Epigenetic Modifiers as Potential Therapeutic Targets in Diabetic Kidney Disease


 ,
,  and
and
Abstract
:1. Diabetic Kidney Disease Outcomes: An Unmet Medical Need
2. Epigenetic Regulation of Gene Expression
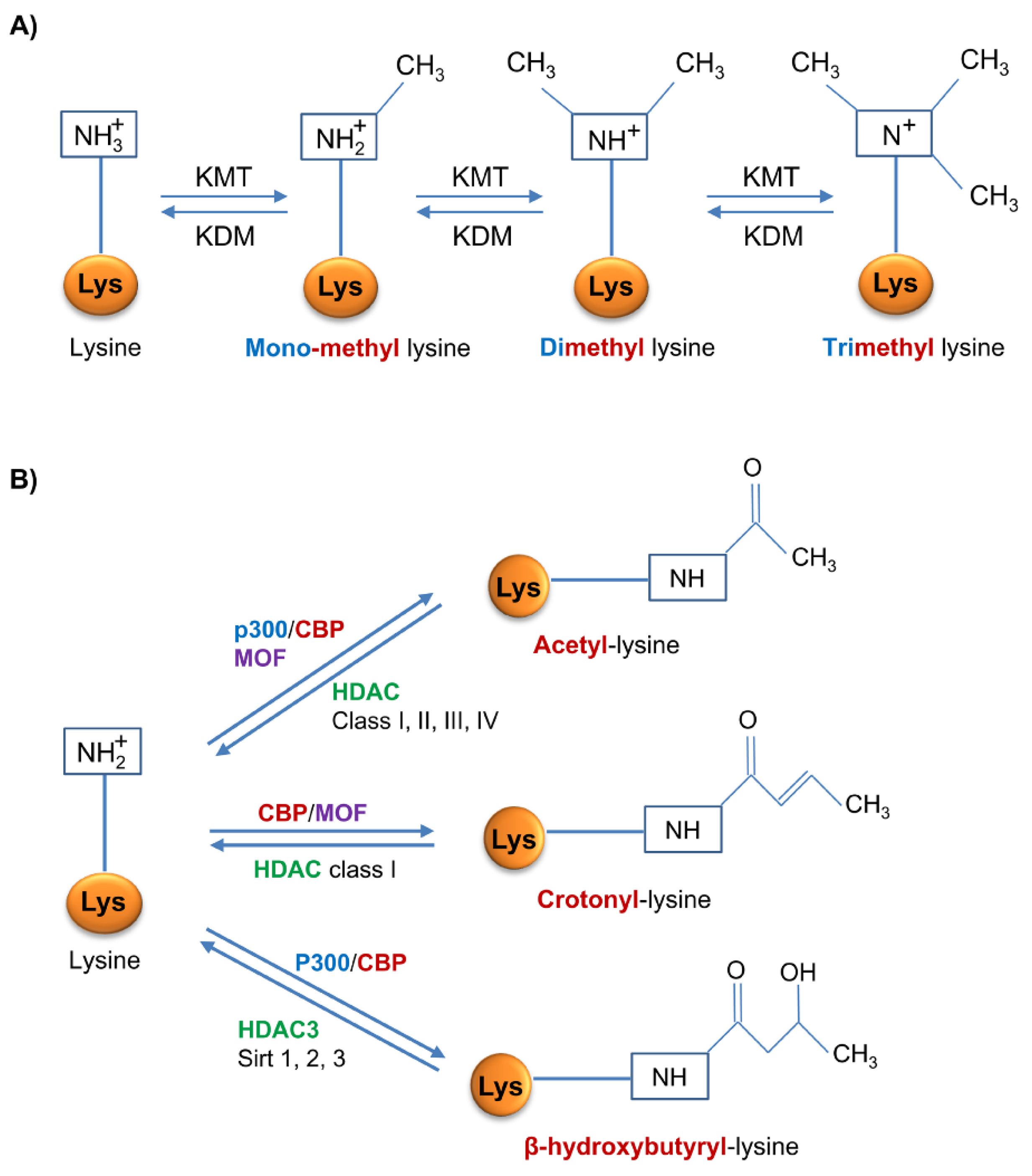
2.1. DNA Methylation
2.2. Histone Methylation
2.3. Histone Acetylation
2.4. Histone Crotonylation
2.5. Histone β-Hydroxybutyrylation
2.6. Epigenetic Readers
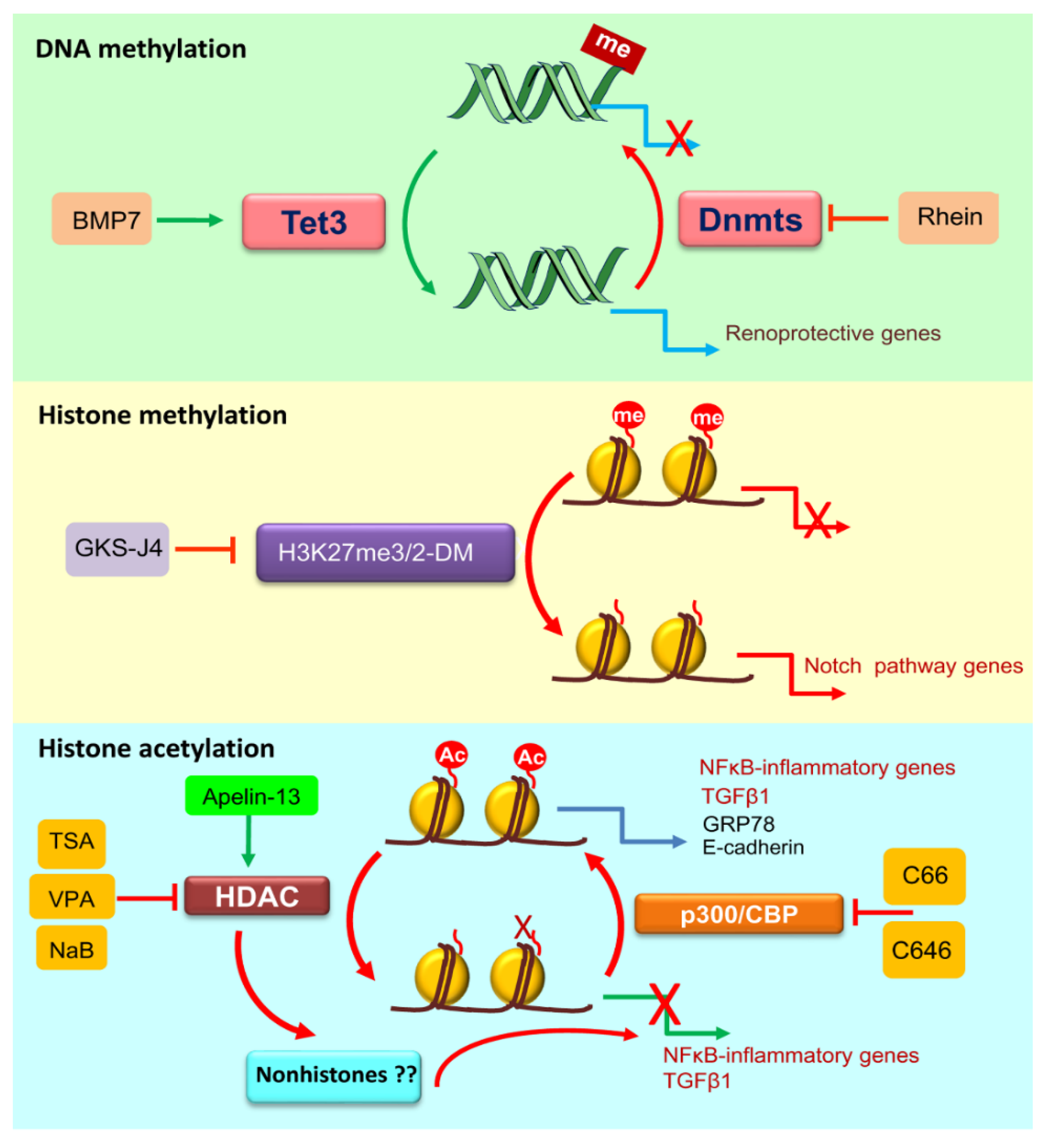
3. DNA Methylation in Diabetic Kidney Disease
4. Histone Post-Translational Modifications in Diabetic Kidney Disease
4.1. Histone Methylation
4.2. Histone Acetylation
4.3. Other Histone Modifications
4.3.1. Crotonylation
4.3.2. β-Hydroxybutyrylation
4.4. Epigenetic Reader Modifiers
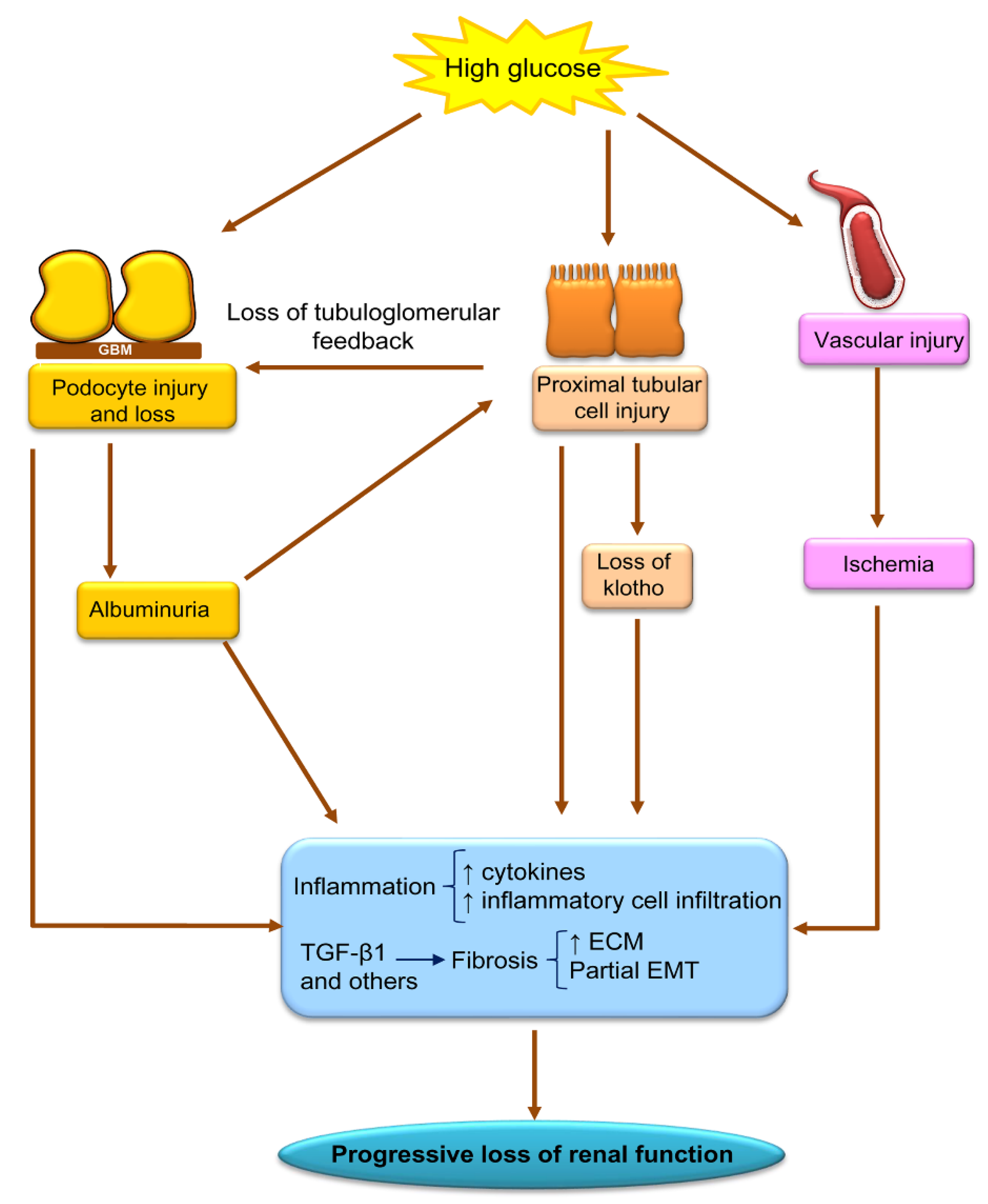
5. Relationship of Epigenetic Modifications to Key Pathogenic Processes in DKD
6. Epigenetic Modifiers as Therapeutic Agents or Targets in Clinical Diabetic Kidney Disease
7. SGLT2 Inhibitors and Epigenetics
8. Summary and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- The Lancet. GBD 2017: A fragile world. Lancet 2018, 392, 1683. [Google Scholar] [CrossRef]
- Thomas, B. The Global Burden of Diabetic Kidney Disease: Time Trends and Gender Gaps. Curr. Diabetes Rep. 2019, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.W.; et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Fernandez, B.; Fernandez-Prado, R.; Górriz, J.L.; Martinez-Castelao, A.; Navarro-González, J.F.; Porrini, E.; Soler, M.J.; Ortiz, A. Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation and Study of Diabetic Nephropathy with Atrasentan: What was learned about the treatment of diabetic kidney disease with canagliflozin and atrasentan? Clin. Kidney J. 2019, 12, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Gomez, M.V.; Sanchez-Niño, M.D.; Sanz, A.B.; Martín-Cleary, C.; Ruiz-Ortega, M.; Egido, J.; Navarro-González, J.F.; Ortiz, A.; Fernandez-Fernandez, B. Horizon 2020 in Diabetic Kidney Disease: The Clinical Trial Pipeline for Add-On Therapies on Top of Renin Angiotensin System Blockade. J. Clin. Med. 2015, 4, 1325–1347. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Sanz, A.B.; Sanchez-Lopez, E.; Ruiz-Ortega, M.; Benito-Martin, A.; Saleem, M.A.; Mathieson, P.W.; Mezzano, S.; Egido, J.; Ortiz, A. HSP27/HSPB1 as an adaptive podocyte antiapoptotic protein activated by high glucose and angiotensin II. Lab. Investig. 2012, 92, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Sanz, A.B.; Ihalmo, P.; Lassila, M.; Holthofer, H.; Mezzano, S.; Aros, C.; Groop, P.H.; Saleem, M.A.; Mathieson, P.W.; et al. The MIF receptor CD74 in diabetic podocyte injury. J. Am. Soc. Nephrol. 2009, 20, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Navarro-González, J.F.; Sánchez-Niño, M.D.; Donate-Correa, J.; Martín-Núñez, E.; Ferri, C.; Pérez-Delgado, N.; Górriz, J.L.; Martínez-Castelao, A.; Ortiz, A.; Mora-Fernández, C. Effects of Pentoxifylline on Soluble Klotho Concentrations and Renal Tubular Cell Expression in Diabetic Kidney Disease. Diabetes Care 2018, 41, 1817–1820. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol. Dial. Transplant. 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Sanz, A.B.; Lorz, C.; Gnirke, A.; Rastaldi, M.P.; Nair, V.; Egido, J.; Ruiz-Ortega, M.; Kretzler, M.; Ortiz, A. BASP1 promotes apoptosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Niño, M.D.; Bozic, M.; Córdoba-Lanús, E.; Valcheva, P.; Gracia, O.; Ibarz, M.; Fernandez, E.; Navarro-Gonzalez, J.F.; Ortiz, A.; Valdivielso, J.M. Beyond proteinuria: VDR activation reduces renal inflammation in experimental diabetic nephropathy. Am. J. Physiol.-Ren. Physiol. 2012, 302, F647–F657. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.; Ferro, C.J.; Morales, E.; Ortiz, A.; Malyszko, J.; Hojs, R.; Khazim, K.; Ekart, R.; Valdivielso, J.; Fouque, D.; et al. SGLT-2 inhibitors and GLP-1 receptor agonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. A consensus statement by the EURECA-m and the DIABESITY working groups of the ERA-EDTA. Nephrol. Dial. Transplant. 2019, 34, 208–230. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Association, A.D. 11. Microvascular Complications and Foot Care in Diabetes—2019. Diabetes Care 2019, 42, S124–S138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Niño, M.D.; Valino-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1875–1886. [Google Scholar] [CrossRef]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Moreno, J.A.; Ruiz-Ortega, M.; Ramos, A.M.; Sanz, A.B.; Ortiz, A. Downregulation of kidney protective factors by inflammation: Role of transcription factors and epigenetic mechanisms. Am. J. Physiol.-Ren. Physiol. 2016, 311, F1329–F1340. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Susztak, K. Understanding the epigenetic syntax for the genetic alphabet in the kidney. J. Am. Soc. Nephrol. 2014, 25, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.G.; Wu, X.; Li, A.X.; Pfeifer, G.P. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011, 39, 5015–5024. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Agborbesong, E.; Zhang, L.; Li, X. Investigation of epigenetics in kidney cell biology. Methods Cell Biol. 2019, 153, 255–278. [Google Scholar] [PubMed]
