Inherited Genetic Mutations and Polymorphisms in Malignant Mesothelioma: A Comprehensive Review


1
Department of Respiratory Medicine, Odense University Hospital, 5000 Odense, Denmark
2
Department of Respiratory Medicine, Aalborg University Hospital, 9000 Aalborg, Denmark
3
Clinical Institute, Aalborg University Hospital, 9000 Aalborg, Denmark
4
Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology, 7491 Trondheim, Norway
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(12), 4327; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124327
Submission received: 18 May 2020
/
Revised: 9 June 2020
/
Accepted: 11 June 2020
/
Published: 17 June 2020
(This article belongs to the Special Issue Molecular Players in Diagnostics and Therapeutics of Malignant Pleural Mesothelioma)
Abstract
:Malignant mesothelioma (MM) is mainly caused by air-born asbestos but genetic susceptibility is also suspected to be a risk factor. Recent studies suggest an increasing number of candidate genes that may predispose to MM besides the well-characterized BRCA1-associated protein-1 gene. The aim of this review is to summarize the most important studies on germline mutations for MM. A total of 860 publications were retrieved from Scopus, PubMed and Web of Science, of which 81 met the inclusion criteria and were consider for this review. More than 50% of the genes that are reported to predispose to MM are involved in DNA repair mechanisms, and the majority of them have a role in the homologous recombination pathway. Genetic alterations in tumor suppressor genes involved in chromatin, transcription and hypoxia regulation have also been described. Furthermore, we identified several single nucleotide polymorphisms (SNPs) that may promote MM tumorigenesis as a result of an asbestos–gene interaction, including SNPs in DNA repair, carcinogen detoxification and other genes previously associated with other malignancies. The identification of inherited mutations for MM and an understanding of the underlying pathways may allow early detection and prevention of malignancies in high-risk individuals and pave the way for targeted therapies.
1. Introduction
Malignant mesothelioma (MM) is an aggressive tumor of the lining of the body cavities. It most often presents in the pleura, malignant pleural mesothelioma (MPM) and less often in the peritoneum, pericardium, tunica vaginalis testis and hernial sacs [1]. The histopathological subtypes are three in number, the most common epithelioid, the more rare sarcomatoid and the biphasic, that has both components [2]. MPM has a poor survival of 12–16 months for the epithelioid, and of only 4–6 months for the sarcomatoid subtype, while the five-year survival is less than 5% [3,4]. MPM is characterized by a high rate of innate and acquired chemoresistance but long-term survivors have been described both after multimodal treatment, including surgery, and chemotherapy alone [3,5,6,7]. Immunotherapy and chemoimmunotherapy are promising modalities [8]. There are no validated biomarkers that are useful for predicting the treatment response and survival in MPM.
The main cause of MPM is exposure to air-born asbestos [9]. Asbestos is a set of six minerals classified in two major groups, the amphiboles, consisting of crocidolite, amosite, tremolite, actinolite and anthophyllite, and the serpentines, namely chrysotile [9,10]. All types of asbestos are declared as carcinogens by the World Health Organization and the International Agency for Research on Cancer [9]. The latency between exposure to asbestos and MPM diagnosis has been reported to vary between 20 and 70 years [11]. Asbestos exposure can occur occupationally for asbestos workers or non-occupationally, including domestic and environmental exposure [9,12]. Asbestos was prohibited in most Western countries between 1970 and 2005, except for the USA, where it is only partly banned, and Canada, where the asbestos ban was effectuated in 2018 [13,14]. However, asbestos use and mining is ongoing in developing countries and approximately 2.2 million metric tons are being produced annually worldwide [15].
Both heavy and low-scale exposure to asbestos can cause MPM, as there is no safe threshold for asbestos use, and no linear dose–response relationship between MM and asbestos [12,16]. Nonetheless, some individuals are more susceptible to MPM subsequent to asbestos exposure than others, while there are also MM patients that report no exposure to asbestos or asbestos-like minerals [7,14]. Genetic susceptibility has long been suspected to be a risk factor for MM, providing an explanation for this observation [7]. The prevalence and spectrum of germline mutations in MM patients is not fully determined and the genetics role in causing MM de novo or enhancing asbestos carcinogenicity is yet to be ascertained. However, there have been a few published studies and case reports about genetic predisposition in MM in recent years. The aim of the current review is to summarize and present the most important studies on germline mutations that predispose to MM.
2. Results and Discussion
A total of 860 publications were retrieved through the research databases and four additional articles were identified through the reference lists. After excluding duplicated articles and publications that did not meet our inclusion criteria, there were 81 articles that were manually reviewed for this manuscript (Figure 1). The studies reporting pathogenic or likely pathogenic genetic variants and those that describe single nucleotide polymorphisms (SNPs) in genes are discussed separately due to the lower risk association of the latter with MM.
2.1. Pathogenic or Likely Pathogenic Genetic Variants
The most well-characterized gene that can predispose to MM is the breast cancer gene 1-associated protein 1 (BAP1) [17,18]. Recent studies suggest an increasing number of candidate genes associated with MM (Table 1).
2.1.1. BAP1 Gene
BAP1 is a tumor suppressor gene, located on chromosome 3p21.1 and encodes the BAP1 nuclear protein [26]. This is a deubiquitinating hydrolase, usually part of a protein complex, participating in various cellular processes including chromatin remodeling, cell cycle regulation and growth and DNA damage response [26]. The functional roles of BAP1 are partially through its deubiquitinase activity and synergy with other proteins, such as HCFC1, YY1, OGT, ASXL1/2 and FOXK1/2, but the impact of the distinct BAP1 mutations on the function of these complexes is not fully understood [17,27]. Germline BAP1 mutations underlie the BAP1 tumor predisposition syndrome, associated with uveal (UM) and cutaneous melanoma (CM), MM, renal cell carcinoma (RCC), non-melanoma skin cancer, meningioma and cholangiocarcinoma as well as other cancers [18,28,29,30,31,32,33,34] (Figure 2).
BAP1 genetic alterations appear typically with one mutant allele in all cells, while the somatic inactivation of the second allele results in tumorigenesis [17,35]. The gene–environment interaction is suspected to play an important role in cancer susceptibility for BAP1 mutation carriers [35,36]. The pathogenic BAP1 variants are known with a high penetration and approximately 85% of the mutation carriers are diagnosed with more than one malignancy [17,31]. Beside malignancies, individuals with germline BAP1 mutations often present with BAP1-inactivated nevi, previously called melanocytic BAP1-mutated atypical intradermal tumors (MBAITs), that are atypical melanocytes proliferations with spitzoid morphology [26].
BAP1 mutations are infrequent in the general population and there are no homozygotes [36,37]. However, their frequency has been reported as 1–2% for UM, 0.5% for CM and 0–7% for MM in distinct cases, rising up to 25%, 0.7% and 20%, respectively, in familial cases [4,7,36,38,39,40,41]. Patients carrying BAP1 genetic variations were shown to have a higher incidence of peritoneal versus pleural MM [7]. In comparison with sporadic MM, the BAP1 mutated patients tend to have sevenfold longer overall survival even when they have other cancers as well [17,42].
This is not the case in patients with other BAP1 tumor predisposition syndrome malignancies without MM. Patients with UM and inherited BAP1 mutations present often with a more aggressive and metastatic disease and more advanced tumor staging, and thus worse survival [43,44]. Similar findings apply to RCC and CM according to the literature [45,46,47]. The underlying molecular mechanisms that are responsible for the high variation in tumor aggressiveness in MM and the other cancers are unknown.
2.1.2. Genetic Variants in DNA Repair Genes
More than half of the genes that are reported to predispose to MM are involved in DNA repair mechanisms (Table 1). The majority of the altered DNA repair genes have a role in the homologous recombination (HR) pathway, while the rest participate in the mismatch repair system (MMR), non-homologous end joining (NHEJ) or nucleotide excision repair (NER). Asbestos fibers are known to induce DNA damage, which is repaired by HR and double-strand breaks repair, MMR and NER, thus individuals with defects in the DNA repair processes are more prone to develop MM [4,7,20,23,48,49,50]. Several of the implicated genes are well-known to increase cancer susceptibility for other malignancies and many of them interact with each other.
BRCA1 and BRCA2 are tumor suppressors responsible to maintain genome stability, specifically in the HR pathway for double-strand DNA repair [51]. BARD1 encodes a protein that interacts with BRCA1, forming a stable complex that is essential for tumor suppression [52]. This protein may be the target of various oncogenic mutations, for example in breast and ovarian cancer [53]. PALB2 encodes a tumor suppressor protein that binds to BRCA2, stabilizes its localization and permits its accumulation [54]. TP53 encodes the tumor protein p53 that determines whether DNA will be repaired or the cell will undergo apoptosis subsequent to toxic damage [55,56]. Hence, TP53 and p53 are crucial for regulating DNA repair and cell division and genetic mutations in this gene may predispose to several malignancies [56]. CHEK2 encodes a cell cycle checkpoint regulator and putative tumor suppressor protein, CHK2, which stabilizes p53, leading to cell cycle arrest, and interacts with BRCA1, restoring survival after DNA damage [57,58]. The protein encoded by ATM regulates various tumor suppressor proteins, including p53, BRCA1 and CHK2, and thus it is responsible for the cell response to DNA damage and genome stability [59]. FANCI, FANCC and FANCF are part of the Fanconi anemia complementation group (FANC) that also includes BRCA2 and PALB2 [60]. The members of the FANC group are assembled into a common protein complex that collaborate to repair DNA interstrand crosslinks after exposure to chemicals [60]. MRE11A encodes a protein that forms a complex with the RAD50 homolog, which is required for NHEJ and DNA double-strand break repair [61]. XPC encodes an important protein for NER that responds to DNA damage induced by ultraviolet radiation by recruiting ATR and ATM kinases to the DNA defect sites [62]. MLH1, MSH3, MSH6 and PSM1 are four of the seven DNA mismatch repair proteins [63]. They are necessary in order to maintain genomic stability and defects in the MMR may result in microsatellite instability and or malignant diseases, such as hereditary nonpolyposis colon cancer (HNPCC) and cancers of the NHPCC spectrum [63]. Finally, WT1 is an oncogene that promotes HR- mediated DNA damage repair [64].
