of G-protein biased and unbiased mu opioid receptor ligands in warm water tail-withdrawal and drug discrimination in male and female rats. Neuropharmacology 2019, 150, 200–209. [Google Scholar] [CrossRef]
- Klein Herenbrink, C.; Sykes, D.A.; Donthamsetti, P.; Canals, M.; Coudrat, T.; Shonberg, J.; Scammells, P.J.; Capuano, B.; Sexton, P.M.; Charlton, S.J.; et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 2016, 7, 10842. [Google Scholar] [CrossRef] [PubMed]
- Appleton, K.M.; Luttrell, L.M. Emergent biological properties of arrestin pathway-selective biased agonism. J. Recept. Signal Transduct. 2013, 33, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Schattauer, S.S.; Kuhar, J.R.; Song, A.; Chavkin, C. Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell. Signal. 2017, 32, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattauer, S.S.; Miyatake, M.; Shankar, H.; Zietz, C.; Levin, J.R.; Liu-Chen, L.Y.; Gurevich, V.V.; Rieder, M.J.; Chavkin, C. Ligand directed signaling differences between rodent and human kappa-opioid receptors. J. Biol. Chem. 2012, 287, 41595–41607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 2017, 171, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Negus, S.S.; Banks, M.L. Medications development for opioid abuse. Cold Spring Harb. Perspect. Med. 2013, 3, a012104. [Google Scholar] [CrossRef] [Green Version]
- Banks, M.L.; Czoty, P.W.; Negus, S.S. Utility of nonhuman primates in substance use disorders research. ILAR J. 2017, 58, 202–215. [Google Scholar] [CrossRef]
- Weerts, E.M.; Fantegrossi, W.E.; Goodwin, A.K. The value of nonhuman primates in drug abuse research. Exp. Clin. Psychopharmacol. 2007, 15, 309–327. [Google Scholar] [CrossRef]
- Vallender, E.J.; Priddy, C.M.; Chen, G.L.; Miller, G.M. Human expression variation in the mu-opioid receptor is paralleled in rhesus macaque. Behav. Genet. 2008, 38, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Vallender, E.J.; Ruedi-Bettschen, D.; Miller, G.M.; Platt, D.M. A pharmacogenetic model of naltrexone-induced attenuation of alcohol consumption in rhesus monkeys. Drug Alcohol Depend. 2010, 109, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, D.M.; Rowlett, J.K. Nonhuman primate models of drug and alcohol addiction. In Nonhuman Primates in Biomedical Research, 2nd ed.; Abee, C.R., Mansfield, K., Tardif, S., Morris, T., Eds.; Academic Press: Boston, MA, USA, 2012; pp. 817–839. [Google Scholar]
- Freeman, K.B.; Naylor, J.E.; Prisinzano, T.E.; Woolverton, W.L. Assessment of the kappa opioid agonist, salvinorin A, as a punisher of drug self-administration in monkeys. Psychopharmacology (Berlin) 2014, 231, 2751–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, M.C.; Husbands, S.M. Effects of atypical kappa-opioid receptor agonists on intrathecal morphine-induced itch and analgesia in primates. J. Pharmacol. Exp. Ther. 2009, 328, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Negus, S.S.; Schrode, K.; Stevenson, G.W. Micro/kappa opioid interactions in rhesus monkeys: Implications for analgesia and abuse liability. Exp. Clin. Psychopharmacol. 