MAP/ERK Signaling in Developing Cognitive and Emotional Function and Its Effect on Pathological and Neurodegenerative Processes

 ,
, 

Abstract
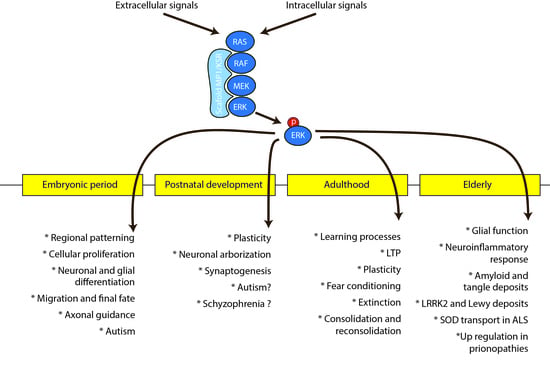
:
1. Introduction
2. Neural Signaling Mechanisms of the MAPK/ERK Pathway
3. The Role of ERK in Emotional Brain Development
3.1. The Role of MAPK/ERK Signaling in Embryonic Development
3.2. The Role of MAPK/ERK Signaling in Late Embryonic and Early Postnatal Development
3.3. The Role of MAPK/ERK Signaling in Adult Emotional and Memory Systems
3.3.1. MAPK/ERK Signaling in Spatial Memory
3.3.2. MAPK/ERK Signaling in Social Behavior
3.3.3. MAPK/ERK Signaling in Fear
4. MAPK/ERK Dysfunction in Neurodegenerative Diseases
4.1. Parkinson’s Disease
4.2. Alzheimer’s Disease
4.3. Amyotrophic Lateral Sclerosis and Huntington’s Disease
4.4. Prion Diseases
5. MAPK/ERK Signaling and Autism Spectrum Disorders
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5HT | Serotonin |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| asd | Asymmetric division |
| ASD | Autism spectrum disorders, |
| Aβ | Amyloid β |
| BDNF | Brain-derived nerve factor |
| CA1–3 | Cornu ammonis fields 1–3 |
| CaM | Calmodulin |
| CGE | Caudal ganglionic eminence |
| CP | Cortical plate |
| CS | Conditioned stimuli |
| CREB | c-AMP response element binding protein |
| DAG | Diacyl glycerol |
| DAPK | Death-associated protein kinase 1 |
| DG | Dentate gyrus |
| Dlx | Distal-less homeobox |
| ER | Endoplasmic reticulum |
| ERK | Extracellular regulated kinase |
| Ets | E26 transformation-specific transcription factor |
| FAK | Focal adhesion kinase |
| FGF8 | Fibroblast growth factor 8 |
| GABA | Gamma amino butyric acid |
| GF | Growth factor |
| GPCR | G-coupled protein receptors |
| Grb | Growth factor receptor bond protein |
| HD | Huntington’s disease |
| HIF1 | Hypoxia-inducible factor 1 transcription factor |
| HO-1 | Heme oxygenase-1 |
| Htt | Huntingtin |
| IEG | Immediate early gene |
| IP3 | Inositol 3-phosphate |
| IPC | Intermediate progenitor cell |
| iPD | Idiopatic Parkinson´s disease |
| IQGAP1 | IQ Motif Containing GTPase Activating Protein 1 |
| IZ | Intermediate zone |
| IκB | Inhibitor of κB |
| JNK | c-jun N-terminal kinase |
| JNKs/SAPK | Jun amino-terminal kinases/stress-activated kinases |
| KSR | Kinase suppressor of Ras1 |
| KSR | Kinase suppressor of Ras1 |
| LID | L-DOPA-induced dyskinesias |
| LRRK2 | Leucine-rich repeat kinase 2 |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MAPK | Microtubule-associated protein kinase |
| MeA | Medial Amygdala |
| MEK | MAP-ERK kinase |
| MGE | Medial ganglionic eminence |
| MKP3 | MAP kinase phosphatase 3 |
| MLCK | Myosin light-chain kinase |
| MP1 | MEK partner 1 |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MZ | Marginal zone |
| NFT | Neurofibrillary tangles |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NPC | Neural progenitor cell |
| OT | Oxytocin |
| OXTR | Oxytocin receptor |
| PD | Parkinson’s disease |
| PKC | Protein kinase C |
| PN1–60 | Postnatal day 1–60 |
| PP | Preplate |
| PRNP | Gene encoding prion protein |
| PrP | Prion protein |
| RAF | Rapidly Accelerated Fibrosarcoma |
| Ras | Rat sarcoma small GTPases protein |
| RAS | Rat sarcoma small GTPases gene |
| RLN3 | Relaxin 3 |
| RTK | Receptor tyrosine kinase |
| SBZ | Subventricular zone |
| sd | Symmetric division |
| Shc | Src homolog and collagen homolog |
| SOD1 | Cu/Zn superoxide dismutase |
| SOS | Son of sevenless homolog a guanine exchange factor |
| SP | Subplate |
| ST | Bed nucleus of the stria terminalis |
| STAT3 | Signal transducer and activator of transcription 3 |