An interesting observation is that MPM patients with inherited mutations in these genes tend to have improved survival compared with those with no genetic alteration, mirroring patients with BRCA1- and BRCA2-associated malignancies [4,7,42,49] (Figure 3). Patients with ovarian, breast or prostate cancer who carry germline BRCA1 or BRCA2 mutations are more likely to respond to cisplatin-based chemotherapy and have better prognosis [65]. These patients have also demonstrated sensitivity to treatment with poly (ADP-ribose) polymerase inhibitors (PARPi). PARPi are proven to be effective for various solid tumors with somatic or germline mutations in HR deficit genes, including breast, prostate, ovarian and pancreatic cancer [66,67,68]. Cisplatin or carboplatin combined with pemetrexed is the cornerstone of MPM chemotherapy but a large part of the patients either do not respond or become resistant to this treatment, while there are no biomarkers in clinical use to identify potential responders [69,70]. The literature suggests that germline mutations in DNA repair and other tumor suppressor genes may be a prognostic biomarker for cisplatin chemotherapy in MPM [4,7,23]. Furthermore, there is evidence that MPM patients, especially those that are not refractory to chemotherapy, could also benefit from PARPi [4,7].
2.1.3. Genetic Variants in Other Genes
A few of the genes that are reported to be mutated for MM patients are involved in chromatin regulation, including NCOR1, ARID1A, ARID2, SMARCE1, SMARCA2 and SMARCA4. The protein encoded by NCOR1 is a transcriptional coregulatory protein which assists nuclear receptors in the downregulation of gene expression [71]. ARID1A, SMARCE1, SMARCA2 and SMARCA4 are part of the ATP-dependent chromatin remodeling complex SNF/SWI, while ARID2 is a subunit of the PBAF (SWI/SNF-B) chromatin remodeling complex [72]. They are responsible for the transcriptional activation of genes that are normally repressed by chromatin and facilitate ligand-dependent transcriptional activation by nuclear receptors [72]. The SNF/SWI complexes can change the position of nucleosomes along DNA, so that binding sites for transcriptional regulators are exposed and gene expression can consequently be controlled [73,74]. Among the genes that are responsible for chromatin modifications, those encoding subunits of the SWI/SNF complexes are the most frequently mutated. Their mutations collectively occur in ∼20% of all human cancer types that have been genomically characterized so far [75]. CREBBP is also involved in the regulation of transcription by coupling chromatin remodeling to transcription factor recognition [76]. SHQ1 and RBM6 are both tumor suppressor genes involved in RNA processing, while NF2 encodes a protein that regulates several key signaling pathways important for controlling cell shape, cell growth and cell adhesion [77,78,79]. CDKN2A and TMEM127 encode both tumor suppressor proteins that are involved in cell growth, proliferation and survival [80,81]. SMO mediates signal transduction in the hedgehog pathway, which is critical for normal development and carcinogenesis [82]. KDR encodes one of the two receptors of the vascular endothelial growth factor and hereby promotes proliferation, survival, migration and differentiation of endothelial cells [83]. Finally, VHL and SDHA are involved in tumorigenesis through impaired hypoxia-inducible factor expression [84,85]. The pathophysiological mechanisms behind MM genesis as a result of these genetic alterations are not yet fully determined.
Most studies describe distinct clinical characteristics that can predict the presence of an inherited mutation, such as limited exposure to asbestos, peritoneal disease, young age and second cancer diagnosis [4,7,23,86]. This observation is of great significance, as it can lead to clinical panel-based genetic testing and the implementation of clinical genetic testing guidelines. Genetic testing would be of high benefit for MM patients and their relatives, as it would allow early detection and prevention of malignancies in high-risk individuals. This could result in the identification and treatment of the malignancies at an earlier stage, and hence improved survival. In addition, a part of these inherited mutations could be clinically significant and the patients may be able to enroll in targeted clinical trials that give them a higher chance of prolonged survival. Most importantly, the patients and their physicians should also be aware of the better survival that mutation-carriers have, as this would have a big impact on their lives and on their treatment considerations and planning.
2.2. Genetic Polymorphisms Associated with MM
2.2.1. Genome-Wide Association Studies
Genome-wide association studies (GWAS) are the most efficient approach in detecting SNPs in MM, as they allow the simultaneous screening of thousands of genetic variants in large panels of MM patients and controls. There have been published two comprehensive GWAS regarding MM; one originating from Australia and one from Italy including 428 MM patients and 778 controls and 407 MM patients and 389 controls, respectively [87,88]. Both groups took asbestos exposure into account, as they hypothesized that MM tumorigenesis was a result of the asbestos–gene interaction. There is no compelling evidence in the two studies that the identified SNPs can cause MM in the absence of asbestos exposure. The Australian study attempted to replicate the most significant SNPs in the Italian study but failed. The heterogeneity of the populations and the different types of asbestos exposures were suggested as potential reasons for the non-replication by the researchers. The SNPs with the highest significance levels from the Australian study were located in the CRTAM, RASGRF2 and SDK1 genes (Table 2). All three genes are associated with cell adhesion, migration and apoptosis and they are suspected to promote carcinogenesis through mechanisms initiated by the human immune system’s response to asbestos fibers [87,89,90,91,92]. The outmost significant signals from the Italian studies were encountered in the PVT1, ETV1, THRB, CEP350, SHC4 and SLC7A14 genes. PVT1, ETV1 and THRB are known oncogenes implicated in several malignancies through transcription regulation, such as prostate cancer, melanoma and breast cancer [88,93,94,95]. CEP350 is required to anchor microtubules at the centrosome and SHC4 regulates cell proliferation; their association with distinct cancers has also been reported [88,96,97]. SLC7A14 is involved in arginine transport and although this gene has not been previously linked to MM, there are indications of neighboring genes involvement due to identified chromosomal gain in this region [88,98,99].
2.2.2. Polymorphisms in Carcinogen Detoxification Genes
It is hypothesized that asbestos toxicity and carcinogenicity may be mediated by reactive oxygen species and free radicals, which occur as a result of inhaled asbestos fibers [100,101]. The four subfamilies of the glutathione S-tranferase supergene (GSTA, GSTM, GSTT and GSTP) play a central role in the detoxification and clearance of reactive oxygen species [102]. Especially the GSTM1 and GSTT1 subfamilies present with homozygous deletion polymorphisms (null genotype) that have been considered as modulators of susceptibility to environmentally induced malignancies [102,103]. The N-Acetyltransferase NAT2 has also an antioxidant function, as it metabolizes aromatic and heterocyclic amine carcinogens, and hence it may modify predisposition to various cancers [104]. EPHX encodes the epoxide hydrolase protein mEH, which is responsible for detoxification and preparation for phase II conjugation reactions [103]. Genetic polymorphisms of EPHX may result in an increase or decrease in enzyme activity, which may promote cancer susceptibility. Manganese superoxide dismutase (MnSOD) is an important antioxidant enzyme in mammalian tissues that is crucial for the response to reactive oxygen species [105]. The most common polymorphism of MnSOD occurs at codon 16 and results in an Alanine (Ala) to Valine amino acid transformation, which alters the protein secondary structure, and thus impairs the transport of the protein into the mitochondria [106]. NAT2 acetylation, EPHX, MnSOD, GSTM1 and GSTT1 null polymorphisms in regard to MPM susceptibility were analyzed by Finnish and Italian researchers [100,107,108,109,110,111]. A Finnish study compared 44 MM patients to 270 controls and concluded that GSTM1 null and the NAT2 slow acetylator increase the MPM risk, with the heavy asbestos-exposed population being at higher risk. An Italian study comprised of 80 MPM patients and 255 healthy individuals and found similar results regarding the GSTM1 null genotype and MPM risk. However, the NAT2 slow acetylator appeared to be a protective factor for the Italian patients. The low-activity-associated EPHX1 genotype was a risk factor for MM in the Italian, but not in the Finnish population. No significant risk was reported for the GSTT1 null genotype in both studies. The nucleotidic change 282C > T within NAT2 was also found to be significantly associated with MPM risk in another Italian study of 50 SNPs within oxidative metabolism enzymes and 75 SNPs in genome stability genes [100]. The allele 282T is connected with the haplotypes NAT2*6 and encodes for the slow acetylator phenotype [100]. Landi et al. genotyped 90 MPM patients versus 395 control subjects and found a higher MPM risk for individuals with a GSTM1 null allele and in those with the Ala/Ala genotypes at codon 16 within MnSOD [107].