- Beckerman, P.; Ko, Y.A.; Susztak, K. Epigenetics: A new way to look at kidney diseases. Nephrol. Dial. Transplant. 2014, 29, 1821–1827. [Google Scholar] [CrossRef] [Green Version]
- Bomsztyk, K.; Denisenko, O.; Wang, Y. DNA methylation yields epigenetic clues into the diabetic nephropathy of Pima Indians. Kidney Int. 2018, 93, 1272–1275. [Google Scholar] [CrossRef]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; An, J.; Pastor, W.A.; Koralov, S.B.; Rajewsky, K.; Rao, A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol. Rev. 2015, 263, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Blackshaw, L.A.; Grundy, D. Responses of vagal efferent fibres to stimulation of gastric mechano- and chemoreceptors in the anaesthetized ferret. J. Auton. Nerv. Syst. 1989, 27, 39–45. [Google Scholar] [CrossRef]
- Black, J.C.; Whetstine, J.R. Tipping the lysine methylation balance in disease. Biopolymers 2013, 99, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, S.; Pili, R. Histone deacetylase inhibitors and epigenetic modifications as a novel strategy in renal cell carcinoma. Cancer J. 2013, 19, 333–340. [Google Scholar] [CrossRef]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef]
- Wei, W.; Mao, A.; Tang, B.; Zeng, Q.; Gao, S.; Liu, X.; Lu, L.; Li, W.; Du, J.X.; Li, J.; et al. Large-Scale Identification of Protein Crotonylation Reveals Its Role in Multiple Cellular Functions. J. Proteom. Res. 2017, 16, 1743–1752. [Google Scholar] [CrossRef]
- Fellows, R.; Denizot, J.; Stellato, C.; Cuomo, A.; Jain, P.; Stoyanova, E.; Balázsi, S.; Hajnády, Z.; Liebert, A.; Kazakevych, J.; et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat. Commun. 2018, 9, 105. [Google Scholar] [CrossRef]
- Sabari, B.R.; Tang, Z.; Huang, H.; Yong-Gonzalez, V.; Molina, H.; Kong, H.E.; Dai, L.; Shimada, M.; Cross, J.R.; Zhao, Y.; et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 2015, 58, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wei, W.; Liu, Y.; Yang, X.; Wu, J.; Zhang, Y.; Zhang, Q.; Shi, T.; Du, J.X.; Zhao, Y.; et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov. 2017, 3, 17016. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Liu, X.; Chen, J.; Gao, S.; Lu, L.; Zhang, H.; Ding, G.; Wang, Z.; Chen, Z.; Shi, T.; et al. Class I histone deacetylases are major histone decrotonylases: Evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017, 27, 898–915. [Google Scholar] [CrossRef]
- Rousseaux, S.; Khochbin, S. Histone Acylation beyond Acetylation: Terra Incognita in Chromatin Biology. Cell J. 2015, 17, 1–6. [Google Scholar] [PubMed]
- Martinez-Moreno, J.M.; Fontecha-Barriuso, M.; Martín-Sánchez, D.; Sánchez-Niño, M.D.; Ruiz-Ortega, M.; Sanz, A.B.; Ortiz, A. The Contribution of Histone Crotonylation to Tissue Health and Disease: Focus on Kidney Health. Front. Pharmacol. 2020, 11, 393. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. Histone lysine crotonylation during acute kidney injury in mice. Dis. Models Mech. 2016, 9, 633–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anson, R.M.; Guo, Z.; de Cabo, R.; Iyun, T.; Rios, M.; Hagepanos, A.; Ingram, D.K.; Lane, M.A.; Mattson, M.P. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc. Natl. Acad. Sci. USA 2003, 100, 6216–6220. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [Green Version]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tang, K.; Ma, J.; Zhou, L.; Liu, J.; Zeng, L.; Zhu, L.; Xu, P.; Chen, J.; Wei, K.; et al. Ketogenesis-generated β-hydroxybutyrate is an epigenetic regulator of CD8. Nat. Cell Biol. 2020, 22, 18–25. [Google Scholar] [CrossRef]
- Dąbek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishitani, S.; Fukuhara, A.; Shin, J.; Okuno, Y.; Otsuki, M.; Shimomura, I. Metabolomic and microarray analyses of adipose tissue of dapagliflozin-treated mice, and effects of 3-hydroxybutyrate on induction of adiponectin in adipocytes. Sci. Rep. 2018, 8, 8805. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Ouchi, N.; Matsuzawa, Y. Anti-inflammatory and anti-atherogenic properties of adiponectin. Biochimie 2012, 94, 2137–2142. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, X.; Li, H. Beyond histone acetylation-writing and erasing histone acylations. Curr. Opin. Struct. Biol. 2018, 53, 169–177. [Google Scholar] [CrossRef]
- Chen, X.F.; Chen, X.; Tang, X. Short-chain fatty acid, acylation and cardiovascular diseases. Clin. Sci. 2020, 134, 657–676. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cao, R.; Niu, J.; Yang, S.; Ma, H.; Zhao, S.; Li, H. Molecular basis for hierarchical histone de-β-hydroxybutyrylation by SIRT3. Cell Discov. 2019, 5, 35. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Z.; Xu, W.W.; Zhu, D.Y.; Zhang, N.; Wang, Y.L.; Ding, M.; Xie, X.M.; Sun, L.L.; Wang, X.X. Specific expression network analysis of diabetic nephropathy kidney tissue revealed key methylated sites. J. Cell Physiol. 2018, 233, 7139–7147. [Google Scholar] [CrossRef]
- Ko, Y.A.; Mohtat, D.; Suzuki, M.; Park, A.S.D.; Izquierdo, M.C.; Han, S.Y.; Kang, H.M.; Si, H.; Hostetter, T.; Pullman, J.M.; et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013, 14, R108. [Google Scholar] [CrossRef] [Green Version]