2008, 16, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Li, Z.; Cvijic, M.E.; Krause, C.; Zhang, L.; Sum, C.S. Measurement of β-Arrestin Recruitment for GPCR Targets. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Caaveiro, J.M.M., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Peterson, Y.K.; Luttrell, L.M. The diverse roles of arrestin scaffolds in G protein-coupled receptor signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S.K.; Lefkowitz, R.J. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Marcus, D.J.; Zee, M.; Hughes, A.; Yuill, M.B.; Hohmann, A.G.; Mackie, K.; Guindon, J.; Morgan, D.J. Tolerance to the antinociceptive effects of chronic morphine requires c-Jun N-terminal kinase. Mol. Pain 2015, 11, s12990-015. [Google Scholar] [CrossRef] [Green Version]
- Melief, E.J.; Miyatake, M.; Bruchas, M.R.; Chavkin, C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 11608–11613. [Google Scholar] [CrossRef] [Green Version]
- Black, J.W.; Leff, P. Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 1983, 220, 141–162. [Google Scholar]
- Hawes, B.E.; Graziano, M.P.; Lambert, D.G. Cellular actions of nociceptin: Transduction mechanisms. Peptides 2000, 21, 961–967. [Google Scholar] [CrossRef]
- Kennedy, N.M.; Schmid, C.L.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Chen, Y.T.; Cameron, M.D.; Bohn, L.M.; Bannister, T.D. Optimization of a Series of Mu Opioid Receptor (MOR) Agonists with High G Protein Signaling Bias. J. Med. Chem. 2018, 61, 8895–8907. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, E.A.; Collins, M.K. Dynamic interaction between the dual specificity phosphatase MKP7 and the JNK3 scaffold protein β-arrestin 2. J. Biol. Chem. 2005, 280, 25651–25658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, J.; Rivero, G.; Baptist, M.; Llorente, J.; Al-Sabah, S.; Krasel, C.; Dewey, W.L.; Bailey, C.P.; Rosethorne, E.M.; Charlton, S.J.; et al. mu-opioid receptors: Correlation of agonist efficacy for signalling with ability to activate internalization. Mol. Pharmacol. 2010, 78, 756–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, G.L.; Kelly, E.; Christopoulos, A.; Canals, M. Novel GPCR paradigms at the mu-opioid receptor. Br. J. Pharmacol. 2015, 172, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Rivero, G.; Llorente, J.; McPherson, J.; Cooke, A.; Mundell, S.J.; McArdle, C.A.; Rosethorne, E.M.; Charlton, S.J.; Krasel, C.; Bailey, C.P.; et al. Endomorphin-2: A biased agonist at the mu-opioid receptor. Mol. Pharmacol. 2012, 82, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of mu-opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef] [Green Version]
