Molecular Epidemiology of B3 and D8 Measles Viruses through Hemagglutinin Phylogenetic History

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Hemagglutinin Phylogenetic and Sequence Analyses
2.2. Overall Selective Pressure Analyses
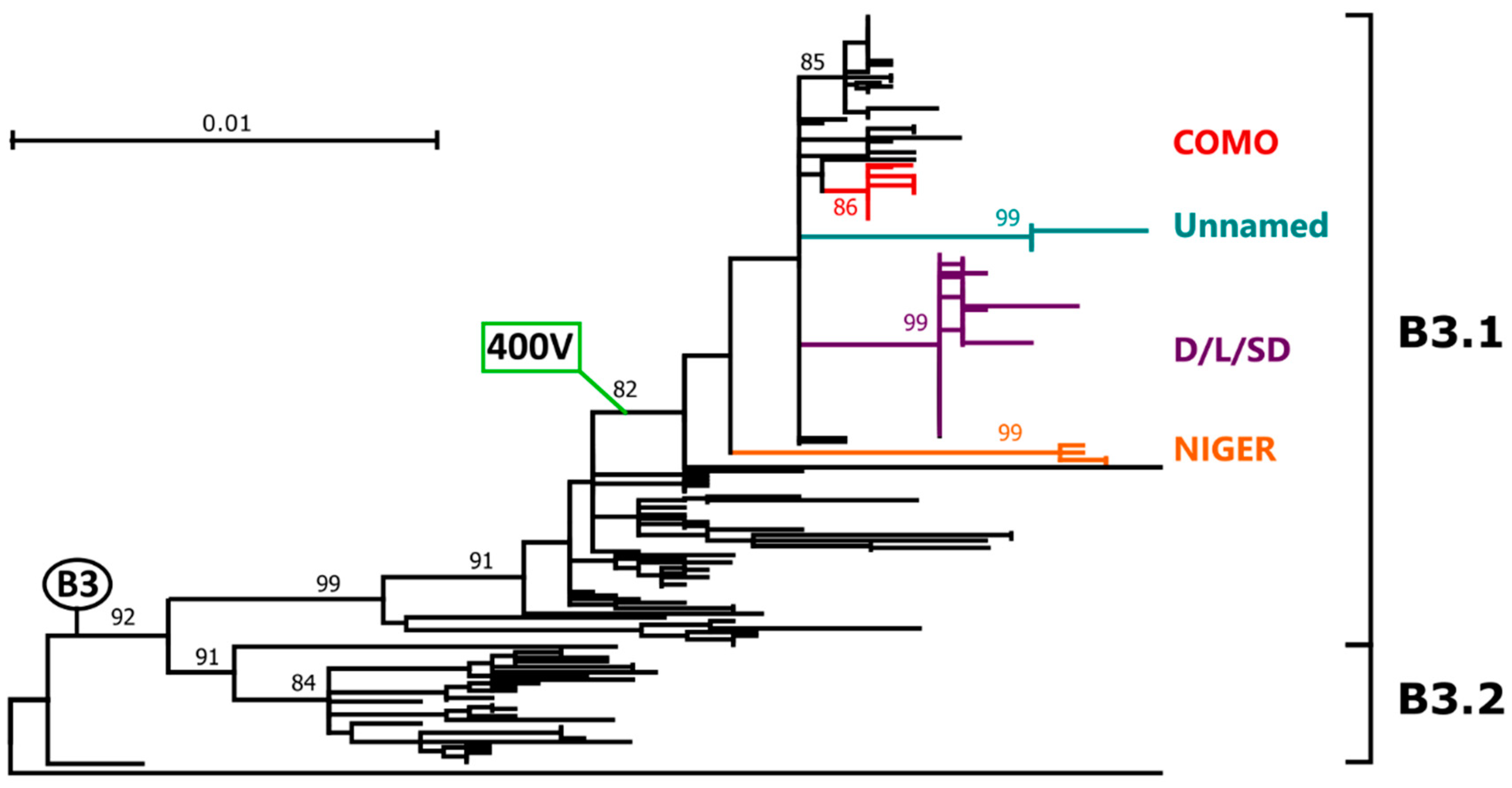
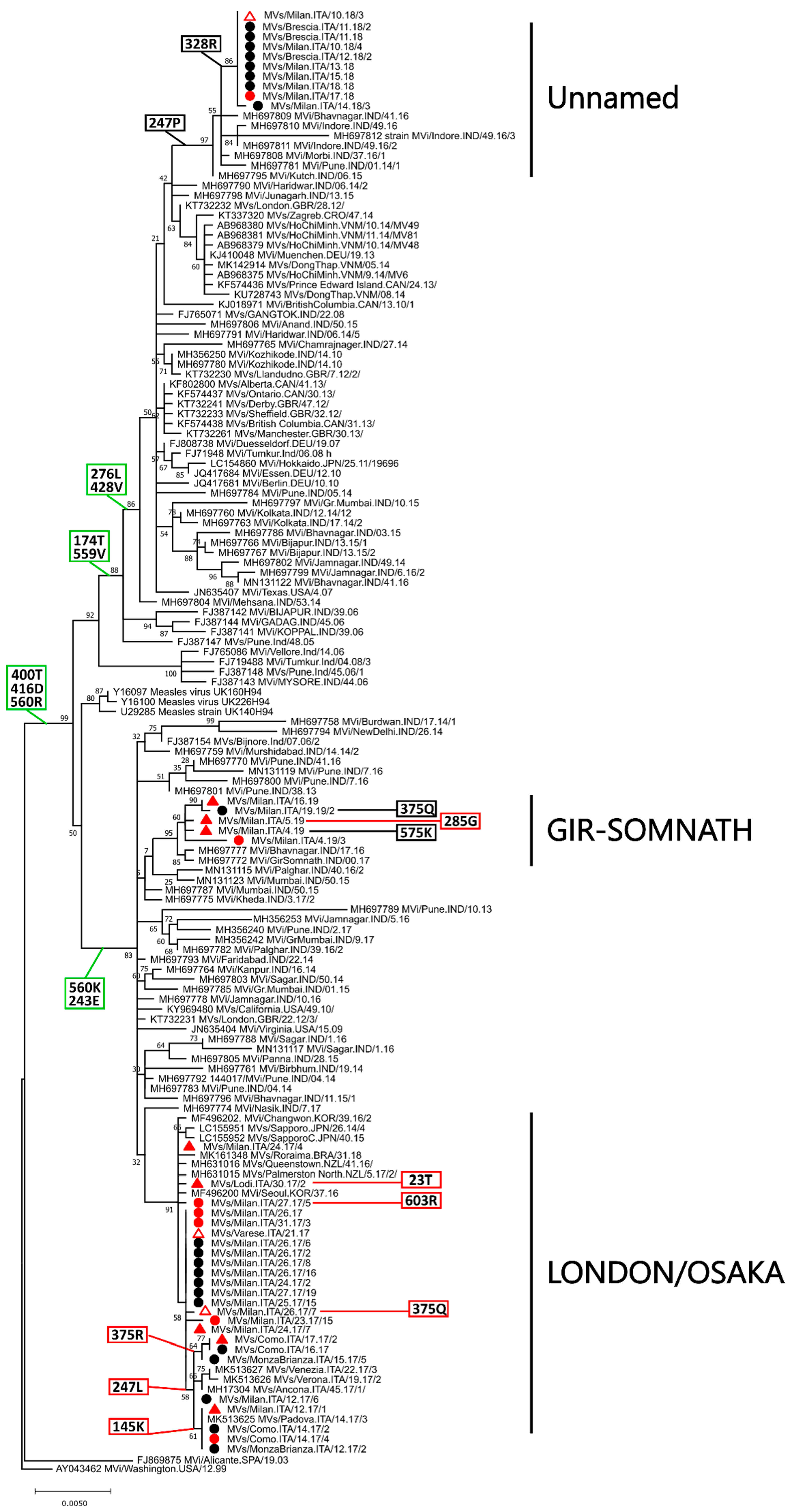
2.3. Genotype B3
2.3.1. Phylogenetic Analyses and Molecular Epidemiology
2.3.2. Genetic Drift and Amino Acid Substitutions
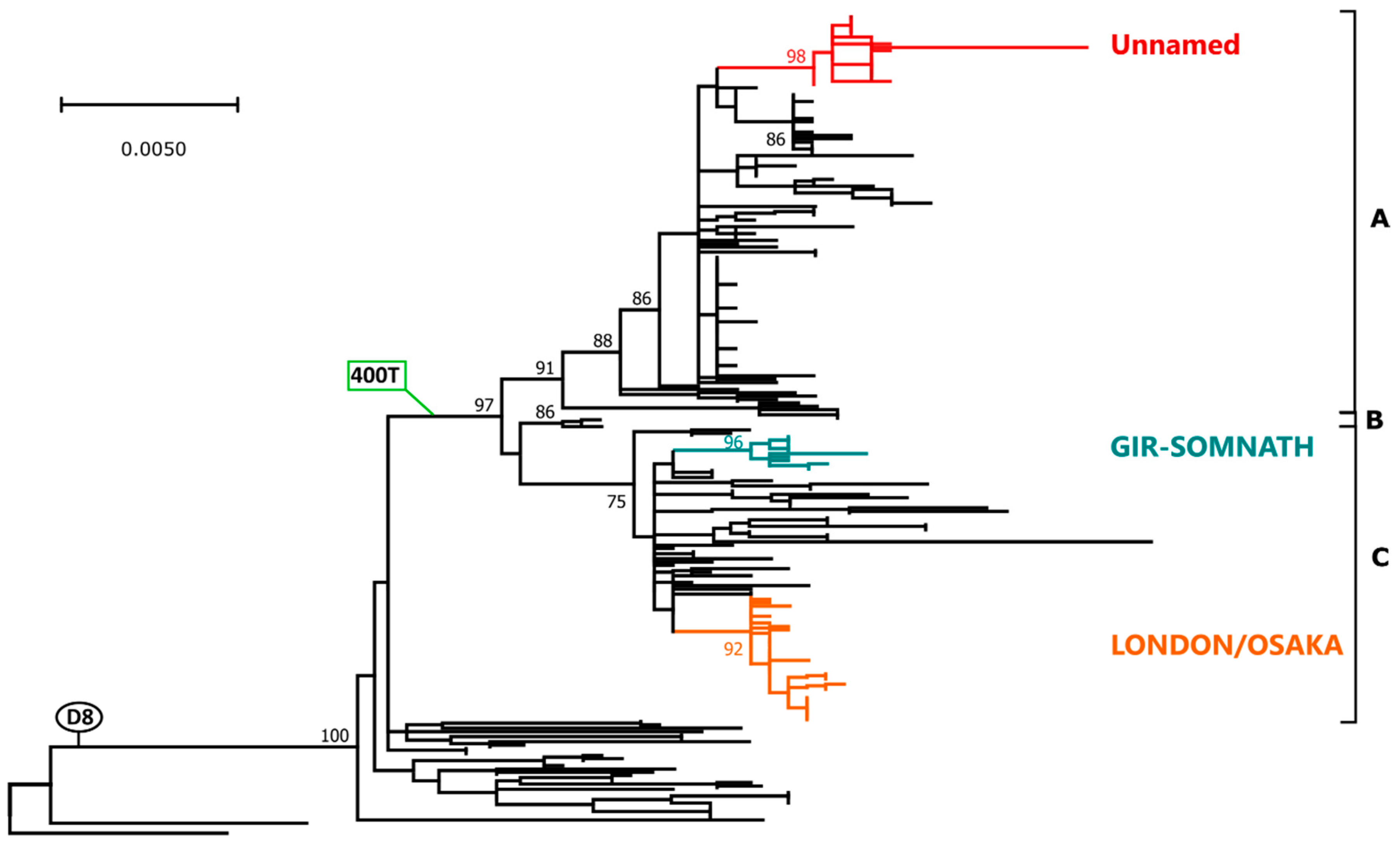
2.4. Genotype D8
2.4.1. Phylogenetic Analyses and Molecular Epidemiology
2.4.2. Genetic Drift and Amino Acid Substitutions
2.5. Analysis of the Immune Epitopes
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Sequencing of the Hemagglutinin Gene
5.2. Phylogenetic Analyses
5.3. Selection Pressure Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDC | Centers for Disease Control and Prevention |
| H | Hemagglutinin |
| HNE | Hemagglutinating and noose epitope |
| LE | Loop epitope |
| MV | Measles virus |
| MeaNS | Measles Nucleotide Surveillance |
| NE | Neutralizing epitope |
| n-450 | 450 nucleotides encoding the carboxyl-terminal 150 amino acids of the nucleoprotein |
| RBE | Receptor-binding epitope |
| SSE | Sugar-shielded epitope |
| WHO | World Health Organization |
References
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef]
- Moss, W.J. Measles. Lancet 2017, 390, 2490–2502. [Google Scholar] [CrossRef]
- Dabbagh, A.; Laws, R.L.; Steulet, C.; Dumolard, L.; Mulders, M.N.; Kretsinger, K.; Alexander, J.P.; Rota, P.A.; Goodson, J.L. Progress Toward Regional Measles Elimination-Worldwide, 2000-2017. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1323–1329. [Google Scholar] [CrossRef]
- Patel, M.; Lee, A.D.; Clemmons, N.S.; Redd, S.B.; Poser, S.; Blog, D.; Zucker, J.R.; Leung, J.; Link-Gelles, R.; Pham, H.; et al. National Update on Measles Cases and Outbreaks-United States, January 1-October 1, 2019. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 893–896. [Google Scholar] [CrossRef]