- Wing, M.R.; Devaney, J.M.; Joffe, M.M.; Xie, D.; Feldman, H.I.; Dominic, E.A.; Guzman, N.J.; Ramezani, A.; Susztak, K.; Herman, J.G.; et al. DNA methylation profile associated with rapid decline in kidney function: Findings from the CRIC study. Nephrol. Dial. Transplant. 2014, 29, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marumo, T.; Yagi, S.; Kawarazaki, W.; Nishimoto, M.; Ayuzawa, N.; Watanabe, A.; Ueda, K.; Hirahashi, J.; Hishikawa, K.; Sakurai, H.; et al. Diabetes Induces Aberrant DNA Methylation in the Proximal Tubules of the Kidney. J. Am. Soc. Nephrol. 2015, 26, 2388–2397. [Google Scholar] [CrossRef]
- Sharma, I.; Dutta, R.K.; Singh, N.K.; Kanwar, Y.S. High Glucose-Induced Hypomethylation Promotes Binding of Sp-1 to Myo-Inositol Oxygenase: Implication in the Pathobiology of Diabetic Tubulopathy. Am. J. Pathol 2017, 187, 724–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Chen, H.; Zhong, F.; Zhang, W.; Lee, K.; He, J.C. Expression of Glutamate Receptor Subtype 3 Is Epigenetically Regulated in Podocytes under Diabetic Conditions. Kidney Dis (Basel) 2019, 5, 34–42. [Google Scholar] [CrossRef]
- Ling, L.; Chen, L.; Zhang, C.; Gui, S.; Zhao, H.; Li, Z. 2018 High glucose induces podocyte epithelial-to-mesenchymal transition by demethylation-mediated enhancement of MMP9 expression. Mol. Med. Rep. 2018, 17, 5642–5651. [Google Scholar]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Azegami, T.; Oguchi, H.; Sakamaki, Y.; Itoh, H. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J. Clin. Investig. 2014, 124, 2523–2537. [Google Scholar] [CrossRef] [Green Version]
- Hishikawa, A.; Hayashi, K.; Abe, T.; Kaneko, M.; Yokoi, H.; Azegami, T.; Nakamura, M.; Yoshimoto, N.; Kanda, T.; Sakamaki, Y.; et al. Decreased KAT5 Expression Impairs DNA Repair and Induces Altered DNA Methylation in Kidney Podocytes. Cell Rep. 2019, 26, 1318–1332. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, A.; Zhang, W.; Huang, Z.; Wang, J.; Yi, B. High glucose-induced cytoplasmic translocation of Dnmt3a contributes to CTGF hypo-methylation in mesangial cells. Biosci. Rep. 2016, 36, e00362. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ren, D.; Shen, Y.; Zheng, X.; Xu, G. Altered DNA methylation of TRIM13 in diabetic nephropathy suppresses mesangial collagen synthesis by promoting ubiquitination of CHOP. EBioMedicine 2020, 51, 102582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gondaliya, P.; Dasare, A.; Srivastava, A.; Kalia, K. Correction: miR29b regulates aberrant methylation in In-Vitro diabetic nephropathy model of renal proximal tubular cells. PLoS ONE 2019, 14, e0211591. [Google Scholar] [CrossRef]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Oba, S.; Ayuzawa, N.; Nishimoto, M.; Kawarazaki, W.; Ueda, K.; Hirohama, D.; Kawakami-Mori, F.; Shimosawa, T.; Marumo, T.; Fujita, T. Aberrant DNA methylation of Tgfb1 in diabetic kidney mesangial cells. Sci Rep. 2018, 8, 16338. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Q.; Wu, Q.; Yu, J.; Mu, J.; Zhang, J.; Zeng, W.; Feng, B. Effect of TET2 on the pathogenesis of diabetic nephropathy through activation of transforming growth factor β1 expression via DNA demethylation. Life Sci. 2018, 207, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Zhang, Q.; Yang, J.; Lin, W.; Li, Y.; Chen, F.; Cao, W. TGFβ-incurred epigenetic aberrations of miRNA and DNA methyltransferase suppress Klotho and potentiate renal fibrosis. Biochim. Biophys. Acta 2017, 1864, 1207–1216. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Q.; Liu, S.; Chen, Y.; Li, R.; Lin, T.; Yu, C.; Zhang, H.; Huang, Z.; Zhao, X.; et al. DNA methyltransferase 1 may be a therapy target for attenuating diabetic nephropathy and podocyte injury. Kidney Int. 2017, 92, 140–153. [Google Scholar] [CrossRef]
- Chang, Y.T.; Yang, C.C.; Pan, S.Y.; Chou, Y.H.; Chang, F.C.; Lai, C.F.; Tsai, M.H.; Hsu, H.L.; Lin, C.H.; Chiang, W.C.; et al. DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J. Clin. Investig. 2016, 126, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Tang, W.; Yuan, Q.; Peng, L.; Yu, P. Epigenetic repression of Krüppel-like factor 4 through Dnmt1 contributes to EMT in renal fibrosis. Int. J. Mol. Med. 2015, 35, 1596–1602. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, W.; Zhong, F.; Das, G.C.; Xie, Y.; Li, Z.; Cai, W.; Jiang, G.; Choi, J.; Sidani, M.; et al. Epigenetic regulation of RCAN1 expression in kidney disease and its role in podocyte injury. Kidney Int. 2018, 94, 1160–1176. [Google Scholar] [CrossRef]
- Chen, G.; Chen, H.; Ren, S.; Xia, M.; Zhu, J.; Liu, Y.; Zhang, L.; Tang, L.; Sun, L.; Liu, H.; et al. Aberrant DNA methylation of mTOR pathway genes promotes inflammatory activation of immune cells in diabetic kidney disease. Kidney Int. 2019, 96, 409–420. [Google Scholar] [CrossRef]