2.2.3. Polymorphisms in DNA Repair Genes
Dianzani et al. focused on four DNA repair genes, XRCC1, XRCC3, XPD and OGG1, hypothesizing that deficient DNA repair mechanisms would fail to protect against the oxidative stress induced by asbestos fibers and eventually result in a higher risk of carcinogenesis [112]. XRCC1 and OGG1 are a part of the base excision repair (BER), while XPD is of the nucleotide excision repair (NER) pathway [113,114,115]. XRRC3 participates in double-strand break repair, where XRCC1 possibly also is involved [115]. The group investigated seven SNPs located in the four genes (i.e., XRCC1-R399Q, XRCC1-R194W, XRCC3-T241M, XRCC3-IVS6-14, XPD-K751Q, XPD-D312N, OGG1-S326C) which have previously been associated with various malignancies and/or impaired DNA repair [115,116,117,118,119]. The study population consisted of 81 MPM patients and 110 gender- and age-matched controls from Casale Monferrato, of which 70 patients and 85 controls had a history of asbestos exposure. Higher risk of MPM was shown for homozygotes and heterozygotes of the XRCC1-R399Q variant, with the risk escalating with the increasing number of XRCC1-399Q alleles. Further analyses were conducted after dividing the genotypes into two subgroups of ‘’risk’’ and ‘’non-risk’’ depending on the functional significance and the frequency distributions of the variants and the epidemiologic evidence. A significant association with MM was noticed for the XRCC1-R399Q Q homozygotes and Q/R heterozygotes versus the R homozygotes and for the XRCC3-T241M T homozygotes and M/T heterozygotes versus the M homozygotes. The haplotype association between two SNPs in XRCC1, XRCC3 and XPD was also calculated, but significance was not reached.
3. Materials and Methods
Electronic searches were conducted using Scopus, PubMed and Web of Science from January 2011 to February 2020. To identify all relevant studies, we combined medical subject headings (MeSH) terms or keywords: mesothelioma AND (“gene AND mutation” OR “germline AND mutation” OR “genetic AND predisposition” OR “genetical AND predisposition” OR “genetical AND alteration” OR “genetic AND alteration” OR “germline AND alteration” OR “genetic AND susceptibility”). The reference lists of all retrieved articles were also reviewed. All publications were limited to human subjects and in the English language. Articles regarding somatic mutations were excluded, as they are out of the scope of this manuscript. Abstracts, case reports, conference presentations, editorials and expert opinions were excluded as well. All potentially relevant articles were manually reviewed.
4. Conclusions
There are only a few large studies investigating germline mutations and polymorphisms in MM. The first MM susceptibility gene was described in 2011, but only in the past couple of years were there published studies that shed more light into the prevalence and spectrum of germline mutations for MM. The rarity of MM together with the inadequate technologies have hampered the conduction of more comprehensive studies. The development of high-throughput technologies that allow faster and cheaper genome sequencing, such as next generation sequencing, enables researches to investigate large gene panels and identify rare genetic variants. By the use of these techniques, underlying mutations can be isolated without the requirement for lengthy genetic mapping studies. However, it is also important that the emerging data are clustered and the pathways analyzed in order to fully comprehend the underlying biological processes. Genetic testing of high-risk individuals may facilitate clinical genetic counseling and help us achieve early cancer detection. Lastly, the identification of inherited mutations and an understanding of the oncogenesis mechanisms may allow us to find potential candidates for targeted therapy, guide the choice of drug treatment and thus give MM patients a better chance of prolonged survival.
Author Contributions
Conceptualization, V.P.; methodology, V.P.; investigation, V.P.; resources, O.D.R.; data curation, V.P.; writing—original draft preparation, V.P.; writing—review and editing, V.P., O.D.R.; supervision, O.D.R.; funding acquisition, O.D.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| MM | Malignant mesothelioma |
| MPM | Malignant pleural mesothelioma |
| SNPs | Single nucleotide polymorphisms |
| MeSH | Medical Subject Headings |
| BAP1 | Breast cancer gene 1-associated protein 1 |
| UV | Uveal melanoma |
| CM | Cutaneous melanoma |
| RCC | Renal cell carcinoma |
| MBAITs | Melanocytic BAP1-mutated atypical intradermal tumors |
| HR | Homologous Recombination |
| MMR | Mismatch repair system |
| NHEJ | Non-homologous recombination end joining |
| NER | Nucleotide excision repair |
| FANC | Fanconi anemia complementation group |
| PARPi | Poly (ADP-ribose) polymerase inhibitors |
| GWAS | Genome-wide association studies |
References
- Robinson, B.W.S.; Musk, A.W.; Lake, R.A. Malignant Mesothelioma. Lancet 2005, 366, 397–408. [Google Scholar] [CrossRef]
- Husain, A.N.; Colby, T.V.; Ordóñez, N.G.; Allen, T.C.; Attanoos, R.L.; Beasley, M.B.; Butnor, K.J.; Chirieac, L.R.; Churg, A.M.; Dacic, S.; et al. Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2018, 142, 89–108. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.C.; Dowell, J.E. Modern Management of Malignant Pleural Mesothelioma. Lung Cancer Targets Ther. 2016, 3, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Morrow, B.; Thomas, A.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Gadiraju, M.; Panou, V.; Gao, S.; Mian, I.; et al. Inherited Predisposition to Malignant Mesothelioma and Overall Survival Following Platinum Chemotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 9008–9013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bovolato, P.; Casadio, C.; Billè, A.; Ardissone, F.; Santambrogio, L.; Ratto, G.B.; Garofalo, G.; Bedini, A.V.; Garassino, M.; Porcu, L.; et al. Does Surgery Improve Survival of Patients with Malignant Pleural Mesothelioma?: A Multicenter Retrospective Analysis of 1365 Consecutive Patients. J. Thorac. Oncol. 2014, 9, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakas, A.; Waller, D. Predictors of Long-Term Survival Following Radical Surgery for Malignant Pleural Mesothelioma. Eur. J. Cardio-thoracic Surg. 2014, 46, 380–385. [Google Scholar] [CrossRef]
- Panou, V.; Gadiraju, M.; Wolin, A.; Weipert, C.M.; Skarda, E.; Husain, A.N.; Patel, J.D.; Rose, B.; Zhang, S.R.; Weatherly, M.; et al. Frequency of Germline Mutations in Cancer Susceptibility Genes in Malignant Mesothelioma. J. Clin. Oncol. 2018, 36, 2863–2871. [Google Scholar] [CrossRef]
- De Gooijer, C.J.; Borm, F.J.; Scherpereel, A.; Baas, P. Immunotherapy in Malignant Pleural Mesothelioma. Front. Oncol. 2020, 10, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Røe, O.D.; Stella, G.M. Malignant Pleural Mesothelioma: History, Controversy and Future of a Manmade Epidemic. Eur. Respir. Rev. 2015, 24, 115–131. [Google Scholar] [CrossRef] [Green Version]
- Stayner, L.T.; Welch, L.; Lemen, R. The Worldwide Pandemic of Asbestos-Related Diseases. SSRN 2013, 34, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Robinson, B.M. Malignant Pleural Mesothelioma: An Epidemiological Perspective. Ann. Cardiothorac. Surg. 2012, 1, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Panou, V.; Vyberg, M.; Meristoudis, C.; Hansen, J.; Bøgsted, M.; Omland, Ø.; Weinreich, U.M.; Røe, O.D. Non-Occupational Exposure to Asbestos Is the Main Cause of Malignant Mesothelioma in Women in North Jutland, Denmark. Scand. J. Work. Environ. Health 2019, 45, 82–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrigan, P.J.; Lemen, R.A. Asbestos Related Diseases in the United States: Historical Trends and Current Situation. Occup. Environ. Med. 2018, 75, 224–225. [Google Scholar]
- Ruff, K. How Canada Changed from Exporting Asbestos to Banning Asbestos: The Challenges That Had to Be Overcome. Environ. Res. Public Heal. 2017, 14, 1135. [Google Scholar] [CrossRef] [Green Version]
- Hashim, D.; Boffetta, P. Occupational and Environmental Exposures and Cancers in Developing Countries. Ann. Glob. Heal. 2014, 80, 393–411. [Google Scholar] [CrossRef] [PubMed]