- Patel, M.K.; Orenstein, W.A. Classification of global measles cases in 2013–17 as due to policy or vaccination failure: A retrospective review of global surveillance data. Lancet 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, T.; Maenaka, K.; Yanagi, Y. Measles Virus Hemagglutinin: Structural Insights into Cell Entry and Measles Vaccine. Front. Microbiol. 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldo, A.; Galanis, E.; Tangy, F.; Herman, P. Biosafety considerations for attenuated measles virus vectors used in virotherapy and vaccination. Hum. Vaccin. Immunother. 2016, 12, 1102–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, M.; Bürckert, J.-P.; Kanou, K.; Maenaka, K.; Muller, C.P.; Takeda, M. Measles Virus Hemagglutinin Protein Epitopes: The Basis of Antigenic Stability. Viruses 2016, 8, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, J.B.; Rota, J.S.; Hickman, C.J.; Sowers, S.B.; Mercader, S.; Rota, P.A.; Bellini, W.J.; Huang, A.J.; Doll, M.K.; Zucker, J.R.; et al. Outbreak of measles among persons with prior evidence of immunity, New York City, 2011. Clin. Infect. Dis. 2014, 58, 1205–1210. [Google Scholar] [CrossRef]
- Rota, J.S.; Hickman, C.J.; Sowers, S.B.; Rota, P.A.; Mercader, S.; Bellini, W.J. Two case studies of modified measles in vaccinated physicians exposed to primary measles cases: High risk of infection but low risk of transmission. J. Infect. Dis. 2011, 204, S559–S563. [Google Scholar] [CrossRef]
- Cherry, J.D.; Zahn, M. Clinical Characteristics of Measles in Previously Vaccinated and Unvaccinated Patients in California. Clin. Inf. Dis. 2018, 67, 1315–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, J.F.; Jacobson, R.M.; Poland, G.A.; Jacobsen, S.J.; Wollan, P.C. Secondary failure rates of measles vaccines: A metaanalysis of published studies. Pediatr. Infect. Dis. J. 1996, 15, 62–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javelle, E.; Colson, P.; Parola, P.; Raoult, D. Measles, the need for a paradigm shift. Eur. J. Epidemiol. 