Reprogramming of Mesothelial-Mesenchymal Transition in Chronic Peritoneal Diseases by Estrogen Receptor Modulation and TGF-β1 Inhibition


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
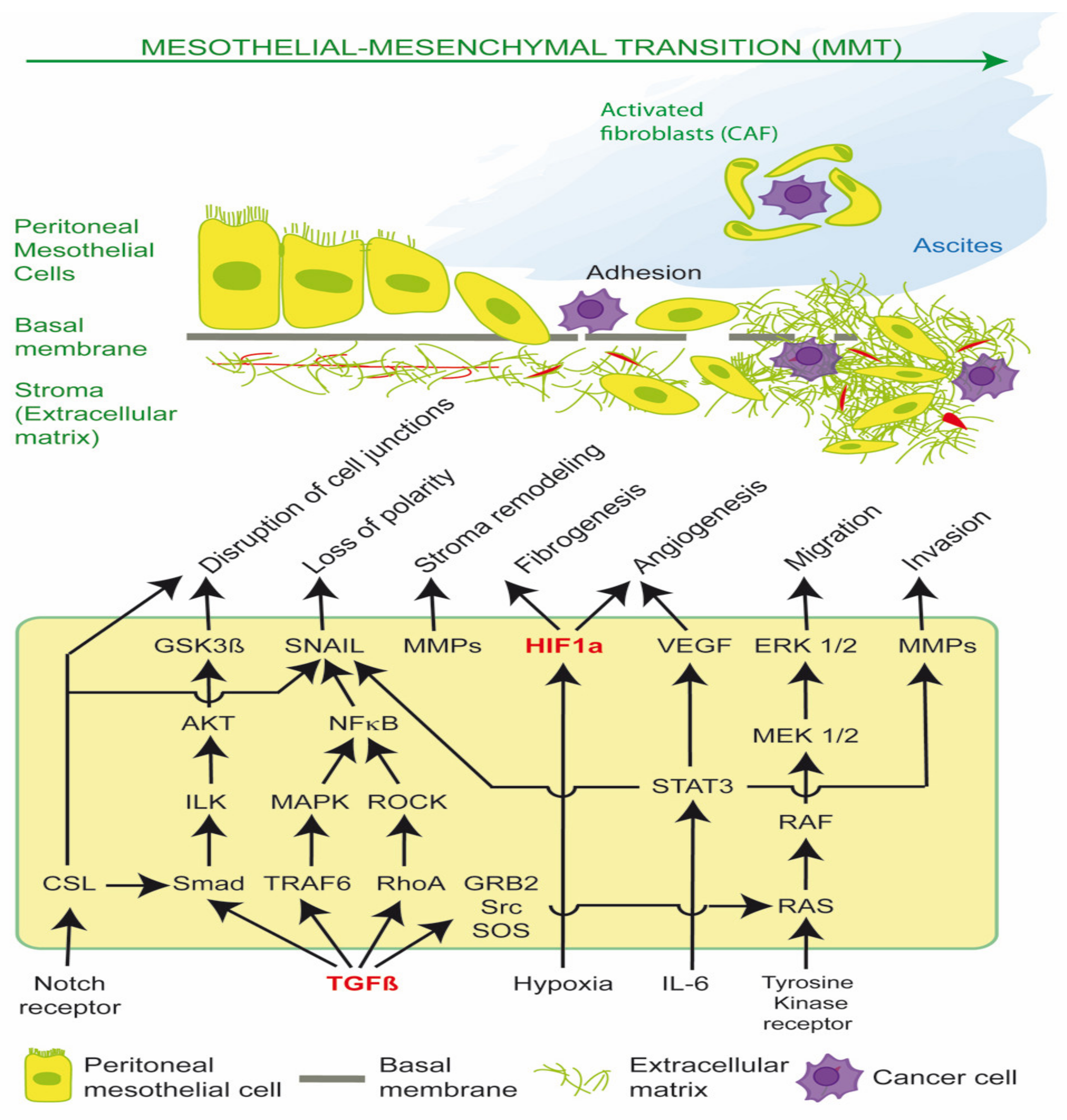
2. TGF-β1, Src and MMT
2.1. Cellular Homeostasis, Cytoplasmic Signaling and Glycolysis
2.2. Role of DAMP Receptors and NF-κB
2.3. Role of HMGB1
3. Generation of Activated Fibroblasts via MMT
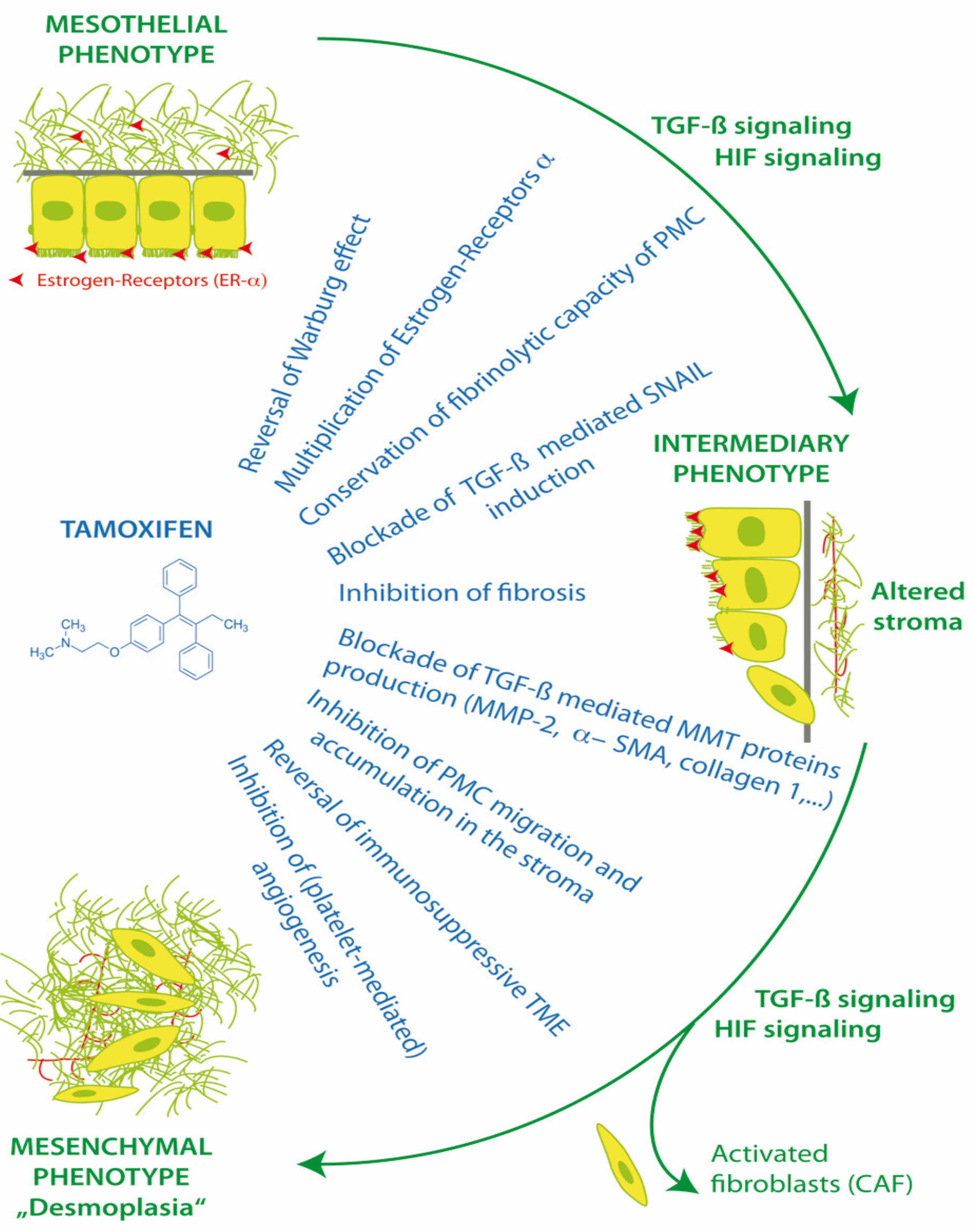
3.1. Therapeutic Approaches in MMT
3.2. Targeting Cancer Associated Fibroblasts
3.3. Tamoxifen and the Peritoneum in Encapsulating Peritoneal Sclerosis (EPS)
3.3.1. In Vitro Experiments
3.3.2. Animal Experiments
3.3.3. Clinical Findings
4. 17β-Estradiol, Hypoxia-Inducible Factor (HIF) and Vascular Endothelial Growth Factor (VEGF)
4.1. Estrogen Receptors (ER-β) and HIF-1β
4.2. 17β-Estradiol (E2) in the Tumor Microenvironment
5. TGF, Platelets, Podoplanin Promote Tumor Progression
5.1. Tamoxifen and Tumor Cell Platelet Activation
5.2. Tamoxifen and Megakaryocytes
5.3. Tamoxifen, Thrombocytosis, and Cancer
5.4. Tamoxifen and HSP90
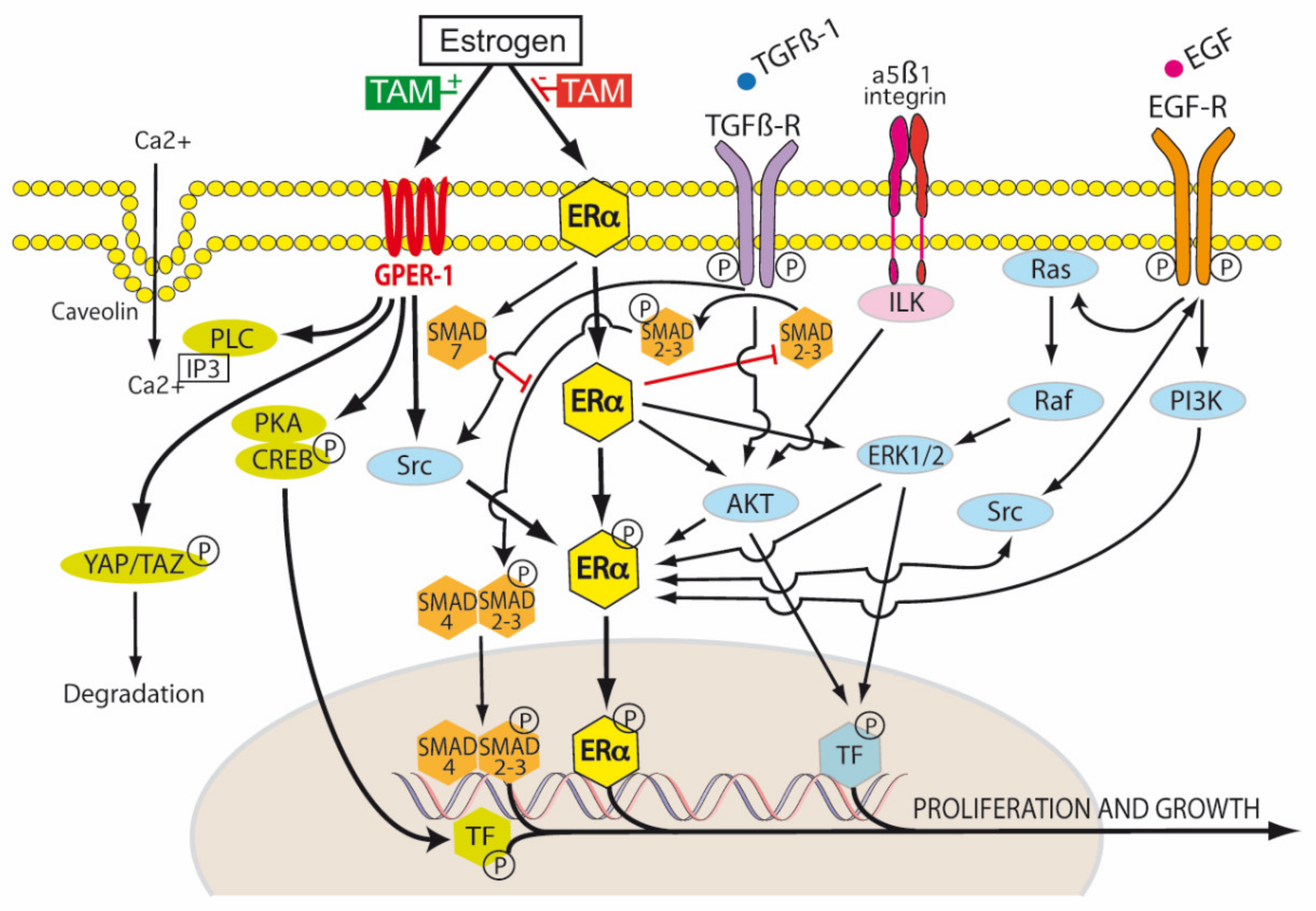
5.5. Tamoxifen and Different Estrogen Receptors (ER-α, ER-β, GPER-1)
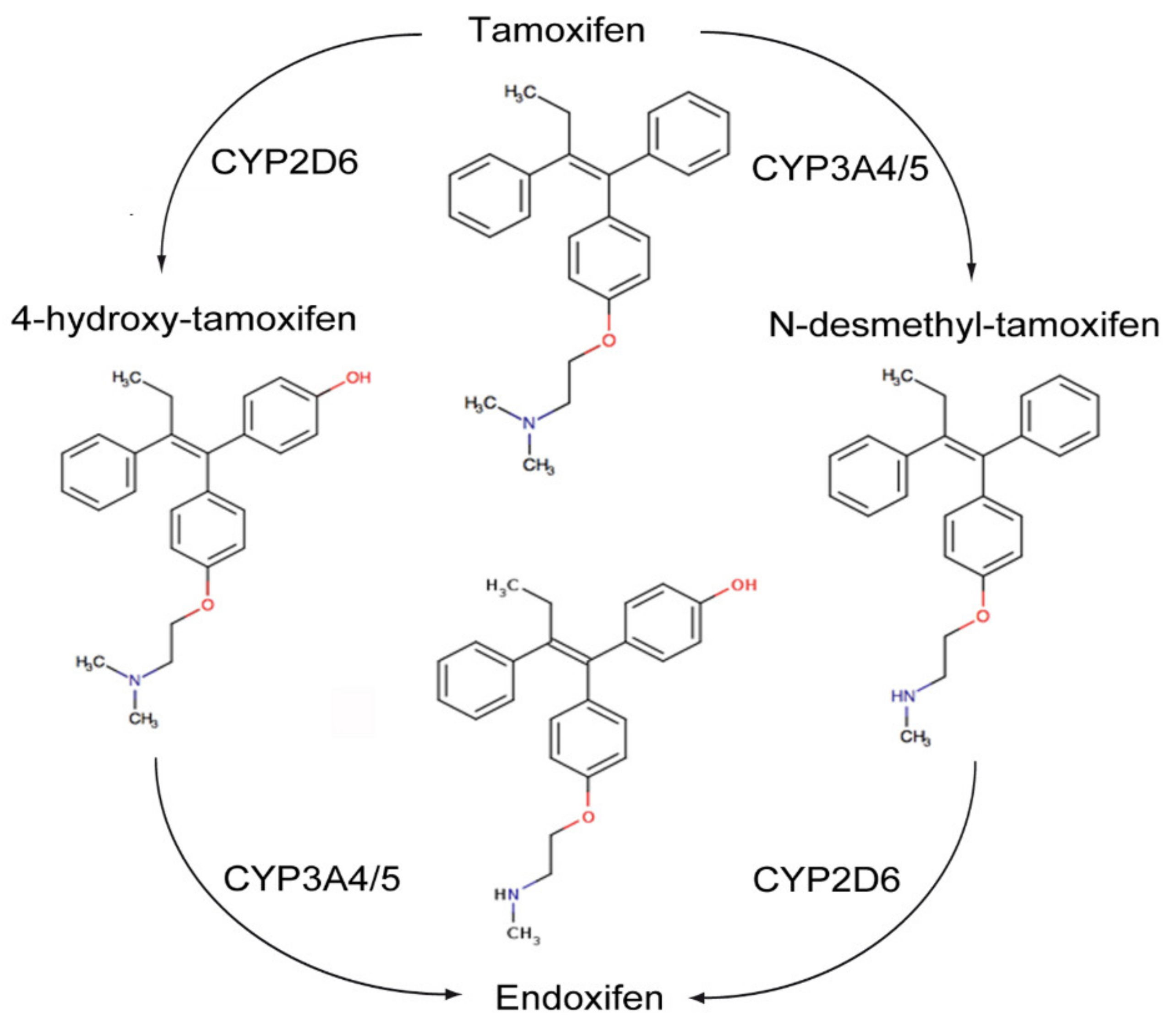
6. Estrogen Independent Effects of Tamoxifen
6.1. Tamoxifen and RAGE
6.1.1. Other Systemic Approaches for Treating MMT
6.1.2. Targeting the TGF-β1 Pathway
6.1.3. Src Inhibition
6.1.4. Connective Tissue Growth Factor Inhibition
6.1.5. Leptin Inhibition in EPS
6.1.6. Glucocorticosteroids (GC)
6.1.7. Vitamin D Receptor Agonists
6.2. Intraperitoneal Chemotherapy (IPC)
6.2.1. PARP Expression
6.2.2. Intraperitoneal Phytoestrogens—Cantrixil
6.2.3. Hyperthermic IntraPeritoneal Chemotherapy (HIPEC)
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| α-SMA | alpha smooth muscle actin |
| ABCC | ATP-binding cassette subfamily C member |
| ADP | adenosine diphosphate |
| AGE | advanced glycation end products |
| Akt | protein kinase B |
| ALK | activin like kinase |
| APA | antiplatelet antibodies |
| ARNT | Aryl hydrocarbon receptor nuclear translocator or HIF-1β |
| ATP | adenosine triphosphate |
| BCL-2 | B cell lymphoma-2 |
| BMP | bone morphogenetic protein |
| BRCA | breast cancer susceptibility gene |
| CAF | cancer associated fibroblast |
| CAPD | continuous ambulatory peritoneal dialysis |
| CCL5 | chemokine (C-C motif) ligand 5 |
| CCN | Cysteine-rich 61, Connective tissue growth factor, Nephroblastoma overexpressed |
| CD | cluster of differentiation |
| CDH1 | E-cadherin gene |
| CDK | cyclin dependent kinase |
| CLEC-2 | C-type lectin-like receptor 2 |
| COX | cyclo-oxygenase |
| CRC | colorectal cancer |
| CRS | cytoreductive surgery |
| CSC | cancer stem cell |
| CTGF | connective tissue growth factor, CCN2 |
| CXCL | C-X-C chemokine ligand |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DAMP | damage-associated molecular pattern |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| EM | endometriosis |
| EMT | epithelial mesenchymal transition |
| EPS | encapsulating peritoneal sclerosis |
| ER-α | estrogen receptor alpha |
| ERE | estrogen response element |
| ERK | extracellular signal-regulated kinase |
| FAK | focal adhesion kinase |
| FGF | fibroblast growth factor |
| FSP-1 | fibroblast specific protein-1 |
| GDP | glucose degradation products |
| GITR | glucocorticoid-induced tumor necrosis factor receptor-related |
| GLUT | membrane glucose transporter |
| GPR30 | G protein coupled estrogen receptor 30 (GPER) |
| HA | hyaluronan |
| HER2 | human epidermal growth factor |
| HGF | hepatocyte growth factor |
| HIF | hypoxia inducible factor |
| HIPEC | hyperthermic intraperitoneal chemotherapy |
| HMGB1 | high-mobility group box 1 protein |
| HRE | hypoxia response element |
| HSP | heat shock protein |
| IFN | interferon |
| IGF-1 | insulin-like growth factor-1 |
| IL | interleukin |
| ILK | integrin linked kinase |
| JNK | c-Jun N-terminal kinase |
| KRAS | Kirsten rat sarcoma virus oncogene |
| LDHA | Lactate dehydrogenase A |
| LMW | low molecular weight |
| MAP3K | mitogen-activating protein kinase kinase kinase |
| MDRT | multidrug resistance transporter |
| MHC | major histocompatibility complex |
| miRNA | microRNA |
| MMP | matrix metalloprotease |
| MMT | mesothelial mesenchymal transition |
| mRNA | messenger ribonucleic acid |
| mTOR | mammalian target of rapamycin |
| MYD88 | myeloid differentiation factor 88 |
| NF-E2 | nuclear factor erythroid-derived 2, Nrf2 |
| NF-κB | nuclear factor kappa light chain enhancer of activated B cells |
| NK | natural killer |
| NOD | nucleotide-binding oligomerization domain |
| NOV | nephroblastoma overexpressed gene |
| PAI-1 | plasminogen activator inhibitor-1 |
| PAMP | pathogen-associated molecular pattern |
| PARP | Poly (ADP-ribose) polymerase |
| peritoneal dialysis fluid | |
| p53 | tumor suppressor protein 53 |
| PDGF | platelet derived growth factor |
| PHD | prolyl hydroxylase |
| PI3K | phosphoinositide 3-kinase |
| PK | pyruvate kinase |
| PKM2 | pyruvate kinase isoenzyme M2 |
| PLCγ2 | phospholipase C gamma 2 |
| PM | peritoneal metastasis |
| PMC | peritoneal mesothelial cell |
| PPAR | peroxisome proliferator activated receptor |
| PPF | proplatelet formation |
| PRRX-1 | Paired Related Homeobox 1 |
| RAGE | receptor for advanced glycosylation end products |
| ROCK | rho-associated, coiled-coil-containing protein kinase |
| ROS | reactive oxygen species |
| RTK | receptor tyrosine kinase |
| SERD | selective estrogen receptor degrader |
| SERM | selective estrogen receptor modulator |
| SLUG | Zinc finger protein SNAI2 |
| SMAD | Small body +mothers against decapentaplegic |
| SOD | Superoxide dismutase |
| SNAIL | Zinc finger protein SNAI1 |
| Src | non receptor sarcoma tyrosine kinase |
| SRC | steroid receptor coactivator |
| Sp-1 | specificity protein 1 |
| STAT3 | signal transducer and activator of transcription |