| TM | Tangential migration from ganglionic eminences |
| TrkB | Tyrosine kinase receptor B |
| VZ | Ventricular zone |
| α7nAChR | α7 nicotinic acetylcholine receptor |
References
- Hausott, B.; Schlick, B.; Vallant, N.; Dorn, R.; Klimaschewski, L. Promotion of neurite outgrowth by fibroblast growth factor receptor 1 overexpression and lysosomal inhibition of receptor degradation in pheochromocytoma cells and adult sensory neurons. Neuroscience 2008, 153, 461–473. [Google Scholar] [CrossRef]
- Morrison, D.K.; Davis, R.J. Regulation of MAP Kinase Signaling Modules by Scaffold Proteins in Mammals. Annu. Rev. Cell Dev. Biol. 2003, 31, 11953–11967. [Google Scholar] [CrossRef]
- Selcher, J.C.; Nekrasova, T.; Paylor, R.; Landreth, G.E.; Sweatt, J.D. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn. Mem. 2001, 8, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, Y.; Endo, S.; Nakata, T.; Kobayashi, Y.; Yamada, K.; Ikeda, T.; Takeuchi, A.; Hiramoto, T.; Watanabe, Y.; Kazama, T. ERK2 Contributes to the Control of Social Behaviors in Mice. J. Neurosci. 2011, 31, 11953–11967. [Google Scholar] [CrossRef] [PubMed]
- Hatano, N.; Mori, Y.; Oh-hora, M.; Kosugi, A.; Fujikawa, T.; Nakai, N.; Niwa, H.; Miyazaki, J.I.; Hamaoka, T.; Ogata, M. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes to Cells 2003, 8, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Terunuma, M. Diversity of structure and function of GABAB receptors: A complexity of GABAB-mediated signaling. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2018, 94, 390–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pin, J.-P.; Bettler, B. Organization and functions of mGlu and GABAB receptor complexes. Nature 2016, 540, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Luján, R.; Shigemoto, R.; López-Bendito, G. Glutamate and GABA receptor signalling in the developing brain. Neuroscience 2005, 130, 567–580. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Raftogianni, A.; Roth, L.C.; García-González, D.; Bus, T.; Kühne, C.; Monyer, H.; Spergel, D.J.; Deussing, J.M.; Grinevich, V. Deciphering the Contributions of CRH Receptors in the Brain and Pituitary to Stress-Induced Inhibition of the Reproductive Axis. Front. Mol. Neurosci. 2018, 11, 305. [Google Scholar] [CrossRef]
- De Souza, E.B.; Battaglia, G. Corticotropin-Releasing Hormone (CRH) Receptors in Brain. In Mechanisms of Physical and Emotional Stress; Springer: Boston, MA, USA, 1988; pp. 123–136. [Google Scholar]
- Ferguson, J.N.; Aldag, J.M.; Insel, T.R.; Young, L.J. Oxytocin in the medial amygdala is essential for social recognition in the mouse. J. Neurosci. 2001, 21, 8278–8285. [Google Scholar] [CrossRef] [PubMed]
- Devost, D.; Wrzal, P.; Zingg, H. Oxytocin receptor signalling. In Advances in Vasopressin and Oxytocin-From Genes to Behaviour to Disease; Elsevier: Amsterdam, The Netherlands, 2008; Vol. 170, pp. 167–176. [Google Scholar]
- Albert-Gascó, H.; García-Avilés, Á.; Moustafa, S.; Sánchez-Sarasua, S.; Gundlach, A.L.; Olucha-Bordonau, F.E.; Sánchez-Pérez, A.M. Central relaxin-3 receptor (RXFP3) activation increases ERK phosphorylation in septal cholinergic neurons and impairs spatial working memory. Brain Struct. Funct. 2017, 222, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert-Gasco, H.; Sanchez-Sarasua, S.; Ma, S.; García-Díaz, C.; Gundlach, A.L.; Sanchez-Perez, A.M.; Olucha-Bordonau, F.E. Central relaxin-3 receptor (RXFP3) activation impairs social recognition and modulates ERK-phosphorylation in specific GABAergic amygdala neurons. Brain Struct. Funct. 2018, 224, 453–469. [Google Scholar] [CrossRef]