- Tampe, B.; Tampe, D.; Müller, C.A.; Sugimoto, H.; LeBleu, V.; Xu, X.; Müller, G.A.; Zeisberg, E.M.; Kalluri, R.; Zeisberg, M. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J. Am. Soc. Nephrol. 2014, 25, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Zhen, Y.Z.; Wei, J.B.; Wei, J.; Dai, J.; Gao, J.L.; Li, K.J.; Hu, G. Rhein lysinate protects renal function in diabetic nephropathy of KK/HlJ mice. Exp. Therapeutic Med. 2017, 14, 5801–5808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, L.; Lin, W.; Yin, S.; Duan, A.; Liu, Z.; Cao, W. Rhein reverses Klotho repression via promoter demethylation and protects against kidney and bone injuries in mice with chronic kidney disease. Kidney Int. 2017, 91, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Fernandez-Fernandez, B.; Ortiz, A. Klotho, the elusive kidney-derived anti-ageing factor. Clin. Kidney J. 2020, 13, 125–127. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Jaisser, F. Pathophysiologic mechanisms in diabetic kidney disease: A focus on current and future therapeutic targets. Diabetes Obes. Metab. 2020, 22, 16–31. [Google Scholar] [CrossRef]
- Ochoa-Rosales, C.; Portilla-Fernandez, E.; Nano, J.; Wilson, R.; Lehne, B.; Mishra, P.P.; Gao, X.; Ghanbari, M.; Rueda-Ochoa, O.L.; Juvinao-Quintero, D.; et al. Epigenetic Link Between Statin Therapy and Type 2 Diabetes. Diabetes Care 2020, 43, 875–884. [Google Scholar] [CrossRef]
- Gondaliya, P.; Dasare, A.; Srivastava, A.; Kalia, K. miR29b regulates aberrant methylation in In-Vitro diabetic nephropathy model of renal proximal tubular cells. PLoS ONE 2018, 13, e0208044. [Google Scholar] [CrossRef]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- Yu, C.; Zhuang, S. Histone Methyltransferases as Therapeutic Targets for Kidney Diseases. Front. Pharmacol. 2019, 10, 1393. [Google Scholar] [CrossRef]
- Miao, F.; Wu, X.; Zhang, L.; Yuan, Y.C.; Riggs, A.D.; Natarajan, R. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J. Biol. Chem. 2007, 282, 13854–13863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, F.; Smith, D.D.; Zhang, L.; Min, A.; Feng, W.; Natarajan, R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: An epigenetic study in diabetes. Diabetes 2008, 57, 3189–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, F.; Chen, Z.; Genuth, S.; Paterson, A.; Zhang, L.; Wu, X.; Li, S.M.; Cleary, P.; Riggs, A.; Harlan, D.M.; et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014, 63, 1748–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninichuk, V.; Kulkarni, O.; Clauss, S.; Anders, H. Tubular atrophy, interstitial fibrosis, and inflammation in type 2 diabetic db/db mice. An accelerated model of advanced diabetic nephropathy. Eur. J. Med. Res. 2007, 12, 351–355. [Google Scholar]
- Sayyed, S.G.; Gaikwad, A.B.; Lichtnekert, J.; Kulkarni, O.; Eulberg, D.; Klussmann, S.; Tikoo, K.; Anders, H.J. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol. Dial. Transplant. 2010, 25, 1811–1817. [Google Scholar] [CrossRef]
- Ninichuk, V.; Clauss, S.; Kulkarni, O.; Schmid, H.; Segerer, S.; Radomska, E.; Eulberg, D.; Buchner, K.; Selve, N.; Klussmann, S.; et al. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3′PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am. J. Pathol. 2008, 172, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Komers, R.; Mar, D.; Denisenko, O.; Xu, B.; Oyama, T.T.; Bomsztyk, K. Epigenetic changes in renal genes dysregulated in mouse and rat models of type 1 diabetes. Lab. Investig. 2013, 93, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Thieme, K.; Batchu, S.N.; Alghamdi, T.A.; Bowskill, B.B.; Kabir, M.G.; Liu, Y.; Advani, S.L.; White, K.E.; Geldenhuys, L.; et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J. Clin. Investig. 2018, 128, 483–499. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Reddy, M.A.; Das, S.; Oh, H.J.; Abdollahi, M.; Yuan, H.; Zhang, E.; Lanting, L.; Wang, M.; Natarajan, R. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-β1-induced gene expression in mesangial cells and diabetic kidney. J. Biol. Chem. 2019, 294, 12695–12707. [Google Scholar] [CrossRef]
- Siddiqi, F.S.; Majumder, S.; Thai, K.; Abdalla, M.; Hu, P.; Advani, S.L.; White, K.E.; Bowskill, B.B.; Guarna, G.; dos Santos, C.C.; et al. The Histone Methyltransferase Enzyme Enhancer of Zeste Homolog 2 Protects against Podocyte Oxidative Stress and Renal Injury in Diabetes. J. Am. Soc. Nephrol. 2016, 27, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.H.; Ho, W.T.; Wang, Y.T.; Chuang, C.T.; Chuang, L.Y.; Guh, J.Y. Histone methyltransferase Suv39h1 attenuates high glucose-induced fibronectin and p21(WAF1) in mesangial cells. Int. J. Biochem. Cell Biol. 2016, 78, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Goru, S.K.; Kadakol, A.; Pandey, A.; Malek, V.; Sharma, N.; Gaikwad, A.B. Histone H2AK119 and H2BK120 mono-ubiquitination modulate SET7/9 and SUV39H1 in type 1 diabetes-induced renal fibrosis. Biochem. J. 2016, 473, 3937–3949. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Reddy, M.A.; Yuan, H.; Lanting, L.; Kato, M.; Natarajan, R. Epigenetic histone methylation modulates fibrotic gene expression. J. Am. Soc. Nephrol. 2010, 21, 2069–2080. [Google Scholar] [CrossRef]
- Li, Y.; Reddy, M.A.; Miao, F.; Shanmugam, N.; Yee, J.K.; Hawkins, D.; Ren, B.; Natarajan, R. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J. Biol. Chem. 2008, 283, 26771–26781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158. [Google Scholar] [CrossRef]
- Reddy, M.A.; Sumanth, P.; Lanting, L.; Yuan, H.; Wang, M.; Mar, D.; Alpers, C.E.; Bomsztyk, K.; Natarajan, R. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. 2014, 85, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, L.M.; Natarajan, R. Epigenetic mechanisms. Contrib. Nephrol. 2011, 170, 57–65. [Google Scholar]