| TAK-1 | TGF-β-activated kinase 1 or MAP3K7 |
| TAM | tumor (tissue) associated macrophages |
| TβR | transforming growth factor beta receptor |
| TF | tissue factor |
| TGF-β1 | transforming growth factor-β1 |
| Th | T helper |
| TKI | tyrosine kinase inhibitor |
| TIMP | tissue inhibitor of metalloproteases |
| TLR2 | toll-like receptor |
| TME | tumor microenvironment |
| TNF-α | tumor necrosis factor alpha |
| TPO | thrombopoietin |
| tPA | tissue plasminogen activator |
| TRAF6 | TNF receptor associated factor |
| Tregs | regulatory T cells |
| TWIST1 | twist basic helix loop helix transcription factor 1 |
| VEGF | vascular endothelial growth factor |
| VTE | venous thromboembolism |
References
- Wilson, R.B. Hypoxia, cytokines and stromal recruitment: Parallels between pathophysiology of encapsulating peritoneal sclerosis, endometriosis and peritoneal metastasis. Pleura Peritoneum 2018, 3, 20180103. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Yu, G.; Maroon, J.C.; D’Agostino, D.P. Press-pulse: A novel therapeutic strategy for the metabolic management of cancer. Nutr. Metab. (Lond.) 2017, 14, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laitala, A.; Erler, J.T. Hypoxic Signalling in Tumour Stroma. Front. Oncol. 2018, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, C.; Gan, G.; Zhang, J.; Wu, J.; Miao, Y.; Zhang, M.; Li, B.; Mi, J. Cancer-associated fibroblasts enhance tumor (18)F-FDG uptake and contribute to the intratumor heterogeneity of PET-CT. Theranostics 2018, 8, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed. Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zahari, M.S.; Renuse, S.; Sahasrabuddhe, N.A.; Chaerkady, R.; Kim, M.S.; Fackler, M.J.; Stampfer, M.; Gabrielson, E.; Sukumar, S.; et al. Quantitative phosphoproteomic analysis reveals reciprocal activation of receptor tyrosine kinases between cancer epithelial cells and stromal fibroblasts. Clin. Proteom. 2018, 15, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archid, R.; Solass, W.; Tempfer, C.; Konigsrainer, A.; Adolph, M.; Reymond, M.A.; Wilson, R.B. Cachexia Anorexia Syndrome and Associated Metabolic Dysfunction in Peritoneal Metastasis. Int. J. Mol. Sci. 2019, 20, 5444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, J.; Korc, M. Pancreatic cancer stroma: Friend or foe? Cancer Cell 2014, 25, 711–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Epps, H.L. Peyton Rous: Father of the tumor virus. J. Exp. Med. 2005, 201, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Davies, K.J.; Forman, H.J. TGFbeta1 rapidly activates Src through a non-canonical redox signaling mechanism. Arch. Biochem. Biophys. 2015, 568, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopa, K.B.; Kusiak, A.A.; Szopa, M.D.; Ferdek, P.E.; Jakubowska, M.A. Pancreatic Cancer and Its Microenvironment-Recent Advances and Current Controversies. Int. J. Mol. Sci. 2020, 21, 3218. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Schiemann, W.P. TGF-β Stimulation of EMT Programs Elicits Non-genomic ER-α Activity and Anti-estrogen Resistance in Breast Cancer Cells. J. Cancer Metastasis Treat. 2017, 3, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoneum, A.; Abdulfattah, A.Y.; Warren, B.O.; Shu, J.; Said, N. Redox Homeostasis and Metabolism in Cancer: A Complex Mechanism and Potential Targeted Therapeutics. Int. J. Mol. Sci. 2020, 21, 3100. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertheloot, D.; Latz, E. HMGB1, IL-1alpha, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Batista, P.R.; Palacios, R.; Martin, A.; Hernanz, R.; Medici, C.T.; Silva, M.A.; Rossi, E.M.; Aguado, A.; Vassallo, D.V.; Salaices, M.; et al. Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS ONE 2014, 9, e104020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, S.; Miao, J.; Zhu, Z.; Xu, E.; Shi, L.; Ai, S.; Wang, F.; Kang, X.; Chen, H.; Lu, X.; et al. NADPH oxidase 4 regulates anoikis resistance of gastric cancer cells through the generation of reactive oxygen species and the induction of EGFR. Cell Death Dis. 2018, 9, 948. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, R.; Kato, M.; Miyagawa, S.; Kamata, T. Nox4-derived ROS signaling contributes to TGF-beta-induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2013, 33, 4431–4438. [Google Scholar] [PubMed]
- Jain, A.; Kaczanowska, S.; Davila, E. IL-1 Receptor-Associated Kinase Signaling and Its Role in Inflammation, Cancer Progression, and Therapy Resistance. Front. Immunol 2014, 5, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Xie, Y.; Zhang, Q.; Hou, W.; Jiang, Q.; Zhu, S.; Liu, J.; Zeng, D.; Wang, H.; Bartlett, D.L.; et al. Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer. Cell Res. 2017, 27, 916–932. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Sung, J.Y.; Park, E.K.; Kho, S.; Koo, K.H.; Park, S.Y.; Goh, S.H.; Jeon, Y.K.; Oh, S.; Park, B.K.; et al. Regulation of anoikis resistance by NADPH oxidase 4 and epidermal growth factor receptor. Br. J. Cancer 2017, 116, 370–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, W.G. The Role of Damage-Associated Molecular Patterns (DAMPs) in Human Diseases: Part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ. Med. J. 2015, 15, e157–e170. [Google Scholar] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef] [PubMed]
- Piyush, T.; Rhodes, J.M.; Yu, L.G. MUC1 O-glycosylation contributes to anoikis resistance in epithelial cancer cells. Cell Death Discov. 2017, 3, 17044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, S.Y.; Lee, K.W.; Shin, D.; An, S.; Cho, K.H.; Kim, S.H. A positive feedback loop bi-stably activates fibroblasts. Nat. Commun. 2018, 9, 3016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.L.; Lin, Y.; Jiang, J.; Tang, Z.; Yang, S.; Lu, L.; Liang, Y.; Liu, X.; Tan, J.; Hu, X.G.; et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J. Pathol. 2017, 243, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Guiu, J.; Hannezo, E.; Yui, S.; Demharter, S.; Ulyanchenko, S.; Maimets, M.; Jorgensen, A.; Perlman, S.; Lundvall, L.; Mamsen, L.S.; et al. Tracing the origin of adult intestinal stem cells. Nature 2019, 570, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Oak, A.S.; Athar, M.; Yusuf, N.; Elmets, C.A. UV and skin: Photocarcinogenesis. In Environment and Skin; Krutmann, H.F., Merk, J., Eds.; Springer International Publishing: Cham, Switzerland, 2018; Chapter 8; pp. 67–103. [Google Scholar]