- Thompson, G.L.; Lane, J.R.; Coudrat, T.; Sexton, P.M.; Christopoulos, A.; Canals, M. Biased Agonism of Endogenous Opioid Peptides at the mu-Opioid Receptor. Mol. Pharmacol. 2015, 88, 335–346. [Google Scholar] [CrossRef] [Green Version]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.T.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Nennig, S.E.; Schank, J.R. The Role of NFkB in drug addiction: Beyond inflammation. Alcohol Alcohol. 2017, 52, 172–179. [Google Scholar] [CrossRef]
- Chen, Y.L.; Law, P.Y.; Loh, H.H. Nuclear factor kappaB signaling in opioid functions and receptor gene expression. J. Neuroimmune Pharmacol. 2006, 1, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, J.; Borner, C.; Giannini, E.; Hollt, V. The role of nuclear factor kappaB in tumor necrosis factor-regulated transcription of the human mu-opioid receptor gene. Mol. Pharmacol. 2003, 64, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Douglas, S.D.; Commons, K.G.; Pleasure, D.E.; Lai, J.; Ho, C.; Bannerman, P.; Williams, M.; Ho, W. A non-peptide substance P antagonist (CP-96,345) inhibits morphine-induced NF-kappa B promoter activation in human NT2-N neurons. J. Neurosci. Res. 2004, 75, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sheng, W.S.; Lokensgard, J.R.; Peterson, P.K. Morphine induces apoptosis of human microglia and neurons. Neuropharmacology 2002, 42, 829–836. [Google Scholar] [CrossRef]
- Sweeney, C.G.; Rando, J.M.; Panas, H.N.; Miller, G.M.; Platt, D.M.; Vallender, E.J. Convergent balancing selection on the mu-opioid receptor in primates. Mol. Biol. Evol. 2017, 34, 1629–1643. [Google Scholar] [CrossRef] [Green Version]
- Standifer, K.M.; Pasternak, G.W. G proteins and opioid receptor-mediated signalling. Cell Signal. 1997, 9, 237–248. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Human | Rhesus Macaque | ||
|---|---|---|---|---|
| LogEC50 | Emax | LogEC50 | Emax | |
| cAMP | ||||
| DAMGO | −8.76 ± 0.07 | 100.0 ± 4.09 | −9.04 ± 0.03 | 100.2 ± 6.06 |
| morphine | −8.33 ± 0.13 | 100.0 ± 5.35 | −8.57 ± 0.08 | 100.9 ± 7.18 |
| met-enkephalin | −8.74 ± 0.07 | 100.4 ± 14.99 | −9.42 ± 0.07 | 100.0 ± 5.09 |
| endomorphin-1 | −8.30 ± 0.10 | 100.4 ± 12.14 | −9.14 ± 0.05 | 99.6 ± 6.72 |
| β-endorphin | −7.70 ± 0.05 | 100.1 ± 3.70 | −7.50 ± 0.03 | 104.2 ± 1.89 |
| TRV130 | −8.85 ± 0.12 | 100.1 ± 6.90 | −9.25 ± 0.07 | 100.0 ± 5.34 |
| NF-ĸB | ||||
| DAMGO | −9.18 ± 0.07 | 100.0 ± 5.22 | −9.23 ± 0.09 | 100.0 ± 7.72 |
| morphine | −8.41 ± 0.05 | 83.2 ± 4.73 | −8.72 ± 0.26 | 93.5 ± 11.34 |
| met-enkephalin | −8.08 ± 0.04 | 104.8 ± 5.06 | −8.67 ± 0.16 | 93.7 ± 4.68 |
| endomorphin-1 | −8.46 ± 0.15 | 104.2 ± 6.43 | −7.54 ± 0.04 | 104.2 ± 5.21 |
| β-endorphin | −7.23 ± 0.07 | 103.0 ± 12.87 | −7.33 ± 0.07 | 103.0 ± 10.71 |
| TRV130 | −6.34 ± 0.10 | 159.6 ± 12.61 | −6.61 ± 0.13 | 132.7 ± 11.57 |
| MAPK/JNK | ||||
| DAMGO | −7.40 ± 0.08 | 101.8 ± 8.47 | −7.88 ± 0.10 | 112.9 ± 10.59 |
| morphine | −8.18 ± 0.07 | 90.8 ± 4.59 | −7.86 ± 0.12 | 91.4 ± 7.10 |
| met-enkephalin | −7.93 ± 0.08 | 72.0 ± 4.58 | −7.53 ± 0.09 | 73.5 ± 5.33 |
| endomorphin-1 | −8.08 ± 0.06 | 90.9 ± 3.84 | −8.99 ± 0.09 | 92.5 ± 3.49 |