2019, 34, 897–915. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Brown, K.; Mankertz, A.; Santibanez, S.; Shulga, S.; Muller, C.P.; Hübschen, J.M.; Siqueira, M.; Beirnes, J.; Ahmed, H.; et al. Global distribution of measles genotypes and measles molecular epidemiology. J. Infect. Dis. 2011, 204, S514–S523. [Google Scholar] [CrossRef]
- Mulders, M.N.; Rota, P.A.; Icenogle, J.P.; Brown, K.E.; Takeda, M.; Rey, G.J.; Ben Mamou, M.C.; Dosseh, A.R.G.A.; Byabamazima, C.R.; Ahmed, H.J.; et al. Global Measles and Rubella Laboratory Network Support for Elimination Goals, 2010-2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 438–442. [Google Scholar] [CrossRef]
- Magurano, F.; Baggieri, M.; Filia, A.; Del Manso, M.; Lazzarotto, T.; Amendola, A.; D’Agaro, P.; Chironna, M.; Ansaldi, F.; Iannazzo, S.; et al. Towards measles elimination in Italy: Virological surveillance and genotypes trend (2013-2015). Virus Res. 2017, 236, 24–29. [Google Scholar] [CrossRef]
- Expanded Programme on Immunization (EPI). Standardization of the nomenclature for describing the genetic characteristics of wild-type measles viruses. Wkly. Epidemiol. Rec. 1998, 73, 265–269. [Google Scholar]
- Finsterbusch, T.; Wolbert, A.; Deitemeier, I.; Meyer, K.; Mosquera, M.M.; Mankertz, A.; Santibanez, S. Measles viruses of genotype H1 evade recognition by vaccine-induced neutralizing antibodies targeting the linear haemagglutinin noose epitope. J. Gen. Virol. 2009, 90, 2739–2745. [Google Scholar] [CrossRef]
- De Swart, R.L.; Yüksel, S.; Osterhaus, A.D.M.E. Relative contributions of measles virus hemagglutinin- and fusion protein-specific serum antibodies to virus neutralization. J. Virol. 2005, 79, 11547–11551. [Google Scholar] [CrossRef] [Green Version]
- Bouche, F.B.; Ertl, O.T.; Muller, C.P. Neutralizing B cell response in measles. Viral Immunol. 2002, 15, 451–471. [Google Scholar] [CrossRef]