- Thomma, B.P.; Nurnberger, T.; Joosten, M.H. Of PAMPs and effectors: The blurred PTI-ETI dichotomy. Plant Cell 2011, 23, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, K.; Tancharoen, S.; Ito, T.; Morimoto-Yamashita, Y.; Miura, N.; Kawahara, K.; Maruyama, I.; Murai, Y.; Tanaka, E. Potential of the angiotensin receptor blockers (ARBs) telmisartan, irbesartan, and candesartan for inhibiting the HMGB1/RAGE axis in prevention and acute treatment of stroke. Int. J. Mol. Sci. 2013, 14, 18899–18924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.S.; Shaked, Y.; Tsai, K.K. Targeting the Interplay between Cancer Fibroblasts, Mesenchymal Stem Cells, and Cancer Stem Cells in Desmoplastic Cancers. Front. Oncol. 2019, 9, 688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gok Yavuz, B.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1(+) TAMs. Sci. Rep. 2019, 9, 3172. [Google Scholar] [CrossRef] [PubMed]
- Stastna, M.; Janeckova, L.; Hrckulak, D.; Kriz, V.; Korinek, V. Human Colorectal Cancer from the Perspective of Mouse Models. Genes (Basel) 2019, 10, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, G.; Hu, C. Molecular Classification of Gastric Adenocarcinoma. Gastroenterol. Res. 2019, 12, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, K.H.; Kim, D.H.; Kim, H.; Lee, D.H.; Cheong, J.H. Differences in TGF-beta1 signaling and clinicopathologic characteristics of histologic subtypes of gastric cancer. BMC Cancer 2016, 16, 60. [Google Scholar] [CrossRef] [Green Version]
- Pak, K.H.; Park, K.C.; Cheong, J.H. VEGF-C induced by TGF- beta1 signaling in gastric cancer enhances tumor-induced lymphangiogenesis. BMC Cancer 2019, 19, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, H.; Lee, N.Y. Multifaceted roles of TAK1 signaling in cancer. Oncogene 2020, 39, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Worzfeld, T.; Pogge von Strandmann, E.; Huber, M.; Adhikary, T.; Wagner, U.; Reinartz, S.; Muller, R. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front. Oncol 2017, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, V.J.; Ahmad, S.F.; Duncan, W.C.; Horne, A.W. The role of TGF-beta in the pathophysiology of peritoneal endometriosis. Hum. Reprod. Update 2017, 23, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, B.H.; Chon, S.J.; Choi, Y.S.; Cho, S.; Lee, B.S.; Seo, S.K. Pathophysiology of Endometriosis: Role of High Mobility Group Box-1 and Toll-Like Receptor 4 Developing Inflammation in Endometrium. PLoS ONE 2016, 11, e0148165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Mateo, G.T.; Aguirre, A.R.; Loureiro, J.; Abensur, H.; Sandoval, P.; Sanchez-Tomero, J.A.; del Peso, G.; Jimenez-Heffernan, J.A.; Ruiz-Carpio, V.; Selgas, R.; et al. Rapamycin Protects from Type-I Peritoneal Membrane Failure Inhibiting the Angiogenesis, Lymphangiogenesis, and Endo-MT. Biomed. Res. Int. 2015, 2015, 989560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, F.M.O.; Costalonga, E.C.; Silva, C.; Carreira, A.C.O.; Gomes, S.A.; Sogayar, M.C.; Fanelli, C.; Noronha, I.L. Tamoxifen and bone morphogenic protein-7 modulate fibrosis and inflammation in the peritoneal fibrosis model developed in uremic rats. Mol. Med. 2019, 25, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gónzalez-Mateo, G.; Gallardo, J.M.; Sánchez-Tomero, J.A.; Majano, P.; Flores-Maldonado, E.; Paniagua, R.; Selgas, R.; López-Cabrera, M.; Aguilera, A. Pharmacological Preservation of Peritoneal Membrane in Peritoneal Dialysis, Some Special Problems in Peritoneal Dialysis; Robert, E., Ed.; IntechOpen: London, UK, 2016. [Google Scholar] [CrossRef] [Green Version]
- Sekiguchi, Y.; Zhang, J.; Patterson, S.; Liu, L.; Hamada, C.; Tomino, Y.; Margetts, P.J. Rapamycin inhibits transforming growth factor beta-induced peritoneal angiogenesis by blocking the secondary hypoxic response. J. Cell Mol. Med. 2012, 16, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Liappas, G.; Gonzalez-Mateo, G.T.; Sanchez-Diaz, R.; Lazcano, J.J.; Lasarte, S.; Matesanz-Marin, A.; Zur, R.; Ferrantelli, E.; Ramirez, L.G.; Aguilera, A.; et al. Immune-Regulatory Molecule CD69 Controls Peritoneal Fibrosis. J. Am. Soc. Nephrol. 2016, 27, 3561–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, J.; Sandoval, P.; del Peso, G.; Gonzalez-Mateo, G.; Fernandez-Millara, V.; Santamaria, B.; Bajo, M.A.; Sanchez-Tomero, J.A.; Guerra-Azcona, G.; Selgas, R.; et al. Tamoxifen ameliorates peritoneal membrane damage by blocking mesothelial to mesenchymal transition in peritoneal dialysis. PLoS ONE 2013, 8, e61165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, C.; Morfoisse, F.; Tatin, F.; Zamora, A.; Zahreddine, R.; Henrion, D.; Arnal, J.F.; Lenfant, F.; Garmy-Susini, B. The Impact of Estrogen Receptor in Arterial and Lymphatic Vascular Diseases. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Dellê, H.; Rocha, J.R.C.; Cavaglieri, R.C.; Vieira, J.M.; Malheiros, D.M.; Noronha, I.L. Antifibrotic effect of tamoxifen in a model of progressive renal disease. J. Am. Soc. Nephrol. 2012, 23, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carthy, J.M.; Sundqvist, A.; Heldin, A.; van Dam, H.; Kletsas, D.; Heldin, C.H.; Moustakas, A. Tamoxifen Inhibits TGF-β-Mediated Activation of Myofibroblasts by Blocking Non-Smad Signaling Through ERK1/2. J. Cell. Physiol. 2015, 230, 3084–3092. [Google Scholar] [CrossRef] [PubMed]