- Donovan, E.P.; Donovan, B.L.; McKinley, M.A.; Cowan, D.M.; Paustenbach, D.J. Evaluation of Take Home (Para-Occupational) Exposure to Asbestos and Disease: A Review of the Literature. Crit. Rev. Toxicol. 2012, 42, 703–731. [Google Scholar] [CrossRef]
- Rai, K.; Pilarski, R.; Cebulla, C.M.; Abdel-Rahman, M.H. Comprehensive Review of BAP1 Tumor Predisposition Syndrome with Report of Two New Cases. Clin. Genet. 2016, 89, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 Mutations Predispose to Malignant Mesothelioma. Nat. Genet. 2012, 43, 1022–1025. [Google Scholar] [CrossRef] [Green Version]
- Betti, M.; Casalone, E.; Ferrante, D.; Aspesi, A.; Morleo, G.; Biasi, A.; Sculco, M.; Mancuso, G.; Guarrera, S.; Righi, L.; et al. Germline Mutations in DNA Repair Genes Predispose Asbestos-Exposed Patients to Malignant Pleural Mesothelioma. Cancer Lett. 2017, 405, 38–45. [Google Scholar] [CrossRef]
- Guo, R.; DuBoff, M.; Jayakumaran, G.; Kris, M.G.; Ladanyi, M.; Robson, M.E.; Mandelker, D.; Zauderer, M.G. Novel Germline Mutations in DNA Damage Repair in Patients with Malignant Pleural Mesotheliomas. J. Thorac. Oncol. 2020, 15, 655–660. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Biasi, A.; Casalone, E.; Ferrante, D.; Ogliara, P.; Gironi, L.C.; Giorgione, R.; Farinelli, P.; Grosso, F.; et al. CDKN2A and BAP1 Germline Mutations Predispose to Melanoma and Mesothelioma. Cancer Lett. 2016, 378, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.M.; Alston, R.D.; McNally, R.J.; Evans, D.G.; Kelsey, A.M.; Harris, M.; Eden, O.B.; Varley, J.M. Relative Frequency and Morphology of Cancers in Carriers of Germline TP53 Mutations. Oncogene 2001, 20, 4621–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorino, S.; Yoshikawa, Y.; Pass, H.I.; Emi, M.; Nasu, M.; Pagano, I.; Takinishi, Y.; Yamamoto, R.; Minaai, M.; Hashimoto-Tamaoki, T.; et al. A Subset of Mesotheliomas With Improved Survival Occurring in Carriers of BAP1 and Other Germline Mutations. J. Clin. Oncol. 2018, 35, 3485–3494. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Milchgrub, S.; Khatri, G.; Gopal, P. Metachronous Uterine Endometrioid Adenocarcinoma and Peritoneal Mesothelioma in Lynch Syndrome: A Case Report. Int. J. Surg. Pathol. 2017, 25, 253–257. [Google Scholar] [CrossRef]
- Karamurzin, Y.; Zeng, Z.; Stadler, Z.K.; Zhang, L.; Ouansafi, I.; Al-Ahmadie, H.A.; Sempoux, C.; Saltz, L.B.; Soslow, R.A.; O’Reilly, E.M.; et al. Unusual DNA Mismatch Repair-Deficient Tumors in Lynch Syndrome: A Report of New Cases and Review of the Literature. Hum. Pathol. 2012, 43, 1677–1687. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and Cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef]
- White, A.E.; Harper, J.W. Emerging Anatomy of the BAP1 Tumor Suppressor System. Science 2012, 337, 1463–1464. [Google Scholar] [CrossRef]
- Popova, T.; Hebert, L.; Jacquemin, V.; Gad, S.; Caux-Moncoutier, V.; Dubois-D’Enghien, C.; Richaudeau, B.; Renaudin, X.; Sellers, J.; Nicolas, A.; et al. Germline BAP1 Mutations Predispose to Renal Cell Carcinomas. Am. J. Hum. Genet. 2013, 92, 974–980. [Google Scholar] [CrossRef] [Green Version]
- Pilarski, R.; Cebulla, C.M.; Massengill, J.B.; Rai, K.; Rich, T.; Strong, L.; Mcgillivray, B.; Asrat, M.J.; Davidorf, F.H.; Abdel-Rahman, M.H. Expanding the Clinical Phenotype of Hereditary BAP1 Cancer Predisposition Syndrome, Reporting Three New Cases. Genes Chromosom. Cancer 2014, 53, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Rahman, M.H.; Pilarski, R.; Cebulla, C.M.; Massengill, J.B.; Christopher, B.N.; Boru, G.; Hovland, P.; Davidorf, F.H. Germline BAP1 Mutation Predisposes to Uveal Melanoma, Lung Adenocarcinoma, Meningioma, and Other Cancers. J. Med. Genet 2011, 856–859. [Google Scholar] [CrossRef]
- Walpole, S.; Pritchard, A.L.; Cebulla, C.M.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; De La Fouchardière, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Santagata, S. BAP1 Mutations in High-Grade Meningioma: Implications for Patient Care. Neuro. Oncol. 2017, 19, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, R.V.; Watson, G.F.; Maher, A.M.; McCarthy, S.W.; Thompson, J.F.; Scolyer, R.A. Germline BAP1 Mutations Also Predispose to Cutaneous Squamous Cell Carcinoma. Pathology 2017, 49, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline Mutations in BAP1 Predispose to Melanocytic Tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef] [Green Version]
- Klebe, S.; Driml, J.; Nasu, M.; Pastorino, S.; Zangiabadi, A.; Henderson, D.; Carbone, M. BAP1 Hereditary Cancer Predisposition Syndrome: A Case Report and Review of Literature. Biomark. Res. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Chau, C.; van Doorn, R.; van Poppelen, N.M.; van der Stoep, N.; Mensenkamp, A.R.; Sijmons, R.H.; van Paassen, B.W.; van den Ouweland, A.M.W.; Naus, N.C.; van der Hout, A.H.; et al. Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: Path to Identification and a Proposal for Genetic Screening Guidelines. Cancers 2019, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 Human Exomes and Genomes Reveals the Spectrum of Loss-of-Function Intolerance across Human Protein-Coding Genes. bioRxiv 2019, 133, 881–887. [Google Scholar] [CrossRef] [Green Version]
- Sneddon, S.; Leon, J.S.; Dick, I.M.; Cadby, G.; Olsen, N.; Brims, F.; Allcock, R.J.N.; Moses, E.K.; Melton, P.E.; de Klerk, N.; et al. Absence of Germline Mutations in BAP1 in Sporadic Cases of Malignant Mesothelioma. Gene 2015, 563, 103–105. [Google Scholar] [CrossRef] [Green Version]
- Rusch, A.; Ziltener, G.; Nackaerts, K.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Prevalence of BRCA-1 Associated Protein 1 Germline Mutation in Sporadic Malignant Pleural Mesothelioma Cases. Lung Cancer 2015, 87, 77–79. [Google Scholar] [CrossRef]
- Repo, P.; Järvinen, R.S.; Jäntti, J.E.; Markkinen, S.; Täll, M.; Raivio, V.; Turunen, J.A.; Kivelä, T.T. Population-Based Analysis of BAP1 Germline Variations in Patients with Uveal Melanoma. Hum. Mol. Genet. 2019, 28, 2415–2426. [Google Scholar] [CrossRef]
- Gupta, M.P.; Lane, A.M.; DeAngelis, M.M.; Mayne, K.; Crabtree, M.; Gragoudas, E.S.; Kim, I.K. Clinical Characteristics of Uveal Melanoma in Patients with Germline BAP1 Mutations. JAMA Ophthalmol. 2015, 133, 881–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, F.; Flores, E.; Napolitano, A.; Kanodia, S.; Taioli, E.; Pass, H.; Yang, H.; Carbone, M. Mesothelioma Patients with Germline BAP1 Mutations Have 7-Fold Improved Long-Term Survival. Carcinogenesis 2015, 36, 76–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalirai, H.; Dodson, A.; Faqir, S.; Damato, B.E.; Coupland, S.E. Lack of BAP1 Protein Expression in Uveal Melanoma Is Associated with Increased Metastatic Risk and Has Utility in Routine Prognostic Testing. Br. J. Cancer 2014, 111, 1373–1380. [Google Scholar] [CrossRef] [Green Version]
- Njauw, C.N.J.; Kim, I.; Piris, A.; Gabree, M.; Taylor, M.; Lane, A.M.; DeAngelis, M.M.; Gragoudas, E.; Duncan, L.M.; Tsao, H. Germline BAP1 Inactivation Is Preferentially Associated with Metastatic Ocular Melanoma and Cutaneous-Ocular Melanoma Families. PLoS ONE 2012, 7, e35295. [Google Scholar] [CrossRef] [Green Version]
- Kapur, P.; Peña-Llopis, S.; Christie, A.; Zhrebker, L.; Pavía-Jiménez, A.; Rathmell, W.K.; Xie, X.J.; Brugarolas, J. Effects on Survival of BAP1 and PBRM1 Mutations in Sporadic Clear-Cell Renal-Cell Carcinoma: A Retrospective Analysis with Independent Validation. Lancet Oncol. 2013, 14, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Murali, R.; Wilmott, J.S.; Jakrot, V.; Al-Ahmadie, H.A.; Wiesner, T.; McCarthy, S.W.; Thompson, J.F.; Scolyer, R.A. BAP1 Expression in Cutaneous Melanoma: A Pilot Study. Pathology 2013, 45, 606–609. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Ostrovnaya, I.; Reva, B.; Schultz, N.; Chen, Y.B.; Gonen, M.; Liu, H.; Takeda, S.; Voss, M.H.; Tickoo, S.K.; et al. Adverse Outcomes in Clear Cell Renal Cell Carcinoma with Mutations of 3p21 Epigenetic Regulators BAP1 and SETD2: A Report by MSKCC and the KIRC TCGA Research Network. Clin. Cancer Res. 2013, 19, 3259–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toumpanakis, D.; Theocharis, S.E. DNA Repair Systems in Malignant Mesothelioma. Cancer Letters 2011, 143–149. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Sculco, M.; Matullo, G.; Magnani, C.; Dianzani, I. Genetic Predisposition for Malignant Mesothelioma: A Concise Review. Mutat. Res. Rev. Mutat. Res. 2019, 781, 1–10. [Google Scholar] [CrossRef]