- De Swart, R.L.; Yüksel, S.; Langerijs, C.N.; Muller, C.P.; Osterhaus, A.D.M.E. Depletion of measles virus glycoprotein-specific antibodies from human sera reveals genotype-specific neutralizing antibodies. J. Gen. Virol. 2009, 90, 2982–2989. [Google Scholar] [CrossRef]
- Tahara, M.; Ohno, S.; Sakai, K.; Ito, Y.; Fukuhara, H.; Komase, K.; Brindley, M.A.; Rota, P.A.; Plemper, R.K.; Maenaka, K.; et al. The receptor-binding site of the measles virus hemagglutinin protein itself constitutes a conserved neutralizing epitope. J. Virol. 2013, 87, 3583–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zheng, J.; Huang, H.; Hu, Y.; Bian, J.; Xu, D.; Li, F. Measles incidence rate and a phylogenetic study of contemporary genotype H1 measles strains in China: Is an improved measles vaccine needed? Virus Genes 2011, 43, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, G.; Canuti, M.; Bianchi, S.; Gori, M.; Piralla, A.; Colzani, D.; Libretti, M.; Frati, E.R.; Baggieri, M.; Lai, A.; et al. Genetic variability of the measles virus hemagglutinin gene in B3 genotype strains circulating in Northern Italy. Infect. Genet. Evol. 2019, 75. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Jin, L.; Knowles, W.A.; Rota, P.A.; Bellini, W.J.; Brown, D.W. Genetic and antigenic characterisation of the haemagglutinin protein of measles virus strains recently circulating in the UK. Virus Res. 1998, 55, 107–113. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar]
- 8th Meeting of the European Regional Verification Commission for Measles and Rubella Elimination (RVC) (2019). Available online: http://www.euro.who.int/en/health-topics/communicable-diseases/measles-and-rubella/publications/2019/8th-meeting-of-the-european-regional-verification-commission-for-measles-and-rubella-elimination-rvc-2019 (accessed on 29 April 2020).
- Magurano, F.; Fortuna, C.; Marchi, A.; Benedetti, E.; Bucci, P.; Baggieri, M.; Nicoletti, L. Molecular epidemiology of measles virus in Italy, 2002–2007. Virol. J. 2012, 9, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amendola, A.; Bianchi, S.; Lai, A.; Canuti, M.; Piralla, A.; Baggieri, M.; Ranghiero, A.; Piatti, A.; Tanzi, E.; Zehender, G.; et al. Measles re-emergence in Northern Italy: Pathways of measles virus genotype D8, 2013-2014. Infect. Genet. Evol. 2017, 48, 120–126. [Google Scholar] [CrossRef]