- de Ávila, C.; Chometton, S.; Lenglos, C.; Calvez, J.; Gundlach, A.L.; Timofeeva, E. Differential effects of relaxin-3 and a selective relaxin-3 receptor agonist on food and water intake and hypothalamic neuronal activity in rats. Behav. Brain Res. 2018, 336, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.M.; Walker, A.W.; Hosken, I.T.; Chua, B.E.; Zhang, C.; Haidar, M.; Gundlach, A.L. Relaxin-3/RXFP3 networks: An emerging target for the treatment of depression and other neuropsychiatric diseases? Front. Pharmacol. 2014, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Ganella, D.E.; Ryan, P.J.; Bathgate, R.A.D.; Gundlach, A.L. Increased feeding and body weight gain in rats after acute and chronic activation of RXFP3 by relaxin-3 and receptor-selective peptides: Functional and therapeutic implications. Behav. Pharmacol. 2012, 23, 516–525. [Google Scholar] [CrossRef]
- Haidar, M.; Guèvremont, G.; Zhang, C.; Bathgate, R.A.D.; Timofeeva, E.; Smith, C.M.; Gundlach, A.L. Relaxin-3 inputs target hippocampal interneurons and deletion of hilar relaxin-3 receptors in “floxed-RXFP3” mice impairs spatial memory. Hippocampus 2017, 27, 529–546. [Google Scholar] [CrossRef]
- Van der Westhuizen, E.T.; Werry, T.D.; Sexton, P.M.; Summers, R.J. The relaxin family peptide receptor 3 activates extracellular signal-regulated kinase 1/2 through a protein kinase C-dependent mechanism. Mol. Pharmacol. 2007, 71, 1618–1629. [Google Scholar] [CrossRef]
- Kania, A.; Gugula, A.; Grabowiecka, A.; de Ávila, C.; Blasiak, T.; Rajfur, Z.; Lewandowski, M.H.; Hess, G.; Timofeeva, E.; Gundlach, A.L.; et al. Inhibition of oxytocin and vasopressin neuron activity in rat hypothalamic paraventricular nucleus by relaxin-3-RXFP3 signalling. J. Physiol. 2017, 595, 3425–3447. [Google Scholar] [CrossRef] [Green Version]
- Rosas, M.; Porru, S.; Giugliano, V.; Antinori, S.; Scheggi, S.; Fadda, P.; Fratta, W.; Acquas, E.; Fattore, L. Sex-specific differences in cannabinoid-induced extracellular-signal-regulated kinase phosphorylation in the cingulate cortex, prefrontal cortex, and nucleus accumbens of Lister Hooded rats. Behav. Pharmacol. 2018, 29, 473–481. [Google Scholar] [CrossRef]
- Plum, L.; Schubert, M.; Brüning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.M. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J. Neurosci. 1988, 8, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Scholl, A.M.; Kuhn, E.N.; Kuhn, E.B.; Stadt, H.A.; Decker, J.R.; Pegram, K.; Hutson, M.R.; Kirby, M.L. FGF8 signaling is chemotactic for cardiac neural crest cells. Dev. Biol. 2011, 354, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Sanchez, C.; Franco, D.; Bonet, F.; Garcia-Lopez, V.; Aranega, A.; Garcia-Martinez, V. Reciprocal repression between Fgf8 and miR-133 regulates cardiac induction through Bmp2 signaling. Data Br. 2015, 5, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, H.; Omi, M.; Sato, T.; Nakamura, H. Pea3 determines the isthmus region at the downstream of Fgf8-Ras-ERK signaling pathway. Dev. Growth Differ. 2015, 57, 657–666. [Google Scholar] [CrossRef] [Green Version]
- Dee, A.; Li, K.; Heng, X.; Guo, Q.; Li, J.Y.H. Regulation of self-renewing neural progenitors by FGF/ERK signaling controls formation of the inferior colliculus. Development 2016, 143, 3661–3673. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Nakamura, H. The Fgf8 signal causes cerebellar differentiation by activating the Ras-ERK signaling pathway. Development 2004, 131, 4275–4285. [Google Scholar] [CrossRef] [Green Version]