- Villeneuve, L.M.; Natarajan, R. The role of epigenetics in the pathology of diabetic complications. Am. J. Physiol.-Ren. Physiol. 2010, 299, F14–F25. [Google Scholar] [CrossRef] [Green Version]
- Tonna, S.; El-Osta, A.; Cooper, M.E.; Tikellis, C. Metabolic memory and diabetic nephropathy: Potential role for epigenetic mechanisms. Nat. Rev. Nephrol. 2010, 6, 332–341. [Google Scholar] [CrossRef]
- Hills, C.E.; Squires, P.E. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am. J. Nephrol. 2010, 31, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, I.; Wolf, G. Epithelial-to-Mesenchymal Transition in Diabetic Nephropathy: Fact or Fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Kolset, S.O.; Reinholt, F.P.; Jenssen, T. Diabetic nephropathy and extracellular matrix. J. Histochem. Cytochem. 2012, 60, 976–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Luo, M.; Wu, H.; Wu, H.; Kong, L.; Xin, Y.; Cui, W.; Zhao, Y.; Wang, J.; Liang, G.; et al. Novel curcumin analog C66 prevents diabetic nephropathy via JNK pathway with the involvement of p300/CBP-mediated histone acetylation. Biochim. Biophys. Acta 2015, 1852, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Reddy, M.A.; Sun, G.; Lanting, L.; Wang, M.; Kato, M.; Natarajan, R. Involvement of p300/CBP and epigenetic histone acetylation in TGF-β1-mediated gene transcription in mesangial cells. Am. J. Physiol.-Ren.Physiol. 2013, 304, F601–F613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, F.; Ghosh-Choudhury, N.; Venkatesan, B.; Li, X.; Mahimainathan, L.; Choudhury, G.G. Akt kinase targets association of CBP with SMAD 3 to regulate TGFbeta-induced expression of plasminogen activator inhibitor-1. J. Cell Physiol. 2008, 214, 513–527. [Google Scholar] [CrossRef]
- Kato, M.; Dang, V.; Wang, M.; Park, J.T.; Deshpande, S.; Kadam, S.; Mardiros, A.; Zhan, Y.; Oettgen, P.; Putta, S.; et al. TGF-β induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci. Signal. 2013, 6, ra43. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Bhattacharyya, S.; Lafyatis, R.; Farina, G.; Yu, J.; Thimmapaya, B.; Wei, J.; Varga, J. p300 is elevated in systemic sclerosis and its expression is positively regulated by TGF-β: Epigenetic feed-forward amplification of fibrosis. J. Investig. Dermatol 2013, 133, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Kanamaru, Y.; Nakao, A.; Tanaka, Y. Involvement of p300 in TGF-beta/Smad-pathway-mediated alpha2(I) collagen expression in mouse mesangial cells. Nephron Exp. Nephrol. 2003, 95, e36–e42. [Google Scholar] [CrossRef]
- Malek, V.; Sharma, N.; Gaikwad, A.B. Histone Acetylation Regulates Natriuretic Peptides and Neprilysin Gene Expressions in Diabetic Cardiomyopathy and Nephropathy. Curr. Mol. Pharmacol. 2019, 12, 61–71. [Google Scholar] [CrossRef]
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 2004, 279, 18091–18097. [Google Scholar] [CrossRef] [Green Version]
- De Marinis, Y.; Cai, M.; Bompada, P.; Atac, D.; Kotova, O.; Johansson, M.E.; Garcia-Vaz, E.; Gomez, M.F.; Laakso, M.; Groop, L. Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int. 2016, 89, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, J.; Jiao, L.; Petersen, R.B.; Li, J.; Peng, A.; Zheng, L.; Huang, K. Apelin inhibits the development of diabetic nephropathy by regulating histone acetylation in Akita mouse. J. Physiol. 2014, 592, 505–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Q.; Zhang, L.; Das, B.; Li, Z.; Liu, B.; Cai, G.; Chen, X.; Chuang, P.Y.; He, J.C.; Lee, K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 2018, 93, 1330–1343. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Oh, E.Y.; Seo, J.Y.; Yu, M.R.; Kim, Y.O.; Ha, H.; Lee, H.B. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am. J. Physiol.-Ren. Physiol. 2009, 297, F729–F739. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.Y.; Qin, H.J.; Zhang, Z.; Xu, Y.; Yang, X.C.; Zhao, D.M.; Li, X.N.; Sun, L.K. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum stress-induced apoptosis. Mol. Med. Rep. 2016, 13, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikoo, K.; Meena, R.L.; Kabra, D.G.; Gaikwad, A.B. Change in post-translational modifications of histone H3, heat-shock protein-27 and MAP kinase p38 expression by curcumin in streptozotocin-induced type I diabetic nephropathy. Br. J. Pharmacol. 2008, 153, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, K.; Liang, Y.; Chen, Y.; Chen, Y.; Gong, Y. Histone acetyltransferase inhibitor C646 reverses epithelial to mesenchymal transition of human peritoneal mesothelial cells via blocking TGF-β1/Smad3 signaling pathway in vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 2746–2754. [Google Scholar]
- Sanz, A.B.; Ruiz-Andres, O.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ramos, A.M.; Ortiz, A. Out of the TWEAKlight: Elucidating the Role of Fn14 and TWEAK in Acute Kidney Injury. Semin. Nephrol. 2016, 36, 189–198. [Google Scholar] [CrossRef]
- Obokata, M.; Negishi, K.; Sunaga, H.; Ishida, H.; Ito, K.; Ogawa, T.; Iso, T.; Ando, Y.; Kurabayashi, M. Association Between Circulating Ketone Bodies and Worse Outcomes in Hemodialysis Patients. J. Am. Heart Assoc. 2017, 6, e006885. [Google Scholar] [CrossRef] [Green Version]