- Magurano, F.; Baggieri, M.; Fortuna, C.; Bella, A.; Filia, A.; Rota, M.C.; Benedetti, E.; Bucci, P.; Marchi, A.; Nicoletti, L. Measles elimination in Italy: Data from laboratory activity, 2011-2013. J. Clin. Virol. 2015, 64, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Filia, A.; Amendola, A.; Faccini, M.; Del Manso, M.; Senatore, S.; Bianchi, S.; Borrini, B.M.; Ciampelli, A.; Tanzi, E.; Filipponi, M.T.; et al. Outbreak of a new measles B3 variant in the Roma/Sinti population with transmission in the nosocomial setting, Italy, November 2015 to April 2016. Euro Surveill. 2016, 21. [Google Scholar] [CrossRef]
- Amendola, A.; Bianchi, S.; Frati, E.R.; Ciceri, G.; Faccini, M.; Senatore, S.; Colzani, D.; Lamberti, A.; Baggieri, M.; Cereda, D.; et al. Ongoing large measles outbreak with nosocomial transmission in Milan, northern Italy, March-August 2017. Euro Surveill. 2017, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, S.; Frati, E.R.; Lai, A.; Colzani, D.; Ciceri, G.; Baggieri, M.; Lamberti, A.; Senatore, S.; Faccini, M.; Mazzilli, F.; et al. Genetic characterisation of Measles virus variants identified during a large epidemic in Milan, Italy, March-December 2017. Epidemiol. Infect. 2019, 147. [Google Scholar] [CrossRef] [Green Version]
- Moss, W. Measles in Vaccinated Individuals and the Future of Measles Elimination. Clin. Infect. Dis. 2018, 67, 1320–1321. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Hierarchical Structure of Proteins. In Molecular Cell Biology, 4th ed.; W. H. FReeman: New York, NY, USA, 2000. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention Genetic Analysis of Measles Virus. Available online: https://www.cdc.gov/measles/lab-tools/genetic-analysis.html (accessed on 11 April 2020).