- Suzuki-Hirano, A.; Harada, H.; Sato, T.; Nakamura, H. Activation of Ras-ERK pathway by Fgf8 and its downregulation by Sprouty2 for the isthmus organizing activity. Dev. Biol. 2010, 337, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.Y.; Snider, W.D. Different signaling pathways mediate regenerative versus developmental sensory axon growth. J. Neurosci. 2001, 21, RC164. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Boxer, L.M.; Latchman, D.S. Activation of the Bcl-2 promoter by nerve growth factor is mediated by the p42/p44 MAPK cascade. Nucleic Acids Res. 1999, 27, 2086–2090. [Google Scholar] [CrossRef] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, C.K.; Kelly, Á.M. Differential BDNF signaling in dentate gyrus and perirhinal cortex during consolidation of recognition memory in the rat. Hippocampus 2012, 22, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, T.W.; Shi, X.; Sun, W.L.; McGinty, J.F. The suppressive effect of an intra-prefrontal cortical infusion of BDNF on cocaine-seeking is TRK receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J. Neurosci. 2011, 31, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.S.; Kang, S.; Liu, W.T.; Li, M.; Liu, Y.; Yu, C.; Chen, J.; Chi, Z.Q.; He, L.; Liu, J.G. Extinction of aversive memories associated with morphine withdrawal requires ERK-mediated epigenetic regulation of brain-derived neurotrophic factor transcription in the rat ventromedial prefrontal cortex. J. Neurosci. 2012, 32, 13763–13775. [Google Scholar] [CrossRef] [Green Version]
- Plata-Salamán, C.R. Epidermal growth factor and the nervous system. Peptides 1991, 12, 653–663. [Google Scholar] [CrossRef]
- Mazzucchelli, C.; Vantaggiato, C.; Ciamei, A.; Fasano, S.; Pakhotin, P.; Krezel, W.; Welzl, H.; Wolfer, D.P.; Pagès, G.; Valverde, O.; et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 2002, 34, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Samuels, I.S.; Karlo, J.C.; Faruzzi, A.N.; Pickering, K.; Herrup, K.; Sweatt, J.D.; Saitta, S.C.; Landreth, G.E. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. 2008, 28, 6983–6995. [Google Scholar] [CrossRef]
- Satoh, Y.; Kobayashi, Y.; Takeuchi, A.; Pagès, G.; Pouysségur, J.; Kazama, T. Deletion of ERK1 and ERK2 in the CNS causes cortical abnormalities and neonatal lethality: Erk1 deficiency enhances the impairment of neurogenesis in Erk2-deficient mice. J. Neurosci. 2011, 31, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Roux, D.; Lenormand, P.; Dowd, S.; Keyse, S.; Pouysségur, J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999, 18, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetterkind, S.; Poythress, R.H.; Lin, Q.Q.; Morgan, K.G. Hierarchical scaffolding of an ERK1/2 activation pathway. Cell Commun. Signal. 2013, 11, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrick, C.; Fischer, A.; Srivastava, D.P.; Tronson, N.C.; Penzes, P.; Radulovic, J. N-Cadherin Regulates Cytoskeletally Associated IQGAP1/ERK Signaling and Memory Formation. Neuron 2007, 55, 786–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.L.; Ralay Ranaivo, H.; Patel, F.; Chrzaszcz, M.; Venkatesan, C.; Wainwright, M.S. Albumin causes increased myosin light chain kinase expression in astrocytes via p38 mitogen-activated protein kinase. J. Neurosci. Res. 2011, 89, 852–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, W.; Wu, Y.; Xian, W.; Song, L.; Hu, L.; Pan, S.; Liu, M.; Yao, S.; Pei, L.; Shang, Y. DAPK1-ERK signal mediates oxygen glucose deprivation reperfusion induced apoptosis in mouse N2a cells. J. Neurol. Sci. 2018, 387, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Shamloo, M.; Soriano, L.; Wieloch, T.; Nikolich, K.; Urfer, R.; Oksenberg, D. Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J. Biol. Chem. 2005, 280, 42290–42299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.; Sharma, K.; Frost, E.E.; Pillai, P.P. Role of PDGF-A-Activated ERK Signaling Mediated FAK-Paxillin Interaction in Oligodendrocyte Progenitor Cell Migration. J. Mol. Neurosci. 2019, 67, 564–573. [Google Scholar] [CrossRef]