- Bertelsen, B.; Tuxen, I.V.; Yde, C.W.; Gabrielaite, M.; Torp, M.H.; Kinalis, S.; Oestrup, O.; Rohrberg, K.; Spangaard, I.; Santoni-Rugiu, E.; et al. High Frequency of Pathogenic Germline Variants within Homologous Recombination Repair in Patients with Advanced Cancer. npj Genomic Med. 2019, 4. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA Interstrand Crosslink Repair and Cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, H.; Wu, W.; Koike, A.; Kojima, R.; Gomi, H.; Fukuda, M.; Ohta, T. BRCA1-Associated Protein 1 Interferes with BRCA1/BARD1 RING Heterodimer Activity. Cancer Res. 2009, 69, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajska, M.; Antoszewska, E.; Piskorz, A.; Brozek, I.; Borg, Å.; Kusmierek, H.; Biernat, W.; Limon, J. Cancer Predisposing BARD1 Mutations in Breast-Ovarian Cancer Families. Breast Cancer Res. Treat. 2012, 131, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-Cycle Checkpoints and Cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 Mutations in Human Cancers: Functional Selection and Impact on Cancer Prognosis and Outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [Green Version]
- Hirao, A. DNA Damage-Induced Activation of P53 by the Checkpoint Kinase Chk2. Science 2000, 287, 1824–1827. [Google Scholar] [CrossRef]
- Zhou, B.B.S.; Elledge, S.J. The DNA Damage Response: Putting Checkpoints in Perspective. Nature 2000, 433–439. [Google Scholar] [CrossRef]
- Bruno, T.; De Nicola, F.; Iezzi, S.; Lecis, D.; D’Angelo, C.; Di Padova, M.; Corbi, N.; Dimiziani, L.; Zannini, L.; Jekimovs, C.; et al. Che-1 Phosphorylation by ATM/ATR and Chk2 Kinases Activates P53 Transcription and the G2/M Checkpoint. Cancer Cell 2006, 10, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Walden, H.; Deans, A.J. The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef]
- D’Amours, D.; Jackson, S.P. The Mre11 Complex: At the Crossroads of DNA Repair and Checkpoint Signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Milum, K.; Battu, A.; Wani, G.; Wani, A.A. NER Initiation Factors, DDB2 and XPC, Regulate UV Radiation Response by Recruiting ATR and ATM Kinases to DNA Damage Sites. DNA Repair 2013, 12, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltomäki, P. Role of DNA Mismatch Repair Defects in the Pathogenesis of Human Cancer. J. Clin. Oncol. 2003, 21, 1174–1179. [Google Scholar] [CrossRef]
- Bishop, A.J.R.; Schiestl, R.H. Homologous Recombination as a Mechanism of Carcinogenesis. Biochim. Biophys. Acta Rev. Cancer 2001. [Google Scholar] [CrossRef]
- Foulkes, W.D. BRCA1 and BRCA2: Chemosensitivity, Treatment Outcomes and Prognosis. Fam. Cancer 2006, 5, 135–142. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral Poly (ADP-Ribose) Polymerase Inhibitor Olaparib in Patients with BRCA1 or BRCA2 Mutations and Advanced Breast Cancer: A Proof-of-Concept Trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral Poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Patients with BRCA1 or BRCA2 Mutations and Recurrent Ovarian Cancer: A Proof-of-Concept Trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination with Cisplatin versus Cisplatin Alone in Patients with Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Blayney, J.K.; Ceresoli, G.L.; Castagneto, B.; O’Brien, M.E.R.; Hasan, B.; Sylvester, R.; Rudd, R.; Steele, J.; Busacca, S.; Porta, C.; et al. Response to Chemotherapy Is Predictive in Relation to Longer Overall Survival in an Individual Patient Combined-Analysis with Pleural Mesothelioma. Eur. J. Cancer 2012, 48, 2983–2992. [Google Scholar] [CrossRef]
- Catic, A.; Suh, C.Y.; Hill, C.T.; Daheron, L.; Henkel, T.; Orford, K.W.; Dombkowski, D.M.; Liu, T.; Liu, X.S.; Scadden, D.T. Genome-Wide Map of Nuclear Protein Degradation Shows NCoR1 Turnover as a Key to Mitochondrial Gene Regulation. Cell 2013, 155, 1380–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisman, D.; Glaros, S.; Thompson, E.A. The SWI/SNF Complex and Cancer. Oncogene 2009, 28, 1653–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alver, B.H.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W.M. The SWI/SNF Chromatin Remodelling Complex Is Required for Maintenance of Lineage Specific Enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of Action and Regulation of ATP-Dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and Bioinformatic Analysis of Mammalian SWI/SNF Complexes Identifies Extensive Roles in Human Malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Cho, K.S.; Elizondo, L.I.; Boerkoel, C.F. Advances in Chromatin Remodeling and Human Disease. Curr. Opin. Genet. Dev. 2004, 14, 308–315. [Google Scholar] [CrossRef]
- Wang, K.; Ubriaco, G.; Sutherland, L.C. RBM6-RBM5 Transcription-Induced Chimeras Are Differentially Expressed in Tumours. BMC Genomics 2007, 8, 348. [Google Scholar] [CrossRef] [Green Version]
- Bratkovič, T.; Rogelj, B. Biology and Applications of Small Nucleolar RNAs. Cell. Mol. Life Sci. 2011, 68, 3483–3851. [Google Scholar] [CrossRef]
- Zhang, N.; Bai, H.; David, K.K.; Dong, J.; Zheng, Y.; Cai, J.; Giovannini, M.; Liu, P.; Anders, R.A.; Pan, D. The Merlin/NF2 Tumor Suppressor Functions through the YAP Oncoprotein to Regulate Tissue Homeostasis in Mammals. Dev. Cell 2010, 19, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability an Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol 2010, 220–228. [Google Scholar] [CrossRef]
- Qin, Y.; Deng, Y.; Ricketts, C.J.; Srikantan, S.; Wang, E.; Maher, E.R.; Dahia, P.L.M. The Tumorsusceptibilitygene TMEM127 Is Mutated in Renal Cell Carcinomas and Modulates Endolysosomal Function. Hum. Mol. Genet. 2014, 23, 2428–2439. [Google Scholar] [CrossRef] [Green Version]
- Philipp, M.; Caron, M.G. Hedgehog Signaling: Is Smo a G Protein-Coupled Receptor? Curr. Biol. 2009, 19, R125–R127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, S.; Oda, N.; Abe, M.; Terai, Y.; Ito, M.; Shitara, K.; Tabayashi, K.; Shibuya, M.; Sato, Y. Roles of Two VEGF Receptors, Flt-1 and KDR, in the Signal Transduction of VEGF Effects in Human Vascular Endothelial Cells. Oncogene 2000, 19, 2138–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, P.H.; Wlesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The Tumour Suppressor Protein VHL Targets Hypoxia-Inducible Factors for Oxygen-Dependent Proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Frezza, C.; Gottlieb, E. Mitochondria in Cancer: Not Just Innocent Bystanders. Semin. Cancer Biol. 2009, 19, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Ohar, J.A.; Cheung, M.; Talarchek, J.; Howard, S.E.; Howard, T.D.; Hesdorffer, M.; Peng, H.; Rauscher, F.J.; Testa, J.R. Germline BAP1 Mutational Landscape of Asbestos-Exposed Malignant Mesothelioma Patients with Family History of Cancer. Cancer Res. 2016, 76, 206–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadby, G.; Mukherjee, S.; Musk, A.W.; Reid, A.; Garlepp, M.; Dick, I.; Robinson, C.; Hui, J.; Fiorito, G.; Guarrera, S.; et al. A Genome-Wide Association Study for Malignant Mesothelioma Risk. Lung Cancer 2013, 82, 1–8. [Google Scholar] [CrossRef]
- Matullo, G.; Guarrera, S.; Betti, M.; Fiorito, G.; Ferrante, D.; Voglino, F.; Cadby, G.; Di Gaetano, C.; Rosa, F.; Russo, A.; et al. Genetic Variants Associated with Increased Risk of Malignant Pleural Mesothelioma: A Genome-Wide Association Study. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Garay, E.; Patiño-López, G.; Islas, S.; Alarcón, L.; Canche-Pool, E.; Valle-Rios, R.; Medina-Contreras, O.; Granados, G.; Chávez-Munguía, B.; Juaristi, E.; et al. CRTAM: A Molecule Involved in Epithelial Cell Adhesion. J. Cell. Biochem. 2010, 111, 111–112. [Google Scholar] [CrossRef]
- Yoon, S.; Woo, S.U.; Kang, J.H.; Kim, K.; Shin, H.J.; Gwak, H.S.; Park, S.; Chwae, Y.J. NF-ΚB and STAT3 Cooperatively Induce IL6 in Starved Cancer Cells. Oncogene 2012, 31, 3467–3481. [Google Scholar] [CrossRef] [Green Version]