- Korte, M.R.; Sampimon, D.E.; Lingsma, H.F.; Fieren, M.W.; Looman, C.W.; Zietse, R.; Weimar, W.; Betjes, M.G.; Dutch Multicenter, E.P.S.S. Risk factors associated with encapsulating peritoneal sclerosis in Dutch EPS study. Perit. Dial. Int. 2011, 31, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.A.; Bargman, J.; van Biesen, W.; Chang, M.Y.; Finkelstein, F.O.; Hurst, H.; Johnson, D.W.; Kawanishi, H.; Lambie, M.; de Moraes, T.P.; et al. Length of Time on Peritoneal Dialysis and Encapsulating Peritoneal Sclerosis - Position Paper for ISPD: 2017 Update. Perit. Dial. Int. 2017, 37, 362–374. [Google Scholar] [CrossRef] [PubMed]
- George, A.L.; Rajoria, S.; Suriano, R.; Mittleman, A.; Tiwari, R.K. Hypoxia and estrogen are functionally equivalent in breast cancer-endothelial cell interdependence. Mol. Cancer 2012, 11, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Mandl, M.; Lieberum, M.K.; Depping, R. A HIF-1alpha-driven feed-forward loop augments HIF signalling in Hep3B cells by upregulation of ARNT. Cell Death Dis. 2016, 7, e2284. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Park, Y.; Cho, J.; Park, C.; Park, J.; Park, Y.K.; Park, H.; Lee, Y. Estrogen receptor beta inhibits transcriptional activity of hypoxia inducible factor-1 through the downregulation of arylhydrocarbon receptor nuclear translocator. Breast Cancer Res. 2011, 13, R32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberger, N.J.; Somasundaram, A.; Stabile, L.P. The Role of the Estrogen Pathway in the Tumor Microenvironment. Int. J. Mol. Sci. 2018, 19, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, A.; Miyata, K.; Fujita, N. Platelet-activating factor podoplanin: From discovery to drug development. Cancer Metastasis Rev. 2017, 36, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, K.; Kono, H.; Nakata, Y.; Akazawa, Y.; Wakana, H.; Fukushima, H.; Fujii, H. Expression of podoplanin in stromal fibroblasts plays a pivotal role in the prognosis of patients with pancreatic cancer. Surg. Today 2018, 48, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, A.; Okitaka, M.; Takagi, S.; Takami, M.; Sato, S.; Nishio, M.; Okumura, S.; Fujita, N. A critical role of platelet TGF-beta release in podoplanin-mediated tumour invasion and metastasis. Sci. Rep. 2017, 7, 42186. [Google Scholar] [CrossRef] [PubMed]
- Alsina-Sanchis, E.; Figueras, A.; Lahiguera, A.; Gil-Martin, M.; Pardo, B.; Piulats, J.M.; Marti, L.; Ponce, J.; Matias-Guiu, X.; Vidal, A.; et al. TGFbeta Controls Ovarian Cancer Cell Proliferation. Int. J. Mol. Sci. 2017, 18, 1658. [Google Scholar] [CrossRef] [PubMed]
- Alexander, E.T.; Minton, A.R.; Peters, M.C.; van Ryn, J.; Gilmour, S.K. Thrombin inhibition and cisplatin block tumor progression in ovarian cancer by alleviating the immunosuppressive microenvironment. Oncotarget 2016, 7, 85291–85305. [Google Scholar] [CrossRef] [PubMed]
- Bogatkevich, G.S.; Ludwicka-Bradley, A.; Nietert, P.J.; Akter, T.; van Ryn, J.; Silver, R.M. Antiinflammatory and antifibrotic effects of the oral direct thrombin inhibitor dabigatran etexilate in a murine model of interstitial lung disease. Arthritis Rheum. 2011, 63, 1416–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placke, T.; Orgel, M.; Schaller, M.; Jung, G.; Rammensee, H.G.; Kopp, H.G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placke, T.; Salih, H.R.; Kopp, H.G. GITR ligand provided by thrombopoietic cells inhibits NK cell antitumor activity. J. Immunol. 2012, 189, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erices, R.; Cubillos, S.; Aravena, R.; Santoro, F.; Marquez, M.; Orellana, R.; Ramirez, C.; Gonzalez, P.; Fuenzalida, P.; Bravo, M.L.; et al. Diabetic concentrations of metformin inhibit platelet-mediated ovarian cancer cell progression. Oncotarget 2017, 8, 20865–20880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.B.; Lee, S.K.; Bach, D.H. The Role of Platelets in the Tumor-Microenvironment and the Drug Resistance of Cancer Cells. Cancers (Basel) 2019, 11, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.E.; Forward, J.A.; Tippy, M.D.; Ceglowski, J.R.; El-Husayni, S.; Kulenthirarajan, R.; Machlus, K.R.; Mayer, E.L.; Italiano Jr, J.E.; Battinelli, E.M. Tamoxifen Directly Inhibits Platelet Angiogenic Potential and Platelet-Mediated Metastasis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Antiplatelet agents for cancer treatment: A real perspective or just an echo from the past? Cancer Metastasis Rev. 2017, 36, 305–329. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Yoshikawa, J.; Hashimoto, A.; Yamamoto, M.; Payne, A.H.; Todokoro, K. Proplatelet formation of megakaryocytes is triggered by autocrine-synthesized estradiol. Genes Dev. 2003, 17, 2864–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Xu, Y.; Yang, K.; Chen, S.; Wang, X.; Wang, S.; Wang, C.; Shen, M.; Chen, F.; Chen, M.; et al. Estrogen promotes megakaryocyte polyploidization via estrogen receptor beta-mediated transcription of GATA1. Leukemia 2017, 31, 945–956. [Google Scholar] [CrossRef]
- Khetawat, G.; Faraday, N.; Nealen, M.L.; Vijayan, K.V.; Bolton, E.; Noga, S.J.; Bray, P.F. Human megakaryocytes and platelets contain the estrogen receptor beta and androgen receptor (AR): Testosterone regulates AR expression. Blood 2000, 95, 2289–2296. [Google Scholar] [CrossRef]