- Poplawski, M.M.; Mastaitis, J.W.; Isoda, F.; Grosjean, F.; Zheng, F.; Mobbs, C.V. Reversal of diabetic nephropathy by a ketogenic diet. PLoS ONE 2011, 6, e18604. [Google Scholar] [CrossRef]
- Khan, S.; Jena, G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol. 2014, 73, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Zoja, C.; Zanchi, C.; Corna, D.; Villa, S.; Bolognini, S.; Novelli, R.; Perico, L.; Remuzzi, G.; Benigni, A.; et al. Manipulating Sirtuin 3 pathway ameliorates renal damage in experimental diabetes. Sci. Rep. 2020, 10, 8418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, J.; Zhou, F.; Wang, W.; Chen, N. PGC-1α ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol. Med. Rep. 2018, 17, 4490–4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Rollin, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006, 69, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef] [Green Version]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Med. Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Alvarez, B.; Rodriguez, R.M.; Ruiz-Ortega, M.; Lopez-Larrea, C. BET Proteins: An Approach to Future Therapies in Transplantation. Am. J. Transplant. 2017, 17, 2254–2262. [Google Scholar] [CrossRef] [Green Version]
- Amir-Zilberstein, L.; Ainbinder, E.; Toube, L.; Yamaguchi, Y.; Handa, H.; Dikstein, R. regulation of NF-kappaB by elongation factors is determined by core promoter type. Mol. Cell. Biol. 2007, 27, 5246–5259. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Yang, X.D.; Zhou, M.M.; Ozato, K.; Chen, L.F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.F. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 2014, 33, 2395–2404. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Alvarez, B.; Morgado-Pascual, J.L.; Rayego-Mateos, S.; Rodriguez, R.M.; Rodrigues-Diez, R.; Cannata-Ortiz, P.; Sanz, A.B.; Egido, J.; Tharaux, P.L.; Ortiz, A.; et al. Inhibition of Bromodomain and Extraterminal Domain Family Proteins Ameliorates Experimental Renal Damage. J. Am. Soc. Nephrol. 2017, 28, 504–519. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Benito-Martin, A.; Ortiz, A. New paradigms in cell death in human diabetic nephropathy. Kidney Int. 2010, 78, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Lavoz, C.; Rayego-Mateos, S.; Orejudo, M.; Opazo-Ríos, L.; Marchant, V.; Marquez-Exposito, L.; Tejera-Muñoz, A.; Navarro-González, J.F.; Droguett, A.; Ortiz, A.; et al. Could IL-17A Be a Novel Therapeutic Target in Diabetic Nephropathy? J. Clin. Med. 2020, 9, 272. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.J.; Shah, A.; Apostolopolou, H.; Bhushan, A. BET Proteins Are Required for Transcriptional Activation of the Senescent Islet Cell Secretome in Type 1 Diabetes. Int. J. Mol. Sci. 2019, 20, 4776. [Google Scholar] [CrossRef] [Green Version]
- Deeney, J.T.; Belkina, A.C.; Shirihai, O.S.; Corkey, B.E.; Denis, G.V. BET Bromodomain Proteins Brd2, Brd3 and Brd4 Selectively Regulate Metabolic Pathways in the Pancreatic β-Cell. PLoS ONE 2016, 11, e0151329. [Google Scholar] [CrossRef] [Green Version]
- Huijbregts, L.; Petersen, M.B.K.; Berthault, C.; Hansson, M.; Aiello, V.; Rachdi, L.; Grapin-Botton, A.; Honore, C.; Scharfmann, R. Bromodomain and Extra Terminal Protein Inhibitors Promote Pancreatic Endocrine Cell Fate. Diabetes 2019, 68, 761–773. [Google Scholar] [CrossRef]
- Wang, J.; Hu, J.; Chen, X.; Huang, C.; Lin, J.; Shao, Z.; Gu, M.; Wu, Y.; Tian, N.; Gao, W.; et al. BRD4 inhibition regulates MAPK, NF-κB signals, and autophagy to suppress MMP-13 expression in diabetic intervertebral disc degeneration. FASEB J. 2019, 33, 11555–11566. [Google Scholar] [CrossRef]
- Guo, M.; Wang, H.X.; Chen, W.J. BET-inhibition by JQ1 alleviates streptozotocin-induced diabetic cardiomyopathy. Toxicol. Appl. Pharmacol. 2018, 352, 9–18. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, Y.; Li, T.; Liu, L.; Zhao, Y.; Li, L.; Zhang, L.; Meng, Y. Function of BRD4 in the pathogenesis of high glucose-induced cardiac hypertrophy. Mol. Med. Rep. 2019, 19, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Wang, S.; Feng, J.; Liu, X. BRD4 contributes to high-glucose-induced podocyte injury by modulating Keap1/Nrf2/ARE signaling. Biochimie 2019, 165, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Kulikowski, E.; Halliday, C.; Johansson, J.; Sweeney, M.; Lebioda, K.; Wong, N.; Haarhaus, M.; Brandenburg, V.; Beddhu, S.; Tonelli, M.; et al. Apabetalone Mediated Epigenetic Modulation is Associated with Favorable Kidney Function and Alkaline Phosphatase Profile in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2018, 43, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Sumida, K.; Molnar, M.Z.; Potukuchi, P.K.; Thomas, F.; Lu, J.L.; Obi, Y.; Rhee, C.M.; Streja, E.; Yamagata, K.; Kalantar-Zadeh, K.; et al. Prognostic significance of pre-end-stage renal disease serum alkaline phosphatase for post-end-stage renal disease mortality in late-stage chronic kidney disease patients transitioning to dialysis. Nephrol. Dial. Transplant. 2018, 33, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Regidor, D.L.; Kovesdy, C.P.; Mehrotra, R.; Rambod, M.; Jing, J.; McAllister, C.J.; Van Wyck, D.; Kopple, J.D.; Kalantar-Zadeh, K. Serum alkaline phosphatase predicts mortality among maintenance hemodialysis patients. J. Am. Soc. Nephrol. 2008, 19, 2193–2203. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.K.; Nicholls, S.J.; Ginsberg, H.D.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Cummings, J.L.; Sweeney, M.; et al. Effect of selective BET protein inhibitor apabetalone on cardiovascular outcomes in patients with acute coronary syndrome and diabetes: Rationale, design, and baseline characteristics of the BETonMACE trial. Am. Heart J. 2019, 217, 72–83. [Google Scholar] [CrossRef]