- Wang, L.; Ding, Z.; Wang, H.; Li, L.; Pang, Y.; Brown, K.E.; Xu, S.; Zhu, Z.; Rota, P.A.; Featherstone, D.; et al. New Measles Virus Genotype Associated with Outbreak, China. Emerg. Infect. Dis. 2010, 16, 943–947. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Classification 1 | Case 2 | Vaccination 3 | Origin |
|---|---|---|---|---|
| B3-2015 | ||||
| MVs/Lecco.ITA/44.15 | B3 Niger | Sporadic | Unvaccinated | Imported |
| MVs/Pavia.ITA/44.15 | B3 Como | Outbreak | Unvaccinated | Autochthonous |
| MVs/Pavia.ITA/44.15/2 | B3 Como | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/45.15 | B3 Como | Sporadic | Unvaccinated | Autochthonous |
| MVs/Pavia.ITA/46.15 | B3 Como | Sporadic | Unvaccinated | Autochthonous |
| MVs/Lecco.ITA/47.15 | B3 Niger | Sporadic | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/53.15/2 | B3 Como | Sporadic | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/53.15/5 | B3 Como | Outbreak | Unvaccinated | Autochthonous |
| B3-2016 | ||||
| MVs/Milan.ITA/1.16 | B3 Como | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/1.16/2 | B3 Niger | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/1.16/3 | B3 Como | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/2.16 | B3 Niger | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/3.16/2 | B3 Como | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/3.16 | B3 Como | Outbreak | Vaccinated (2) | Autochthonous |
| MVs/Milan.ITA/3.16/4 | B3 Como | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/3.16/6 | B3 Como | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/6.16/3 | B3 Como | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/7.16 | B3 Como | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Pavia.ITA/08.16 | B3 Niger | Sporadic | Vaccinated (1) | Autochthonous |
| B3-2017 | ||||
| MVs/Bergamo.ITA/9.17 | B3 Dublin | Outbreak | Unvaccinated | Imported |
| MVs/Brescia.ITA/12.17 | B3 Dublin | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/17.17/2 | B3 Dublin | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/21.17/3 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/22.17/4 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/23.17/3 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/23.17/4 | B3 Dublin | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/25.17/12 | B3 Dublin | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/26.17/3 | B3 Dublin | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/38.17 | B3 Dublin | Sporadic | Vaccinated (2) | Autochthonous |
| B3-2018 | ||||
| MVs/Milan.ITA/8.18/2 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/10.18/2 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/11.18 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/11.18/3 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/13.18/2 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/14.18 | B3 Dublin | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/14.18/2 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/15.18/3 | B3 Ljubjana | Sporadic | Unvaccinated | Imported |
| MVs/Milan.ITA/15.18/4 | B3 Dublin | Sporadic | Unvaccinated | Imported |
| MVs/Milan.ITA/16.18 | B3 Dublin | Sporadic | Unvaccinated | Autochthonous |
| MVs/Brescia.ITA/17.18 | B3 Dublin | Outbreak | Unvaccinated | Autochthonous |
| MVs/Como.ITA/16.18 | B3 Ljubjana | Sporadic | Unvaccinated | Autochthonous |
| MVs/Como.ITA/17.18 | B3 Ljubjana | Outbreak | Unvaccinated | Autochthonous |