- Daly, M.E. Determinants of platelet count in humans. Haematologica 2011, 96, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelbaset-Ismail, A.; Suszynska, M.; Borkowska, S.; Adamiak, M.; Ratajczak, J.; Kucia, M.; Ratajczak, M.Z. Human haematopoietic stem/progenitor cells express several functional sex hormone receptors. J. Cell. Mol. Med. 2016, 20, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Bottsford-Miller, J.; Choi, H.J.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Haemmerle, M.; Nick, A.M.; Pradeep, S.; Zand, B.; Previs, R.A.; et al. Differential platelet levels affect response to taxane-based therapy in ovarian cancer. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2015, 21, 602–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voutsadakis, I.A. Thrombocytosis as a prognostic marker in gastrointestinal cancers. World J. Gastrointest. Oncol. 2014, 6, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-beta Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Love, R.R.; Surawicz, T.S.; Williams, E.C. Antithrombin III level, fibrinogen level, and platelet count changes with adjuvant tamoxifen therapy. Arch. Intern. Med. 1992, 152, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Vitseva, O.; Flockhart, D.A.; Jin, Y.; Varghese, S.; Freedman, J.E. The effects of tamoxifen and its metabolites on platelet function and release of reactive oxygen intermediates. J. Pharm. Exp. 2005, 312, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Dhamad, A.E.; Zhou, Z.; Zhou, J.; Du, Y. Systematic Proteomic Identification of the Heat Shock Proteins (Hsp) that Interact with Estrogen Receptor Alpha (ERalpha) and Biochemical Characterization of the ERalpha-Hsp70 Interaction. PLoS ONE 2016, 11, e0160312. [Google Scholar] [CrossRef] [PubMed]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitesell, L.; Santagata, S.; Mendillo, M.L.; Lin, N.U.; Proia, D.A.; Lindquist, S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. USA 2014, 111, 18297–18302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Deng, H.; Zou, F.; Fu, Z.; Chen, Y.; Wang, Z.; Liu, L. ER-alpha36-mediated gastric cancer cell proliferation via the c-Src pathway. Oncol. Lett. 2013, 6, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Liu, R.; Yan, Y.; Pan, X.; Wang, M.; Han, X.; Ren, H.; Zhang, Z. Expression of estrogen receptors and androgen receptor and their clinical significance in gastric cancer. Oncotarget 2017, 8, 40765–40777. [Google Scholar] [CrossRef] [Green Version]
- van der Post, R.S.; Gullo, I.; Oliveira, C.; Tang, L.H.; Grabsch, H.I.; O’Donovan, M.; Fitzgerald, R.C.; van Krieken, H.; Carneiro, F. Histopathological, Molecular, and Genetic Profile of Hereditary Diffuse Gastric Cancer: Current Knowledge and Challenges for the Future. Adv. Exp. Med. Biol. 2016, 908, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Do, I.G.; Jang, J.; Kim, S.T.; Kim, K.M.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; et al. Anti-tumor efficacy of fulvestrant in estrogen receptor positive gastric cancer. Sci. Rep. 2014, 4, 7592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwart, W.; de Leeuw, R.; Rondaij, M.; Neefjes, J.; Mancini, M.A.; Michalides, R. The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. J. Cell Sci. 2010, 123, 1253–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkhem, T.; Carlsson, B.; Nilsson, Y.; Enmark, E.; Gustafsson, J.; Nilsson, S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol. Pharm. 1998, 54, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Rondon-Lagos, M.; Villegas, V.E.; Rangel, N.; Sanchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Subramaniam, M.; Grygo, S.B.; Sun, Z.; Negron, V.; Lingle, W.L.; Goetz, M.P.; Ingle, J.N.; Spelsberg, T.C.; Hawse, J.R. Estrogen receptor-beta sensitizes breast cancer cells to the anti-estrogenic actions of endoxifen. Breast Cancer Res. 2011, 13, R27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Schuler-Toprak, S.; Moehle, C.; Skrzypczak, M.; Ortmann, O.; Treeck, O. Effect of estrogen receptor beta agonists on proliferation and gene expression of ovarian cancer cells. BMC Cancer 2017, 17, 319. [Google Scholar] [CrossRef] [PubMed]
- Nass, N.; Bromme, H.J.; Hartig, R.; Korkmaz, S.; Sel, S.; Hirche, F.; Ward, A.; Simm, A.; Wiemann, S.; Lykkesfeldt, A.E.; et al. Differential response to alpha-oxoaldehydes in tamoxifen resistant MCF-7 breast cancer cells. PLoS ONE 2014, 9, e101473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Hilakivi-Clarke, L.; Clarke, R. Molecular mechanisms of tamoxifen-associated endometrial cancer (Review). Oncol. Lett. 2015, 9, 1495–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, E.; Lachowski, D.; Robinson, B.; Sarper, M.; Teppo, J.S.; Thorpe, S.D.; Lieberthal, T.J.; Iwamoto, K.; Lee, D.A.; Okada-Hatakeyama, M.; et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. 2019, 20, e46557. [Google Scholar] [CrossRef] [PubMed]
- Cortes, E.; Sarper, M.; Robinson, B.; Lachowski, D.; Chronopoulos, A.; Thorpe, S.D.; Lee, D.A.; Del Rio Hernandez, A.E. GPER is a mechanoregulator of pancreatic stellate cells and the tumor microenvironment. EMBO Rep. 2019, 20, e46556. [Google Scholar] [CrossRef] [PubMed]
- Pein, M.; Oskarsson, T. Tamoxifen calms down the distressed PDAC stroma. EMBO Rep. 2019, 20, e47334. [Google Scholar] [CrossRef] [PubMed]
- Yoneura, N.; Takano, S.; Yoshitomi, H.; Nakata, Y.; Shimazaki, R.; Kagawa, S.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Miyazaki, M.; et al. Expression of annexin II and stromal tenascin C promotes epithelial to mesenchymal transition and correlates with distant metastasis in pancreatic cancer. Int. J. Mol. Med. 2018, 42, 821–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.; Vukelja, S.J.; Ann Holmes, F.; Blum, J.L.; McIntyre, K.J.; Lindquist, D.L.; Osborne, C.R.; Sanchez, I.J.; Goldschmidt, J.H.; Wang, Y.; et al. Randomized phase-II evaluation of letrozole plus dasatinib in hormone receptor positive metastatic breast cancer patients. NPJ Breast Cancer 2019, 5, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowell, L.N.; Graham, J.D.; Bouton, A.H.; Clarke, C.L.; O’Neill, G.M. Tamoxifen treatment promotes phosphorylation of the adhesion molecules, p130Cas/BCAR1, FAK and Src, via an adhesion-dependent pathway. Oncogene 2006, 25, 7597–7607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yu, D. Targeting Src family kinases in anti-cancer therapies: Turning promise into triumph. Trends Pharm. Sci. 2012, 33, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekele, R.T.; Venkatraman, G.; Liu, R.Z.; Tang, X.; Mi, S.; Benesch, M.G.