- Bosco, N.; Pelliccia, F.; Rocchi, A. Characterization of FRA7B, a Human Common Fragile Site Mapped at the 7p Chromosome Terminal Region. Cancer Genet. Cytogenet. 2010, 202, 47–52. [Google Scholar] [CrossRef]
- Calvo, F.; Sanz-Moreno, V.; Agudo-Ibáñez, L.; Wallberg, F.; Sahai, E.; Marshall, C.J.; Crespo, P. RasGRF Suppresses Cdc42-Mediated Tumour Cell Movement, Cytoskeletal Dynamics and Transformation. Nat. Cell Biol. 2011, 13, 819–826. [Google Scholar] [CrossRef]
- Park, J.W.; Zhao, L.; Willingham, M.; Cheng, S.Y. Oncogenic Mutations of Thyroid Hormone Receptor β. Oncotarget 2015, 6, 8115–8131. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; You, L.; Ren, X.; Zhao, W.; Liao, Q.; Zhao, Y. Long Non-Coding RNA PVT1 and Cancer. Biochem. Biophys. Res. Commun. 2016, 471, 10–14. [Google Scholar] [CrossRef]
- Jané-Valbuena, J.; Widlund, H.R.; Perner, S.; Johnson, L.A.; Dibner, A.C.; Lin, W.M.; Baker, A.C.; Nazarian, R.M.; Vijayendran, K.G.; Sellers, W.R.; et al. An Oncogenic Role for ETV1 in Melanoma. Cancer Res. 2010, 70, 2075–2084. [Google Scholar] [CrossRef] [Green Version]
- Tunesi, S.; Ferrante, D.; Mirabelli, D.; Andorno, S.; Betti, M.; Fiorito, G.; Guarrera, S.; Casalone, E.; Neri, M.; Ugolini, D.; et al. Gene-Asbestos Interaction in Malignant Pleural Mesothelioma Susceptibility. Carcinogenesis 2015, 36, 1129–1135. [Google Scholar] [CrossRef] [Green Version]
- Fagiani, E.; Giardina, G.; Luzi, L.; Cesaroni, M.; Quarto, M.; Capra, M.; Germano, G.; Bono, M.; Capillo, M.; Pelicci, P.G.; et al. RaLP, a New Member of the Src Homology and Collagen Family, Regulates Cell Migration and Tumor Growth of Metastatic Melanomas. Cancer Res. 2007, 67, 3064–3073. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.B.; Huang, X.F.; Lv, J.N.; Xiang, L.; Li, D.Q.; Chen, J.; Huang, C.; Wu, J.; Lu, F.; Qu, J. SLC7A14 Linked to Autosomal Recessive Retinitis Pigmentosa. Nat. Commun. 2014, 5, 3517. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.G.; Fennell, D.A.; Mutti, L.; O’Byrne, K.J. In Arrayed Ranks Array Technology in the Study of Mesothelioma Steven. J. Thorac. Oncol. 2009, 4, 411–425. [Google Scholar] [CrossRef]
- Gemignani, F.; Neri, M.; Bottari, F.; Barale, R.; Canessa, P.A.; Canzian, F.; Ceppi, M.; Spitaleri, I.; Cipollini, M.; Ivaldi, G.P.; et al. Risk of Malignant Pleural Mesothelioma and Polymorphisms in Genes Involved in the Genome Stability and Xenobiotics Metabolism. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 671, 76–83. [Google Scholar] [CrossRef]
- Neri, M.; Ugolini, D.; Dianzani, I.; Gemignani, F.; Landi, S.; Cesario, A.; Magnani, C.; Mutti, L.; Puntoni, R.; Bonassi, S. Genetic Susceptibility to Malignant Pleural Mesothelioma and Other Asbestos-Associated Diseases. Mutat. Res. Rev. Mutat. Res. 2008, 659, 126–136. [Google Scholar] [CrossRef]
- Ambrosone, C.B.; Sweeney, C.; Coles, B.F.; McClure, G.Y.; Fares, M.Y.; Stone, A.; Kadlubar, F.F.; Korourian, S.; Hutchins, L.F.; Thompson, P.A. Polymorphisms in Glutathione S-Transferases (GSTM1 and GSTT1) and Survival after Treatment for Breast Cancer. Cancer Res. 2001, 61, 7130–7135. [Google Scholar]
- Spurdle, A.B.; Chang, J.H.; Byrnes, G.B.; Chen, X.; Dite, G.S.; McCredie, M.R.E.; Giles, G.G.; Southey, M.C.; Chenevix-Trench, G.; Hopper, J.L. A Systematic Approach to Analysing Gene-Gene Interactions: Polymorphisms at the Microsomal Epoxide Hydrolase EPHX and Glutathione S-Transferase GSTM1, GSTT1, and GSTP1 Loci and Breast Cancer Risk. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 769–774. [Google Scholar] [CrossRef] [Green Version]
- Hein, D.W.; Doll, M.A.; Fretland, A.J.; Leff, M.A.; Webb, S.J.; Xiao, G.H.; Devanaboyina, U.S.; Nangju, N.A.; Feng, Y. Molecular Genetics and Epidemiology of the NAT1 and NAT2 Acetylation Polymorphisms. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 29–42. [Google Scholar]
- Kahlos, K.; Pääkkö, P.; Kurttila, E.; Soini, Y.; Kinnula, V.L. Manganese Superoxide Dismutase as a Diagnostic Marker for Malignant Pleural Mesothelioma. Br. J. Cancer 2000, 82, 1022–1029. [Google Scholar] [CrossRef] [Green Version]
- Sutton, A.; Khoury, H.; Prip-Buus, C.; Cepanec, C.; Pessayre, D.; Degoul, F. The Ala16Val Genetic Dimorphism Modulates the Import of Human Manganese Superoxide Dismutase into Rat Liver Mitochondria. Pharmacogenetics 2003, 13, 145–157. [Google Scholar] [CrossRef]
- Landi, S.; Gemignani, F.; Neri, M.; Barale, R.; Bonassi, S.; Bottari, F.; Canessa, P.A.; Canzian, F.; Ceppi, M.; Filiberti, R.; et al. Polymorphisms of Glutathione-S-Transferase M1 and Manganese Superoxide Dismutase Are Associated with the Risk of Malignant Pleural Mesothelioma. Int. J. Cancer 2007, 120, 2739–2743. [Google Scholar] [CrossRef] [Green Version]
- Neri, M.; Filiberti, R.; Taioli, E.; Garte, S.; Paracchini, V.; Bolognesi, C.; Canessa, P.A.; Fontana, V.; Ivaldi, G.P.; Verna, A.; et al. Pleural Malignant Mesothelioma, Genetic Susceptibility and Asbestos Exposure. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 592, 36–44. [Google Scholar] [CrossRef]
- Neri, M.; Taioli, E.; Filiberti, R.; Paolo Ivaldi, G.; Aldo Canessa, P.; Verna, A.; Marroni, P.; Puntoni, R.; Hirvonen, A.; Garte, S. Metabolic Genotypes as Modulators of Asbestos-Related Pleural Malignant Mesothelioma Risk: A Comparison of Finnish and Italian Populations. Int. J. Hyg. Environ. Health 2006, 209, 393–398. [Google Scholar] [CrossRef]
- Hirvonen, A.; Pelin, K.; Tammilehto, L.; Karjalainen, A.; Mattson, K.; Linnainmaa, K. Inherited GSTM1 and NAT2 Defects as Concurrent Risk Modifiers in Asbestos-Related Human Malignant Mesothelioma. Cancer Res. 1995, 55, 2981–2983. [Google Scholar]
- Hirvonen, A.; Saarikoski, S.T.; Linnainmaa, K.; Koskinen, K.; Husgafvel-Pursiainen, K.; Mattson, K.; Vainio, H. Glutathione S-Transferase and N-Acetyltransferase Genotypes and Asbestos-Associated Pulmonary Disorders. J. Natl. Cancer Inst. 1996, 88, 1853–1856. [Google Scholar] [CrossRef] [PubMed]
- Dianzani, I.; Gibello, L.; Biava, A.; Giordano, M.; Bertolotti, M.; Betti, M.; Ferrante, D.; Guarrera, S.; Betta, G.P.; Mirabelli, D.; et al. Polymorphisms in DNA Repair Genes as Risk Factors for Asbestos-Related Malignant Mesothelioma in a General Population Study. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2006, 599, 124–134. [Google Scholar] [CrossRef] [PubMed]
- London, R.E. The Structural Basis of XRCC1-Mediated DNA Repair. DNA Repair (Amst) 2015, 30, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, D.B.; Ghosh, A.; Lu, J.; Bohr, V.A.; Liu, Y. Factors That Influence Telomeric Oxidative Base Damage and Repair by DNA Glycosylase OGG1. DNA Repair (Amst) 2011, 10, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorour, A.; Ayad, M.W.; Kassem, H. The Genotype Distribution of the XRCC1, XRCC3, and XPD DNA Repair Genes and Their Role for the Development of Acute Myeloblastic Leukemia. Genet. Test. Mol. Biomarkers 2013, 17, 195–201. [Google Scholar] [CrossRef]
- Matullo, G.; Palli, D.; Peluso, M.; Guarrera, S.; Carturan, S.; Celentano, E.; Krogh, V.; Munnia, A.; Tumino, R.; Polidoro, S.; et al. XRCC1, XRCC3, XPD Gene Polymorphisms, Smoking and 32P-DNA Adducts in a Sample of Healthy Subjects. Carcinogenesis 2001, 9, 1437–1445. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.Y.; Hildesheim, A.; Chen, C.J.; Hsu, M.M.; Chen, I.H.; Mittl, B.F.; Levine, P.H.; Liu, M.Y.; Chen, J.Y.; Brinton, L.A.; et al. Nasopharyngeal Carcinoma and Genetic Polymorphisms of DNA Repair Enzymes XRCC1 and HOGG1. Cancer Epidemiol. Biomarkers Prev. 2003, 12, 1100–1104. [Google Scholar]
- Duell, E.J.; Wiencke, J.K.; Varkonyi, A.; Zuo, Z.; Ashok, T.D.; Mark, E.J.; Wain, J.C.; Christiani, D.C.; Kelsey, K.T. Polymorphisms in the DNA Repair Genes XRCC1 and ERCC2 and Biomarkers of DNA Damage in Human Blood Mononuclear Cells. Carcinogenesis 2000, 21, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Hung, R.J.; Brennan, P.; Canzian, F.; Szeszenia-Dabrowska, N.; Zaridze, D.; Lissowska, J.; Rudnai, P.; Fabianova, E.; Mates, D.; Foretova, L.; et al. Large-Scale Investigation of Base Excision Repair Genetic Polymorphisms and Lung Cancer Risk in a Multicenter Study. J. Natl. Cancer Inst. 2005, 97, 567–576. [Google Scholar] [CrossRef]
Figure 1.