- Ray, K.K.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients With Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2020, 323, 1565–1573. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Sanz, A.B.; Carrasco, S.; Saleem, M.A.; Mathieson, P.W.; Valdivielso, J.M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Globotriaosylsphingosine actions on human glomerular podocytes: Implications for Fabry nephropathy. Nephrol. Dial. Transplant. 2011, 26, 1797–1802. [Google Scholar] [CrossRef] [Green Version]
- Aguilera-Correa, J.J.; Madrazo-Clemente, P.; Martínez-Cuesta, M.D.C.; Peláez, C.; Ortiz, A.; Sánchez-Niño, M.D.; Esteban, J.; Requena, T. Lyso-Gb3 modulates the gut microbiota and decreases butyrate production. Sci. Rep.. 2019, 9, 12010. [Google Scholar] [CrossRef] [Green Version]
- van Bommel, E.J.M.; Lytvyn, Y.; Perkins, B.A.; Soleymanlou, N.; Fagan, N.M.; Koitka-Weber, A.; Joles, J.A.; Cherney, D.Z.; van Raalte, D.H. Renal hemodynamic effects of sodium-glucose cotransporter 2 inhibitors in hyperfiltering people with type 1 diabetes and people with type 2 diabetes and normal kidney function. Kidney Int. 2020, 97, 631–635. [Google Scholar] [CrossRef]
- Soler, M.J.; Porrini, E.; Fernandez-Fernandez, B.; Ortiz, A. SGLT2i and postglomerular vasodilation. Kidney Int. 2020, 97, 805–806. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solini, A.; Seghieri, M.; Giannini, L.; Biancalana, E.; Parolini, F.; Rossi, C.; Dardano, A.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. The Effects of Dapagliflozin on Systemic and Renal Vascular Function Display an Epigenetic Signature. J. Clin. Endocrinol. Metab. 2019, 104, 4253–4263. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martín-Sánchez, D.; Martinez-Moreno, J.M.; Carrasco, S.; Ruiz-Andrés, O.; Monsalve, M.; Sanchez-Ramos, C.; Gómez, M.J.; Ruiz-Ortega, M.; Sánchez-Niño, M.D.; et al. PGC-1α deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J. Pathol. 2019, 249, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sánchez-Ramos, C.; Monsalve, M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int. 2015, 89, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Change in DN * | Effect in Target Gene ** | Model | Cell or Tissue | Reference |
|---|---|---|---|---|
| ↓ DNA-methylation | ↑ MIOX | STZ mice | Kidney | [64] |
| HG in human cells | Tubular (HK-2) | |||
| ↑ MMP9 | HG in human cells | Podocytes | [66] | |
| ↑ CLDN-1 | STZ mice | Kidney | [69] | |
| HG in human cells | Renal epithelial cells | |||
| ↑ CTGF | HG in human cells | Mesangial cells | [70] | |
| ↑ TGFB1 | db/db mice | Mesangial cells from diabetic mice | [74,75] | |
| db/db mice | Kidney | |||
| HG in human cells | Mesangial cells | |||
| ↑ Agt, Abcc4, Slco1a1 | db/db mice | Proximal tubules | [63] | |
| ↑ DNA-methylation | ↓ mTOR upstream inhibitors | db/db mice | Immune cells from diabetic mice | [81] |
| ↓ NPHS1 | db/db mice | Kidney | [67,68] | |
| STZ and db/db mice | Kidney and isolated podocytes | |||
| HG in human cells | Podocytes | |||
| ↓ Trim13 | STZ and db/db mice | Kidney | [71] | |
| ↓ KLF4 | TGF-β1 in human cells | HK-2 | [79] | |
| ↓ RCAN1 | HG in human cells | Podocytes | [80] | |
| ↓ Rasal1 | STZ mice | Kidney | [82] | |
| ↓ ESX1, GRIA3 | HG in human cells | Podocytes | [65] |
| Histone Methylation | DN | Model | Sample | Reference | |
|---|---|---|---|---|---|
| Activating marks | H3K4m2 | ↓ global | db/db mice (early time points) | Kidney | [95] |
| ↑ global | Uninephrectomiced db/db mice | ||||
| ↑ in Ccl21, Fsp1 | OVE26 mice (T1D) | Kidney | [97] | ||
| STZ rats | Kidney | ||||
| H3K4m1/2/3 | ↑ in EMT-associated genes | HG in rat cells | Mesangial cells | [103] | |
| H3K4m3 | ↑ global | T1D patients | Blood monocytes | [93] | |
| Repressive mark | H3K9m2 | ↑ in IL1A | HG in human cells | THP-1 monocytes | [91] |
| T1D and T2D patients | Blood monocytes | ||||
| ↑ in CLTA4 | T1D patients | Blood lymphocytes | [92] | ||
| H3K9m3 | ↓ in Fn1, p21 | HG in mouse cells | Mesangial cells | [101] | |
| H3K9m2/3 | ↓ in EMT-associated genes | HG in rat cells | Mesangial cells | [103] | |
| H3K27m2 | ↓ global | Adriamycin mice | Isolated podocytes | [98] | |
| DKD patients | Isolated podocytes | ||||
| ↓ in Pai-1, Ccl2 | STZ rats | Kidney | [99] | ||
| TGF-β1 in rat cells | Mesangial cells | ||||
| ↓ in Pax6 | STZ rats | Kidney | [100] | ||
| HG in mouse cells | Podocytes | ||||
| H3K27m3 | ↓ in Ccl21, Fsp1 | OVE26 mice (T1D) | Kidney | [97] | |
| STZ rats | Kidney | ||||