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, S.; Ohmichi, M.; Kimura, A.; Ikebuchi, Y.; Hisamoto, K.; Arimoto-Ishida, E.; Nishio, Y.; Takahashi, K.; Tasaka, K.; Murata, Y. Tamoxifen inhibits cell proliferation via mitogen-activated protein kinase cascades in human ovarian cancer cell lines in a manner not dependent on the expression of estrogen receptor or the sensitivity to cisplatin. Endocrinology 2004, 145, 1302–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.X.; Tiwari, A.K.; Wu, H.C.; Chen, Z.S. Overexpression of P-glycoprotein induces acquired resistance to imatinib in chronic myelogenous leukemia cells. Chin. J. Cancer 2012, 31, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedukhina, A.S.; Sundaramoorthy, E.; Hara, M.; Kumai, T.; Sato, K. Beyond resistance to PARP inhibition: Mechanisms and effective treatment options. Cancer Cell Microenviron. 2015, 2, e821. [Google Scholar]
- Cheng, R.; Liu, Y.J.; Cui, J.W.; Yang, M.; Liu, X.L.; Li, P.; Wang, Z.; Zhu, L.Z.; Lu, S.Y.; Zou, L.; et al. Aspirin regulation of c-myc and cyclinD1 proteins to overcome tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncotarget 2017, 8, 30252–30264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiavarina, B.; Nokin, M.J.; Bellier, J.; Durieux, F.; Bletard, N.; Sherer, F.; Lovinfosse, P.; Peulen, O.; Verset, L.; Dehon, R.; et al. Methylglyoxal-Mediated Stress Correlates with High Metabolic Activity and Promotes Tumor Growth in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichiule, P.; Chavez, J.C.; Schmidt, A.M.; Vannucci, S.J. Hypoxia-inducible factor-1 mediates neuronal expression of the receptor for advanced glycation end products following hypoxia/ischemia. J. Biol. Chem. 2007, 282, 36330–36340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nass, N.; Ignatov, A.; Andreas, L.; Weissenborn, C.; Kalinski, T.; Sel, S. Accumulation of the advanced glycation end product carboxymethyl lysine in breast cancer is positively associated with estrogen receptor expression and unfavorable prognosis in estrogen receptor-negative cases. Histochem. Cell Biol. 2017, 147, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, T.; Ye, G.; Shen, Z.; Hu, Y.; Mou, T.; Yu, J.; Li, S.; Liu, H.; Li, G. Overexpression of the Receptor for Advanced Glycation Endproducts (RAGE) is associated with poor prognosis in gastric cancer. PLoS ONE 2015, 10, e0122697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Hou, W.; Zhang, Q.; Chen, R.; Lee, Y.J.; Bartlett, D.L.; Lotze, M.T.; Tang, D.; Zeh, H.J. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. 2014, 5, e1480. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, C.T.; Nagaraj, R.H. AGE-RAGE interaction in the TGFbeta2-mediated epithelial to mesenchymal transition of human lens epithelial cells. Glycoconj. J. 2016, 33, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Park, S.; Lakatta, E.G. RAGE signaling in inflammation and arterial aging. Front. Biosci. (Landmark Ed.) 2009, 14, 1403–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Yonekura, H.; Yamagishi, S.; Fujimori, H.; Yamamoto, Y.; Yamamoto, H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J. Biol. Chem. 2000, 275, 25781–25790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Leung, J.C.; Lam, M.F.; Chan, L.Y.; Tsang, A.W.; Lan, H.Y.; Lai, K.N. Smad7 transgene attenuates peritoneal fibrosis in uremic rats treated with peritoneal dialysis. J. Am. Soc. Nephrol. 2007, 18, 2689–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moinuddin, Z.; Summers, A.; Van Dellen, D.; Augustine, T.; Herrick, S.E. Encapsulating peritoneal sclerosis-a rare but devastating peritoneal disease. Front. Physiol. 2014, 5, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, L.; Xu, L.; Shi, Y.; Liu, F.; Qi, H.; Liu, N.; Zhuang, S. Targeting Src attenuates peritoneal fibrosis and inhibits the epithelial to mesenchymal transition. Oncotarget 2017, 8, 83872–83889. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Peng, X.; Liu, F.; Tang, C.; Hu, C.; Xu, X.; Wang, M.; Luo, Y.; Yang, S.; Song, P.; et al. AKT regulation of mesothelial-to-mesenchymal transition in peritoneal dialysis is modulated by Smurf2 and deubiquitinating enzyme USP4. BMC Cell Biol. 2015, 16, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelis, T.; Oreopoulos, D.G. Update on potential medical treatments for encapsulating peritoneal sclerosis; human and experimental data. Int. Urol. Nephrol. 2011, 43, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, K.Y.; Shyu, R.S.; Fang, C.C.; Tsai, C.C.; Lee, P.H.; Tsai, T.J.; Hsieh, B.S. Dipyridamole inhibits human peritoneal mesothelial cell proliferation in vitro and attenuates rat peritoneal fibrosis in vivo. Kidney Int. 2001, 59, 2316–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagirdar, R.M.; Bozikas, A.; Zarogiannis, S.G.; Bartosova, M.; Schmitt, C.P.; Liakopoulos, V. Encapsulating Peritoneal Sclerosis: Pathophysiology and Current Treatment Options. Int. J. Mol. Sci. 2019, 20, 5765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Fan, Y.; Zhang, X.; Huang, W.; Ma, J. 1,25(OH)2D3 treatment attenuates high glucoseinduced peritoneal epithelial to mesenchymal transition in mice. Mol. Med. Rep. 2017, 16, 3817–3824. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tan, Y.; Yu, W.; Zheng, S.; Zhang, S.; Sun, L.; Ding, K. Small role with big impact: miRNAs as communicators in the cross-talk between cancer-associated fibroblasts and cancer cells. Int. J. Biol. Sci. 2017, 13, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.B.; Solass, W.; Archid, R.; Weinreich, F.J.; Konigsrainer, A.; Reymond, M.A. Resistance to anoikis in transcoelomic shedding: The role of glycolytic enzymes. Pleura Peritoneum 2019, 4, 20190003. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Li, Y.; Zhao, J.; Li, Q.; Yang, B.; Wang, Y.; Zhu, Z.; Sun, H.; Zhai, Z. Transforming growth factor-beta1 contributes to oxaliplatin resistance in colorectal cancer via epithelial to mesenchymal transition. Oncol. Lett. 2017, 14, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Auer, K.; Bachmayr-Heyda, A.; Sukhbaatar, N.; Aust, S.; Schmetterer, K.G.; Meier, S.M.; Gerner, C.; Grimm, C.; Horvat, R.; Pils, D. Role of the immune system in the peritoneal tumor spread of high grade serous ovarian cancer. Oncotarget 2016, 7, 61336–61354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rynne-Vidal, A.; Au-Yeung, C.L.; Jimenez-Heffernan, J.A.; Perez-Lozano, M.L.; Cremades-Jimeno, L.; Barcena, C.; Cristobal-Garcia, I.; Fernandez-Chacon, C.; Yeung, T.L.; Mok, S.C.; et al. Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J. Pathol. 2017, 242, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Barron, L.; Hinck, C.S.; Petrunak, E.M.; Cano, K.E.; Thangirala, A.; Iskra, B.; Brothers, M.; Vonberg, M.; Leal, B.; et al. An engineered transforming growth factor beta (TGF-beta) monomer that functions as a dominant negative to block TGF-beta signaling. J. Biol. Chem. 2017, 292, 7173–7188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Tran, T.; Dwabe, S.; Sarkissyan, M.; Kim, J.; Nava, M.; Clayton, S.; Pietras, R.; Farias-Eisner, R.; Vadgama, J.V. A83-01 inhibits TGF-beta-induced upregulation of Wnt3 and epithelial to mesenchymal transition in HER2-overexpressing breast cancer cells. Breast Cancer Res. Treat. 2017, 163, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartscht, T.; Rosien, B.; Rades, D.; Kaufmann, R.; Biersack, H.; Lehnert, H.; Gieseler, F.; Ungefroren, H. Dasatinib blocks transcriptional and promigratory responses to transforming growth factor-beta in pancreatic adenocarcinoma cells through inhibition of Smad signalling: Implications for in vivo mode of action. Mol. Cancer 2015, 14, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yingling, J.M.; McMillen, W.T.; Yan, L.; Huang, H.; Sawyer, J.S.; Graff, J.; Clawson, D.K.; Britt, K.S.; Anderson, B.D.; Beight, D.W.; et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget 2018, 9, 6659–6677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, S.; Kudo, M.; Shitara, K.; Yamauchi, M.; Doi, T.; Matsumura, Y. TGF-β inhibitor LY2157299 (galunisertib) in combination with standard chemotherapy and inhibition of signaling to pSmad and EMT and suppression of tumor growth in gastric cancer. J. Clin. Oncol. 2016, 34, 50. [Google Scholar] [CrossRef]
- Serova, M.; Tijeras-Raballand, A.; Dos Santos, C.; Albuquerque, M.; Paradis, V.; Neuzillet, C.; Benhadji, K.A.; Raymond, E.; Faivre, S.; de Gramont, A. Effects of TGF-beta signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget 2015, 6, 21614–21627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.K.; Wang, N.; Wang, W.D.; Du, X.N.; Wen, X.Y.; Wang, L.Y.; Deng, Y.Y.; Wang, D.P.; Lin, H.L. Blocking Posttranslational Core Fucosylation Ameliorates Rat Peritoneal Mesothelial Cell Epithelial-Mesenchymal Transition. Chin. Med. J. (Engl.) 2017, 130, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- De Vriese, A.S.; Tilton, R.G.; Mortier, S.; Lameire, N.H. Myofibroblast transdifferentiation of mesothelial cells is mediated by RAGE and contributes to peritoneal fibrosis in uraemia. Nephrol. Dial. Transplant. Publ. Eur. Dial. Transplant. Assoc.—Eur. Ren. Assoc. 2006, 21, 2549–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakawa, M.; Takimoto, R.; Tamura, F.; Yoshida, M.; Ono, M.; Murase, K.; Sato, Y.; Osuga, T.; Sato, T.; Iyama, S.; et al. Fucosylated TGF-beta receptors transduces a signal for epithelial-mesenchymal transition in colorectal cancer cells. Br. J. Cancer 2014, 110, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fukuda, T.; Isaji, T.; Lu, J.; Im, S.; Hang, Q.; Gu, W.; Hou, S.; Ohtsubo, K.; Gu, J. Loss of alpha1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J. 2015, 29, 3217–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herynk, M.H.; Beyer, A.R.; Cui, Y.; Weiss, H.; Anderson, E.; Green, T.P.; Fuqua, S.A. Cooperative action of tamoxifen and c-Src inhibition in preventing the growth of estrogen receptor-positive human breast cancer cells. Mol. Cancer 2006, 5, 3023–3031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, L.; Figueroa, C.D.; Bhoola, K.D.; Ehrenfeld, P. GPER-1/GPR30 a novel estrogen receptor sited in the cell membrane: Therapeutic coupling to breast cancer. Expert Opin. Targets 2017, 21, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Lima, N.C.; Atkinson, E.; Bunney, T.D.; Katan, M.; Huang, P.H. Targeting the Src Pathway Enhances the Efficacy of Selective FGFR Inhibitors in Urothelial Cancers with FGFR3 Alterations. Int. J. Mol. Sci. 2020, 21, 3214. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Barnes, J.L.; Varani, J. Balanced regulation of the CCN family of matricellular proteins: A novel approach to the prevention and treatment of fibrosis and cancer. J. Cell Commun. Signal. 2015, 9, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubink, I.; Verhaar, E.R.; Kranenburg, O.; Goldschmeding, R. A potential role for CCN2/CTGF in aggressive colorectal cancer. J. Cell Commun. Signal. 2016, 10, 223–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, N.; Nakamura, M.; Lipson, K.E.; Miyake, T.; Kamikawa, Y.; Sagara, A.; Shinozaki, Y.; Kitajima, S.; Toyama, T.; Hara, A.; et al. Inhibition of CTGF ameliorates peritoneal fibrosis through suppression of fibroblast and myofibroblast accumulation and angiogenesis. Sci. Rep. 2017, 7, 5392. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ren, S.; Macarak, E.; Rosenbloom, J. Pathobiological mechanisms of peritoneal adhesions: The mesenchymal transition of rat peritoneal mesothelial cells induced by TGF-beta1 and IL-6 requires activation of Erk1/2 and Smad2 linker region phosphorylation. Matrix Biol. 2016, 51, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borazanci, E.H.; Jameson, G.S.; Borad, M.J.; Ramanathan, R.K.; Korn, R.L.; Caldwell, L.; Ansaldo, K.; Hendrickson, K.; Marceau, K.; Hoff, D.D.V. A phase II pilot trial of nivolumab (N) + albumin bound paclitaxel (AP) + paricalcitol (P) + cisplatin (C) + gemcitabine (G) (NAPPCG) in patients with previously untreated metastatic pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2018, 36, 358. [Google Scholar] [CrossRef]