| MVs/Como.ITA/18.18 | B3 Ljubjana | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Como.ITA/18.18/2 | B3 Ljubjana | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/24.18/n | B3 Dublin | Outbreak | Vaccinated (2) | Autochthonous |
| MVs/Milan.ITA/26.18 | B3 Unnamed 4 | Sporadic | Vaccinated (2) | Imported |
| MVs/Milan.ITA/27.18 | B3 Unnamed | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/27.18/2 | B3 Unnamed | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/27.18/4 | B3 Unnamed | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/28.18 | B3 Unnamed | Outbreak | Unvaccinated | Autochthonous |
| MVs/MonzaBrianza.ITA/28.18 | B3 Unnamed | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/28.18/2 | B3 Unnamed | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/47.18 | B3 Saint Denis | Outbreak | Unvaccinated | Autochthonous |
| D8-2017 | ||||
| MVs/Como.ITA/16.17 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/12.17/1 | D8 Osaka | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/12.17/6 | D8 Osaka | Sporadic | Unvaccinated | Autochthonous |
| MVs/MonzaBrianza.ITA/12.17/2 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Como.ITA/14.17/2 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Como.ITA/14.17/4 | D8 Osaka | Outbreak | Vaccinated (2) | Autochthonous |
| MVs/MonzaBrianza.ITA/15.17/5 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Como.ITA/17.17/2 | D8 Osaka | Sporadic | Vaccinated (1) | Autochthonous |
| MVs/Varese.ITA/21.17 | D8 Osaka | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/24.17/2 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/24.17/4 | D8 London | Sporadic | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/24.17/7 | D8 Osaka | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/25.17/15 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/26.17/2 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/26.17/6 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/26.17 | D8 Osaka | Outbreak | Vaccinated (2) | Autochthonous |
| MVs/Milan.ITA/26.17/7 | D8 Osaka | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/26.17/8 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/26.17/16 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/27.17/5 | D8 London | Outbreak | Vaccinated (2) | Autochthonous |
| MVs/Milan.ITA/27.17/19 | D8 Osaka | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/23.17/15 | D8 Osaka | Sporadic | Vaccinated (2) | Autochthonous |
| MVs/Lodi.ITA/30.17/2 | D8 London | Outbreak | Vaccinated (1) | Autochthonous |
| MVs/Milan.ITA/31.17/3 | D8 Osaka | Outbreak | Vaccinated (2) | Autochthonous |
| D8-2018 | ||||
| MVs/Milan.ITA/10.18/4 | D8 Unnamed | Sporadic | Unvaccinated | Autochthonous |
| MVs/Brescia.ITA/11.18 | D8 Unnamed | Outbreak | Unvaccinated | Imported |
| MVs/Brescia.ITA/11.18/2 | D8 Unnamed | Sporadic | Unvaccinated | Autochthonous |
| MVs/Brescia.ITA/12.18/2 | D8 Unnamed | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/13.18 | D8 Unnamed | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/15.18 | D8 Unnamed | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/17.18 | D8 Unnamed | Sporadic | Vaccinated (2) | Imported |
| MVs/Milan.ITA/18.18 | D8 Unnamed | Sporadic | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/14.18/ | D8 Unnamed | Outbreak | Unvaccinated | Autochthonous |
| MVs/Milan.ITA/10.18/3 | D8 Unnamed | Sporadic | Vaccinated (1) | Autochthonous |
| D8-2019 | ||||
| Mvs/Milan.ITA/4.19 | D8 Gir-Somnath | Sporadic | Vaccinated (1) | Imported |
| Mvs/Milan.ITA/4.19/3 | D8 Gir-Somnath | Sporadic | Vaccinated (2) | Imported |
| Mvs/Milan.ITA/5.19 | D8 Gir-Somnath | Sporadic | Vaccinated (1) | Autochthonous |