Search strategy for identifying scientific publications for this comprehensive review paper.
Figure 1.
Search strategy for identifying scientific publications for this comprehensive review paper.

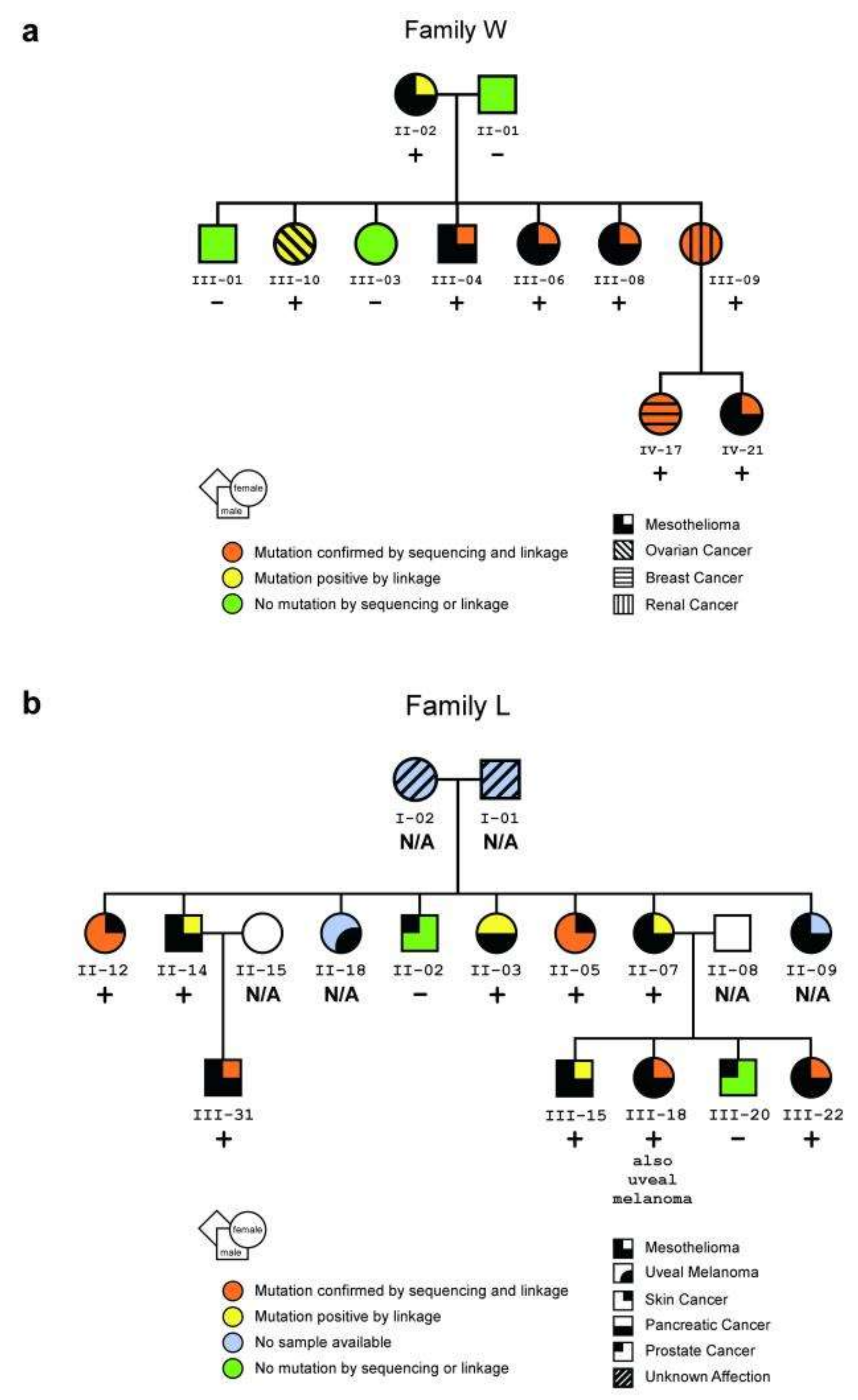
Figure 2.
Pedigrees of two U.S. mesothelioma families. (a,b) Pedigrees showing family members with a germline mutation in BAP1, as confirmed by both sequencing and linkage analyses (orange) or by linkage analysis alone (yellow, i.e., no DNA was available for sequencing); individuals without the mutation (green) and individuals for whom DNA was unavailable (blue) are also shown. Presence or absence of germline BAP1 mutation is also indicated with + or − symbols, respectively. (a) Pedigree of family W showing the presence or absence of a germline mutation at the BAP1 consensus splice acceptor site. (b) Pedigree of family L showing the presence or absence of a germline nonsense mutation. The development of other tumor types in these families may also be related to BAP1 germline mutations. In family W, the presence of a breast cancer before age 45 and an ovarian cancer suggests that the BAP1 mutation is associated with a hereditary form of breast/ovarian cancer, as might be expected given BAP1′s relationship with the breast/ovarian cancer susceptibility gene product, BRCA115. In family L, the skin cancers shown were squamous cell carcinomas. Reprinted with permission from Springer Nature Genetics (Germline BAP1 Mutations Predispose to Malignant Mesothelioma by Testa et al.) [18], Copyright © 2012.
Figure 2.
Pedigrees of two U.S. mesothelioma families. (a,b) Pedigrees showing family members with a germline mutation in BAP1, as confirmed by both sequencing and linkage analyses (orange) or by linkage analysis alone (yellow, i.e., no DNA was available for sequencing); individuals without the mutation (green) and individuals for whom DNA was unavailable (blue) are also shown. Presence or absence of germline BAP1 mutation is also indicated with + or − symbols, respectively. (a) Pedigree of family W showing the presence or absence of a germline mutation at the BAP1 consensus splice acceptor site. (b) Pedigree of family L showing the presence or absence of a germline nonsense mutation. The development of other tumor types in these families may also be related to BAP1 germline mutations. In family W, the presence of a breast cancer before age 45 and an ovarian cancer suggests that the BAP1 mutation is associated with a hereditary form of breast/ovarian cancer, as might be expected given BAP1′s relationship with the breast/ovarian cancer susceptibility gene product, BRCA115. In family L, the skin cancers shown were squamous cell carcinomas. Reprinted with permission from Springer Nature Genetics (Germline BAP1 Mutations Predispose to Malignant Mesothelioma by Testa et al.) [18], Copyright © 2012.

Figure 3.
Survival of patients with mesothelioma treated with platinum-based chemotherapy, by patient’s genotype and primary site of tumor. Survival of patients with an inherited damaging mutation in any targeted gene is indicated in blue; survival of patients with no inherited mutation is indicated in red. (A) All mesothelioma patients with versus without inherited mutations. Median survival: 8.0 vs. 2.9 y, p = 0.0006. (B) Pleural mesothelioma patients with versus without inherited mutations. Median survival: 7.9 vs. 2.4 y, p = 0.0012. (C) Peritoneal mesothelioma patients with versus without inherited mutation. Median survival: 8.2 vs. 5.4 y, p = 0.47. Reprinted with permission from Proceedings of the National Academy of Sciences of the United States of America (Inherited predisposition to malignant mesothelioma and overall survival following platinum chemotherapy by Hassan et al.) [4], Copyright © 2019.
Figure 3.
Survival of patients with mesothelioma treated with platinum-based chemotherapy, by patient’s genotype and primary site of tumor. Survival of patients with an inherited damaging mutation in any targeted gene is indicated in blue; survival of patients with no inherited mutation is indicated in red. (A) All mesothelioma patients with versus without inherited mutations. Median survival: 8.0 vs. 2.9 y, p = 0.0006. (B) Pleural mesothelioma patients with versus without inherited mutations. Median survival: 7.9 vs. 2.4 y, p = 0.0012. (C) Peritoneal mesothelioma patients with versus without inherited mutation. Median survival: 8.2 vs. 5.4 y, p = 0.47. Reprinted with permission from Proceedings of the National Academy of Sciences of the United States of America (Inherited predisposition to malignant mesothelioma and overall survival following platinum chemotherapy by Hassan et al.) [4], Copyright © 2019.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Pathogenic or likely pathogenic germline mutations associated with malignant mesothelioma*.