| Histone Modification | Change in DN * | Effect in Target Gene ** | Model | Sample | Ref. |
|---|---|---|---|---|---|
| Acetylation | ↑ H2BK5Ac | ↑ Mme | STZ rats | Kidney | [119] |
| ↑ H3K9Ac | Global | Uninephrectomiced db/db mice | Kidney | [95] | |
| Akita mice (T1D) | Kidney | [122] | |||
| HG in rat cells | Mesangial | ||||
| T1D patients | Blood monocytes | [93] | |||
| ↑ TXNIP | Sur1-E1506K+/+ mice (T2D) | Kidney | [121] | ||
| HG in human, murine cells | Mesangial | ||||
| ↑ H3K9/14Ac | ↑ Pai-1 and p21 | HG in rat cells | Mesangial | [114] | |
| ↑ Fn1, Ctgf, Pai-1 | STZ mice | Kidney | [113] | ||
| ↑ Ets1 | db/db mice | Kidney | [116] | ||
| ↑ TNF, COX2 | HG in human cells | THP-1 monocytes | [120] | ||
| ↑ H3K18Ac | ↑ global | Akita mice (T1D) | Kidney | [122] | |
| HG in rat cells | Mesangial | ||||
| ↑ Mme | STZ rats | Kidney | [119] | ||
| ↑ H3K23Ac | ↓ global | db/db mice | Kidney | [95] | |
| ↑ global | Uninephrectomiced db/db mice | Kidney | |||
| ↑ H4Ac | ↑ Grp78, Chop Atf4 | STZ in rats | Kidney | [125] | |
| ↑ H4K5/8/12Ac | ↑ TNF, COX2 | HG in human cells | THP-1 monocytes | [120] | |
| Crotony-lation | N/A (increased, global and Ppargc1a and Sirt3 genes in AKI) | N/A (Crotonylation increased PGC-1α and SIRT3, and decreased CCL2 expression ***) | N/A (AKI mice) | N/A (Kidney and tubular cells) | N/A [43] |
| β-hydroxy-butyrylation | ↑ H3K9bhb | Global | STZ mice | Liver | [47] |
| Fasted mice | Kidney | ||||
| ↑ H3K18bhb | Global | STZ mice | Liver | ||
| ↑ H4K8bhb | Global | Fasted mice | Kidney |
| Injury | DNA or Protein Modification | Effect in Target Genes | Relation between Epigenetic Modification and Gene Target * | Sample/Model or Treatment | Ref. |
|---|---|---|---|---|---|
| Podocyte injury | ↑ DNA-methylation | ↓ GRIA3 | Causal | Human and murine podocytes/HG | [65] |
| ↑ DNA-methylation | ↓ Nphs1 | Association | Kidney/db/db mice Kidney/STZ rat and db/db mice | [67] [68] | |
| ↓ H3K27m2 | ↑ Pax6 | Causal | Murine podocytes/HG Kidney/STZ rats | [100] | |
| Inflammation | ↑ DNA-methylation | ↓ negative regulators of mTOR | Association | PBMCs/db/db mice | [81] |
| ↓ H3K27m2 | ↑ CCL2 | Association | Rat mesangial cells/ TGF-β1 Kidney/OVE26 mice (T1D) | [99] [97] | |
| ↑ H3K4 m1/m2/m3 | ↑ inflammatory genes ↑ inflammatory genes ↑ Mcp-1 | Causal Association Causal | Macrohages/diabetic mice Monocytes /diabetic patients Kidney/OVE26 mice (T1D) | [104] [105] [97] | |
| ↑ H2BK5ac | ↑ Mme | Association | Kidney/STZ rats | [119] | |
| ↑ H3K18ac | |||||
| ↑ H3K9/14ac | ↑ TNFα and COX-1 | Association | Monocytes/Diabetic human | [120] | |
| ↑ Txnip | Association | Kidney/Diabetic mice | [121] | ||
| Human and mouse mesangial cells/HG | |||||
| ↑ Mcp-1 | Association | Kidney/db/db mice Murine mesangial cells/HG | [106] | ||
| ↑ H4K5/8/12ac | ↑ TNFα and COX-1 | Association | THP-1 monocytes/ HG | [120] | |
| Fibrosis ↑ EMT | ↓ DNA-methylation | ↑ MMP9 | Association | Human podocytes/HG Kidney/STZ rat | [66] |
| ↑ DNA-methylation | ↓ KLF4 | Causal | Human proximal tubular cells/ TGF-β1 | [79] | |
| ↑ H3K4m2 | ↑ Fsp1 | Association | Kidney/STZ rats | [97] | |
| ↓ H3K27m3 | |||||
| Fibrosis ↑ ECM | ↑ DNA-methylation | ↓ Trim13 | Causal | Kidney/STZ rat and db/db mice | [71] |
| ↓ DNA-methylation | ↑ MIOX | Association | Human proximal tubular cells/HG Kidney/STZ rat | [64] | |
| ↓ H3K9m3 | ↑ Fn-1, p21 | Causal | Murine mesangial cells/HG | [101] | |
| ↑ H3K4m1/2/3 ↓ H3K9m2/3 | ↑ ECM-associated genes | Causal Association | Rat mesangial cell/HG | [103] | |
| ↑ H3K9/14ac | ↑ Fn-1 and Pai-1 | Association | Kidney/STZ-induced diabetic mice | [113] | |
| ↑ Pai-1 and p21 | Association | Rat mesangial cells/HG | [114,115] | ||
| ↑ Pai-1 and Rage | Causal | Kideny/db/db mice Murine mesangial cells/HG | [106] | ||
| ↑ H4Kac | ↑ Col1A2 | Causal | Murine mesangial cells/TGF-β | [118] | |
| Fibrosis (TGFβ1) | ↓ DNA-methylation | ↑ Tgf-β1 | Association | Kidney/db/db mice | [74] |
| Causal | Kidney/db/db mice Human mesangial cells/HG | [75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Moreno, J.M.; Fontecha-Barriuso, M.; Martin-Sanchez, D.; Guerrero-Mauvecin, J.; Goma-Garces, E.; Fernandez-Fernandez, B.; Carriazo, S.; Sanchez-Niño, M.D.; Ramos, A.M.; Ruiz-Ortega, M.; et al. Epigenetic Modifiers as Potential Therapeutic Targets in Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 4113. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114113
Martinez-Moreno JM, Fontecha-Barriuso M, Martin-Sanchez D, Guerrero-Mauvecin J, Goma-Garces E, Fernandez-Fernandez B, Carriazo S, Sanchez-Niño MD, Ramos AM, Ruiz-Ortega M, et al. Epigenetic Modifiers as Potential Therapeutic Targets in Diabetic Kidney Disease. International Journal of Molecular Sciences. 2020; 21(11):4113. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114113
Chicago/Turabian StyleMartinez-Moreno, Julio M., Miguel Fontecha-Barriuso, Diego Martin-Sanchez, Juan Guerrero-Mauvecin, Elena Goma-Garces, Beatriz Fernandez-Fernandez, Sol Carriazo, Maria D. Sanchez-Niño, Adrian M. Ramos, Marta Ruiz-Ortega, and et al. 2020. "Epigenetic Modifiers as Potential Therapeutic Targets in Diabetic Kidney Disease" International Journal of Molecular Sciences 21, no. 11: 4113. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114113