| Mvs/Milan.ITA/16.19 | D8 Gir-Somnath | Sporadic | Vaccinated (1) | Autochthonous |
| Mvs/Milan.ITA/19.19/2 | D8 Gir-Somnath | Outbreak | Unvaccinated | Autochthonous |
| Mutation | Overall n (%) 1 | n in Specific Clades/Clusters | Immune Epitope/Recognizing Antibody 2 | PSP 3 |
|---|---|---|---|---|
| 400V | 127 (0.4) | Clade 400V: 126 | HNE | |
| 178T | 120 (8.9) | Clade 400V: 119 | ||
| 307I | 123 (9.1) | Clade 400V: 119 | ||
| 165T | 12 (0.9) | Cluster Niger: 6 | ||
| 222I | 7 (0.5) | Cluster Niger: 6 | ||
| 282D | 35 (2.6) | Cluster Niger: 6 | Yes | |
| 542F | 1 (0.1) | Cluster Niger: 1 | ||
| 601A | 3 (0.2) | Cluster Niger: 3 | ||
| 603E | 50 (3.7) | Cluster Niger: 3 Cluster D/L/SD: 1 | ||
| 605T | 3 (0.2) | Cluster Como: 3 | ||
| 617K | 5 (0.4) | Cluster Como: 3 | ||
| 429T | 1 (0.1) | Cluster Como: 1 | ||
| 71R | 2 (0.1) | Cluster Como: 1 | ||
| 30P | 3 (0.2) | Cluster Como: 1 | ||
| 50V | 8 (0.6) | Cluster Unnamed: 7 | ||
| 192A | 10 (0.7) | Cluster Unnamed: 7 | Ab I-44, BH26 | |
| 412L | 7 (0.5) | Cluster Unnamed: 7 | ||
| 355K | 3 (0.2) | Cluster Unnamed: 1 | ||
| 585I | 52 (3.9) | Cluster D/L/SD: 52 | ||
| 465S | 1 (0.1) | Cluster D/L/SD: 1 | ||
| 509E | 1 (0.1) | Cluster D/L/SD: 1 | ||
| 321F | 1 (0.1) | Cluster D/L/SD: 1 | ||
| 400A | 1004 (74.5) | Cluster D/L/SD: 1 | HNE | |
| 445Q | 1 (0.1) | Cluster D/L/SD: 1 | ||
| 176I | 2 (0.1) | Cluster D/L/SD: 1 |
| Mutation | Overall n (%) 1 | n in Specific Clades/Clusters | Immune Epitope/Recognizing Antibody 2 | PSP 3 |
|---|---|---|---|---|
| 400T | 213 (15.8) | Clade 400T: 209 | HNE | |
| 416D | 951 (70.6) | Clade 400T: 212 | SSE | |
| 560R | 145 (10.8) | Clade 400T: 123 | ||
| 174T | 849 (63.0) | Clade 400T: 115 | ||
| 559V | 131 (9.7) | Clade 400T: 116 | ||
| 276L | 270 (20.0) | Clade 400T: 111 | ||
| 428V | 111 (8.2) | Clade 400T: 110 | ||
| 560K | 1199 (89.0) | Clade 400T: 89 | ||
| 243E | 91 (6.8) | Clade 400T: 89 | ||
| 328R | 35 (2.6) | Cluster Unnamed: 10 | ||
| 247P | 155 (11.5) | Cluster Unnamed: 22 | NE | |
| 285G | 206 (15.3) | Cluster Gir-Somnath: 1 | ||
| 375Q | 1 (0.1) | Cluster Gir-Somnath: 1 Cluster London/Osaka: 1 | ||
| 575K | 68 (5.1) | Cluster Gir-Somnath: 1 | BH26 | Yes |
| 23T | 1 (0.1) | Cluster London/Osaka: 1 | ||
| 603R | 4 (0.3) | Cluster London/Osaka: 1 | ||
| 375R | 6 (0.5) | Cluster London/Osaka: 3 | ||
| 247L | 23 (1.7) | Cluster London/Osaka: 14 | NE | |
| 145K | 8 (0.6) | Cluster London/Osaka: 7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchi, S.; Canuti, M.; Ciceri, G.; Gori, M.; Colzani, D.; Dura, M.; Pennati, B.M.; Baggieri, M.; Magurano, F.; Tanzi, E.; et al. Molecular Epidemiology of B3 and D8 Measles Viruses through Hemagglutinin Phylogenetic History. Int. J. Mol. Sci. 2020, 21, 4435. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124435
Bianchi S, Canuti M, Ciceri G, Gori M, Colzani D, Dura M, Pennati BM, Baggieri M, Magurano F, Tanzi E, et al. Molecular Epidemiology of B3 and D8 Measles Viruses through Hemagglutinin Phylogenetic History. International Journal of Molecular Sciences. 2020; 21(12):4435. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124435
Chicago/Turabian StyleBianchi, Silvia, Marta Canuti, Giulia Ciceri, Maria Gori, Daniela Colzani, Marco Dura, Beatrice Marina Pennati, Melissa Baggieri, Fabio Magurano, Elisabetta Tanzi, and et al. 2020. "Molecular Epidemiology of B3 and D8 Measles Viruses through Hemagglutinin Phylogenetic History" International Journal of Molecular Sciences 21, no. 12: 4435. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124435