Table 1.
Pathogenic or likely pathogenic germline mutations associated with malignant mesothelioma*.
| Gene | Location | Function | Implication in Other Tumors | Reference |
|---|---|---|---|---|
| BAP1 | 3 | Tumor suppressor, DNA repair | Uveal and cutaneous melanoma, renal cell carcinoma, non-melanoma skin cancer, meningioma, cholangiocarcinoma | [18] |
| BRCA1 | 17 | Tumor suppressor, DNA repair | Breast, ovarian, prostate, colon and pancreatic cancer, melanoma | [7,19] |
| BRCA2 | 13 | Tumor suppressor, DNA repair | Breast, ovarian, prostate and pancreatic cancer, melanoma | [7,19] |
| BARD1 | 2 | DNA repair | Breast and ovarian cancer | [20] |
| TP53 | 17 | Tumor suppressor, DNA repair | Lung, head and neck, ovarian, breast, bladder, liver and colorectal cancer, melanoma, osteosarcoma, rhabdomyosarcoma, glioma, adrenocortical carcinoma, cholangiocarcinoma | [7,21,22] |
| PALB2 | 16 | Tumor suppressor, DNA repair | Breast, ovarian and pancreatic cancer | [19] |
| CHEK2 | 22 | DNA repair | Breast, ovarian and prostate cancer, osteosarcoma | [7] |
| ATM | 11 | DNA repair | Breast and bladder cancer, melanoma | [7,19] |
| SLX4 | 16 | DNA repair | Head and neck and pancreatic cancer | [19] |
| FANCC | 9 | DNA repair | Breast, head and neck and pancreatic cancer | [19] |
| FANCF | 11 | DNA repair | Breast, head and neck, pancreatic and prostate cancer | [19] |
| FANCI | 10 | DNA repair | Breast, head and neck, pancreatic and prostate cancer | [19] |
| RAD50 | 5 | DNA repair | Prostate and breast cancer | [23] |
| MRE11A | 11 | DNA repair | Breast and prostate cancer | [7] |
| WT1 | 11 | DNA repair | Wilm´s tumor | [7] |
| RECQL4 | 8 | DNA repair | Osteosarcoma | [20] |
| XPC | 3 | DNA repair | Basal and squamous cell carcinoma, melanoma | [19] |
| SETD2 | 3 | Tumor suppressor, DNA repair, chromatin regulation | Renal cell carcinoma, leukemia | [23] |
| PMS1 | 2 | DNA repair | Colon cancer | [19] |
| MSH3 | 3 | DNA repair | Colon and endometrial cancer | [20] |
| MSH6 | 2 | DNA repair | Colorectal, endometrial and ovarian cancer, leukemia, lymphoma | [7,24,25] |
| MLH1 | 3 | Tumor suppressor, DNA repair | Colorectal, endometrial and ovarian cancer, leukemia, lymphoma | [23] |
| POT1 | 7 | DNA repair, telomere maintenance | Melanoma, glioma | [4] |
| NCOR1 | 17 | Chromatin regulation | - | [23] |
| ARID1A | 1 | Tumor suppressor, chromatin regulation | Ovarian, endometrial, kidney, stomach, bladder, lung, breast and brain cancer, cholangiocarcinoma | [23] |
| SMARCE1 | 17 | Chromatin regulation | - | [23] |
| ARID2 | 12 | Tumor suppressor, chromatin regulation | - | [23] |
| CREBBP | 16 | Tumor suppressor, transcription regulation | Bladder cancer, leukemia | [23] |
| SMARCA4 | 9 | Tumor suppressor, chromatin regulation | Lung cancer, rhabdoid tumor predisposition syndrome type 2 | [23] |
| SMARCA2 | 9 | Tumor suppressor, chromatin regulation | Lung and head and neck cancer | [23] |
| SHQ1 | 3 | Tumor suppressor, ribosomal and telomerase RNA processing | - | [20] |
| RBM6 | 3 | Tumor suppressor, RNA processing | - | [23] |
| NF2 | 22 | Tumor suppressor | Schwannoma | [21] |
| CDKN2A | 9 | Tumor suppressor, cell cycle regulation | Bladder, head and neck, lung, breast and pancreatic cancer, melanoma | [7,21] |
| KDR | 4 | Tyrosine kinase receptor | - | [23] |
| TMEM127 | 2 | Tumor suppressor, rapamycin signaling pathway | - | [7] |
| SMO | 7 | G-protein couple receptor | Basal cell carcinoma | [23] |
| SDHA | 5 | Regulation of hypoxia inducible factor expression | Gastrointestinal stromal tumor | [7] |
| VHL | 3 | Regulation of hypoxia inducible factor expression | Von Hippel–Lindau syndrome | [7] |
* Gene chromosome location, function and implication in other tumors were listed on the basis of gene annotations provided by the National Center for Biotechnology Information’s Online Mendelian Inheritance in Man, available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/omim (accessed on 8 June 2020), The Human Gene Database, Weizmann Institute of Science, available online: https://www.genecards.org (accessed on 8 June 2020) and the Atlas of Genetics and Cytogenetics in Oncology and Haematology, available online: http://atlasgeneticsoncology.org (accessed on 8 June 2020).
Table 2.
The most significant single nucleotide polymorphisms (SNPs) associated with malignant mesothelioma, as identified through genome-wide association studies (GWAS)*.
Table 2.
The most significant single nucleotide polymorphisms (SNPs) associated with malignant mesothelioma, as identified through genome-wide association studies (GWAS)*.
| SNP | Locus | Gene/Neighboring Genes | Gene Function | References |
|---|---|---|---|---|
| rs17228032 | 11q24.1 | CRTAM, JHY, UBASH3B | Adaptive immune response | [87] |
| rs1379270 | 5q13 | RASGRF2, CKMT2, MSH3 | Apoptosis, Rho and Ras protein and small GTPase mediated signal transduction regulation | [87] |
| rs12540101 | 7p22.2 | SDK1, CYP3A54P, CARD11 | Cell adhesion | [87] |
| rs12701229 | 7p22.2 | SDK1, CYP3A54P, CARD11 | Cell adhesion | [87] |
| rs10089418 | 8p21.3 | LOC286114, LINC02153, LZTS1 | - | [87] |
| rs11126523 | 2p12 | C2orf3, LRRTM4 | - | [87] |
| rs13287752 | 9p21.1 | MIR873, C9orf72 | - | [87] |
| rs282718 | 4q12 | IGFBP7, LINC02390, IGFBP7 | - | [87] |
| rs4707427 | 6q15 | SPACA1, AKIRIN2 | - | [87] |
| rs4895337 | 5q23.1 | FTMT, SRFBP1 | - | [87] |
| rs7958488 | 12p13.31 | CD27 | - | [87] |
| rs8142386 | 22q112 | LOC150185 | - | [87] |
| rs9548166 | 13q13.3 | LINC00571, LINC02334 | - | [87] |
| rs7841347 | 8q24.21 | PVT1, MYC, TMEM75 | Oncogene, Transcription regulation | [88] |
| rs3801094 | 7p21.2 | ETV1, ARL4A, DGKB | Oncogene, Transcription regulation | [88] |
| rs9833191 | 3p24.2 | THRB, NR1D2, MIR4792 | Tumor suppressor, Transcription regulation | [88] |
| rs7632718 | 3q26.2 | SLC7A14, CLDN11, RPL22L1 | Amino acid transport | [88] |
| rs4701085 | 5q35.3 | ADAMTS2, ZNF354C, AX747985 | Collagen degradation | [88] |
| rs2501618 | 1q25.2 | CEP350, TOR1AIP1 | Microtubule anchoring | [88] |
| rs10519201 | 15q21.1 | SHC4, EID1, SEDISBP2L | Apoptosis, Regulation of cell proliferation | [88] |
| rs1508805 | 5q23.1 | PRR16, FTMT | - | [88] |
| rs4290865 | 4q22.1 | FAM190A, GRID2 | - | [88] |
| rs5756444 | 22q12.3 | CXF2RB2, C22orf33, TEX33 | - | [88] |
| rs742109 | 6q21 | PRDM1, ATGS | - | [88] |
| rs9536579 | 13q14.3 | OLFMS, MIR1297 | - | [88] |
* Gene location and function were listed on the basis of gene annotations provided by the National Center for Biotechnology Information’s Online Mendelian Inheritance in Man, available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/omim (assessed on 8 June 2020), The Human Gene Database, Weizmann Institute of Science, available online: https://www.genecards.org (assessed on 8 June 2020) and the Ensembl genome browser, available online: https://www.ensembl.org/index.html (assessed on 8 June 2020).
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Panou, V.; Røe, O.D. Inherited Genetic Mutations and Polymorphisms in Malignant Mesothelioma: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 4327. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124327
AMA Style
Panou V, Røe OD. Inherited Genetic Mutations and Polymorphisms in Malignant Mesothelioma: A Comprehensive Review. International Journal of Molecular Sciences. 2020; 21(12):4327. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124327
Chicago/Turabian StylePanou, Vasiliki, and Oluf Dimitri Røe. 2020. "Inherited Genetic Mutations and Polymorphisms in Malignant Mesothelioma: A Comprehensive Review" International Journal of Molecular Sciences 21, no. 12: 4327. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124327
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.