- Salazar, R.; Roepman, P.; Willems, S.M.; Brunen, D.; Kellner, U.; Midgley, R.A.; Bernards, R.; Simon, I.M. Molecular subtyping of colorectal cancer to identify a mesenchymal tumor type that might benefit from TGF-beta pathway inhibition. J. Clin. Oncol. 2014, 32, 456. [Google Scholar] [CrossRef]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.P.; Fan, J.; Wu, Y.J.; Xie, Y.F.; Zha, J.M.; Zhou, X.M. MiR-155 up-regulated by TGF-beta promotes epithelial-mesenchymal transition, invasion and metastasis of human hepatocellular carcinoma cells in vitro. Am. J. Transl. Res. 2017, 9, 2956–2965. [Google Scholar] [PubMed]
- Stasenko, M.; Plegue, M.; Sciallis, A.P.; McLean, K. Clinical response to antiestrogen therapy in platinum-resistant ovarian cancer patients and the role of tumor estrogen receptor expression status. Int. J. Gynecol. Cancer 2015, 25, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Q.; Huang, X.; Zou, F.; Fu, Z.; Chen, Y.; Li, Y.; Wang, Z.; Liu, L. Effects of 17beta-estradiol and tamoxifen on gastric cancer cell proliferation and apoptosis and ER-alpha36 expression. Oncol. Lett. 2017, 13, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmayr-Heyda, A.; Aust, S.; Auer, K.; Meier, S.M.; Schmetterer, K.G.; Dekan, S.; Gerner, C.; Pils, D. Integrative Systemic and Local Metabolomics with Impact on Survival in High-Grade Serous Ovarian Cancer. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2017, 23, 2081–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, V.J.; Brown, J.K.; Maybin, J.; Saunders, P.T.; Duncan, W.C.; Horne, A.W. Transforming growth factor-beta induced Warburg-like metabolic reprogramming may underpin the development of peritoneal endometriosis. J. Clin. Endocrinol. Metab. 2014, 99, 3450–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Heaton, A.; Lilischkis, K.; Garner, J.; Brown, D.M. Pharmacology and toxicology of the novel investigational agent Cantrixil (TRX-E-002-1). Cancer Chemother. Pharm. 2017, 79, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, A.J.; Ager, E.I.; Proctor, M.A.; Skalamera, D.; Heaton, A.; Brown, D.; Gabrielli, B.G. Mechanism of action of the third generation benzopyrans and evaluation of their broad anti-cancer activity in vitro and in vivo. Sci. Rep. 2018, 8, 5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, L.; van der Kuip, H.; Zopf, W.; Winter, S.; Munch, M.; Murdter, T.E.; Thon, K.P.; Steurer, W.; Aulitzky, W.E.; Ulmer, C. A Temperature of 40 degrees C Appears to be a Critical Threshold for Potentiating Cytotoxic Chemotherapy In Vitro and in Peritoneal Carcinomatosis Patients Undergoing HIPEC. Ann. Surg. Oncol. 2015, 22, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; van Leeuwen, C.M.; Ahire, V.R.; Rodermond, H.M.; Ten Cate, R.; Westermann, A.M.; Stalpers, L.J.A.; Crezee, J.; Kok, H.P.; Krawczyk, P.M.; et al. Enhancing synthetic lethality of PARP-inhibitor and cisplatin in BRCA-proficient tumour cells with hyperthermia. Oncotarget 2017, 8, 28116–28124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, L.; Schwab, M.; Ulmer, C.; Heine, S.; Murdter, T.E.; Schmid, J.O.; Sauer, G.; Aulitzky, W.E.; van der Kuip, H. Hyperthermia Synergizes with Chemotherapy by Inhibiting PARP1-Dependent DNA Replication Arrest. Cancer Res. 2016, 76, 2868–2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prada-Villaverde, A.; Esquivel, J.; Lowy, A.M.; Markman, M.; Chua, T.; Pelz, J.; Baratti, D.; Baumgartner, J.M.; Berri, R.; Bretcha-Boix, P.; et al. The American Society of Peritoneal Surface Malignancies evaluation of HIPEC with Mitomycin C versus Oxaliplatin in 539 patients with colon cancer undergoing a complete cytoreductive surgery. J. Surg. Oncol. 2014, 110, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, A.; Sharip, A.; Sharip, A.; Jiang, J.; Yang, Q.; Xie, Y. Reverse the Resistance to PARP Inhibitors. Int. J. Biol. Sci. 2017, 13, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Cesna, V.; Sukovas, A.; Jasukaitiene, A.; Naginiene, R.; Barauskas, G.; Dambrauskas, Z.; Paskauskas, S.; Gulbinas, A. Narrow line between benefit and harm: Additivity of hyperthermia to cisplatin cytotoxicity in different gastrointestinal cancer cells. World J. Gastroenterol. 2018, 24, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, P.M.; Eppink, B.; Essers, J.; Stap, J.; Rodermond, H.; Odijk, H.; Zelensky, A.; van Bree, C.; Stalpers, L.J.; Buist, M.R.; et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc. Natl. Acad. Sci. USA 2011, 108, 9851–9856. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, R.B.; Archid, R.; Reymond, M.A. Reprogramming of Mesothelial-Mesenchymal Transition in Chronic Peritoneal Diseases by Estrogen Receptor Modulation and TGF-β1 Inhibition. Int. J. Mol. Sci. 2020, 21, 4158. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114158
Wilson RB, Archid R, Reymond MA. Reprogramming of Mesothelial-Mesenchymal Transition in Chronic Peritoneal Diseases by Estrogen Receptor Modulation and TGF-β1 Inhibition. International Journal of Molecular Sciences. 2020; 21(11):4158. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114158
Chicago/Turabian StyleWilson, Robert B., Rami Archid, and Marc A. Reymond. 2020. "Reprogramming of Mesothelial-Mesenchymal Transition in Chronic Peritoneal Diseases by Estrogen Receptor Modulation and TGF-β1 Inhibition" International Journal of Molecular Sciences 21, no. 11: 4158. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114158