| β-endorphin | −7.20 ± 0.07 | 102.0 ± 8.43 | −7.71 ± 0.12 | 88.5 ± 7.70 |
| TRV130 | −6.99 ± 0.07 | 101.3 ± 10.05 | −6.93 ± 0.08 | 123.2 ± 8.65 |
| Ligand | Human | Rhesus Macaque | ||||
|---|---|---|---|---|---|---|
| cAMP | ||||||
| DAMGO | 10.76 ± 0.18 | 0.00 ± 0.25 | 0.00 ± 0.36 | 11.04 ± 0.12 | 0.00 ± 0.17 | 0.00 ± 0.24 |
| morphine | 10.33 ± 0.31 | −0.43 ± 0.36 | 0.00 ± 0.51 | 10.59 ± 0.21 | −0.45 ± 0.25 | 0.00 ± 0.35 |
| met-enkephalin | 10.72 ± 0.26 | −0.04 ± 0.32 | 0.00 ± 0.45 | 11.41 ± 0.18 | 0.37 ± 0.21 | 0.00 ± 0.30 |
| endomorphin-1 | 10.30 ± 0.30 | −0.46 ± 0.35 | 0.00 ± 0.49 | 11.09 ± 0.15 | 0.05 ± 0.19 | 0.00 ± 0.27 |
| β-endorphin | 9.70 ± 0.13 | −1.06 ± 0.22 | 0.00 ± 0.32 | 9.50 ± 0.07 | −1.54 ± 0.14 | 0.00 ± 0.20 |
| TRV130 | 10.90 ± 0.30 | 0.14 ± 0.35 | 0.00 ± 0.49 | 11.23 ± 0.19 | 0.18 ± 0.22 | 0.00 ± 0.32 |
| NF-ĸB | ||||||
| DAMGO | 11.18 ± 0.19 | 0.00±0.26 | 0.00 ± 0.37 | 11.23 ± 0.24 | 0.00 ± 0.34 | 0.00 ± 0.38 |
| morphine | 10.33 ± 0.15 | −0.85 ± 0.24 | −0.42 ± 0.43 | 10.69 ± 0.62 | −0.54 ± 0.66 | −0.09 ± 0.71 |
| met-enkephalin | 10.10 ± 0.11 | −1.08 ± 0.22* | −1.04 ± 0.39 | 10.64 ± 0.36 | −0.59 ± 0.43 | −0.95 ± 0.48 |
| endomorphin-1 | 10.48 ± 0.36 | −0.70 ± 0.41 | −0.24 ± 0.53 | 9.55 ± 0.11 | −1.68 ± 0.27 | −1.73 ± 0.33 |
| β-endorphin | 9.24 ± 0.25 | −1.94 ± 0.32 | −0.88 ± 0.39 | 9.34 ± 0.23 | −1.88 ± 0.34 | −0.34 ± 0.37 |
| TRV130 | 8.54 ± 0.26 | −2.64 ± 0.32 | −2.78 ± 0.48 | 8.74 ± 0.34 | −2.49 ± 0.41 | −2.67 ± 0.47 |
| MAPK/JNK | ||||||
| DAMGO | 9.41 ± 0.23 | 0.00 ± 0.32 | 0.00 ± 0.41 | 9.93 ± 0.29 | 0.00 ± 0.41 | 0.00 ± 0.44 |
| morphine | 10.14 ± 0.19 | 0.74 ± 0.29 | 1.16 ± 0.46 | 9.82 ± 0.31 | −0.11 ± 0.42 | 0.34 ± 0.49 |
| met-enkephalin | 9.79 ± 0.22 | 0.39 ± 0.31 | 0.42 ± 0.45 | 9.39 ± 0.25 | −0.54 ± 0.38 | −0.90 ± 0.44 |
| endomorphin-1 | 10.04 ± 0.17 | 0.64 ± 0.28 | 1.10 ± 0.45 | 10.96 ± 0.22 | 1.03 ± 0.36 | 0.98 ± 0.41 |
| β-endorphin | 9.21 ± 0.20 | −0.19 ± 0.31 | 0.87 ± 0.38 | 9.66 ± 0.32 | −0.27 ± 0.43 | 1.27 ± 0.45 |
| TRV130 | 8.99 ± 0.22 | −0.41 ± 0.32 | −0.55 ± 0.47 | 9.02 ± 0.22 | −0.91 ± 0.36 | −1.10 ± 0.42 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Hutchins, S.D.; Blough, B.E.; Vallender, E.J. In Vitro Effects of Ligand Bias on Primate Mu Opioid Receptor Downstream Signaling. Int. J. Mol. Sci. 2020, 21, 3999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113999
Zhang X, Hutchins SD, Blough BE, Vallender EJ. In Vitro Effects of Ligand Bias on Primate Mu Opioid Receptor Downstream Signaling. International Journal of Molecular Sciences. 2020; 21(11):3999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113999
Chicago/Turabian StyleZhang, Xiao, Shaurita D. Hutchins, Bruce E. Blough, and Eric J. Vallender. 2020. "In Vitro Effects of Ligand Bias on Primate Mu Opioid Receptor Downstream Signaling" International Journal of Molecular Sciences 21, no. 11: 3999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113999