- Costa, M.; Marchi, M.; Cardarelli, F.; Roy, A.; Beltram, F.; Maffei, L.; Ratto, G.M. Dynamic regulation of ERK2 nuclear translocation and mobility in living cells. J. Cell Sci. 2006, 119, 4952–4963. [Google Scholar] [CrossRef]
- Plotnikov, A.; Chuderland, D.; Karamansha, Y.; Livnah, O.; Seger, R. Nuclear ERK translocation is mediated by protein kinase CK2 and accelerated by autophosphorylation. Cell. Physiol. Biochem. 2019, 53, 366–387. [Google Scholar]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular, interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef]
- Ota, K.T.; Monsey, M.S.; Wu, M.S.; Young, G.J.; Schafe, G.E. Synaptic plasticity and NO-cGMP-PKG signaling coordinately regulate ERK-driven gene expression in the lateral amygdala and in the auditory thalamus following Pavlovian fear conditioning. Learn. Mem. 2010, 17, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Ito, K. Roles of Ets-1 and p70S6 kinase in chondrogenic and gliogenic specification of mouse mesencephalic neural crest cells. Mech. Dev. 2010, 127, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Hardingham, G.E.; Quinn, D.R.; Bading, H. CBP: A signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science 1998, 281, 1505–1509. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; Pinhero, V.; Vaghi, V.; Tongiorgi, E. Signaling pathways controlling activity-dependent local translation of BDNF and their localization in dendritic arbors. J. Cell Sci. 2016, 129, 2852–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Alshikho, M.J.; Herbert, M.R. Pathway network analyses for autism reveal multisystem involvement, major overlaps with other diseases and convergence upon MAPK and calcium signaling. PLoS ONE 2016, 11, e0153329. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta-Mol. Basis Dis. 2010, 4, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Vithayathil, J.; Pucilowska, J.; Friel, D.; Landreth, G.E. Chronic impairment of ERK signaling in glutamatergic neurons of the forebrain does not affect spatial memory retention and LTP in the same manner as acute blockade of the ERK pathway. Hippocampus 2017, 27, 1239–1249. [Google Scholar] [CrossRef]
- Crespo-Enriquez, I.; Partanen, J.; Martinez, S.; Echevarria, D. Fgf8-related secondary organizers exert different polarizing planar instructions along the mouse anterior neural tube. PLoS ONE 2012, 7, e39977. [Google Scholar] [CrossRef] [Green Version]
- Blak, A.A.; Naserke, T.; Weisenhorn, D.M.V.; Prakash, N.; Partanen, J.; Wurst, W. Expression of Fgf receptors 1, 2, and 3 in the developing mid- and hindbrain of the mouse. Dev. Dyn. 2005, 233, 1023–1030. [Google Scholar] [CrossRef]
- Nowak, M.; MacHate, A.; Yu, S.R.; Gupta, M.; Brand, M. Interpretation of the FGF8 morphogen gradient is regulated by endocytic trafficking. Nat. Cell Biol. 2011, 13, 153–158. [Google Scholar] [CrossRef]
- Böttcher, R.T.; Niehrs, C. Fibroblast growth factor signaling during early vertebrate development. Endocr. Rev. 2005, 26, 63–77. [Google Scholar] [CrossRef]
- Corson, L.B.; Yamanaka, Y.; Lai, K.-M.V.; Rossant, J. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development 2003, 130, 4527–4537. [Google Scholar] [CrossRef] [Green Version]
- Nonomura, K.; Yamaguchi, Y.; Hamachi, M.; Koike, M.; Uchiyama, Y.; Nakazato, K.; Mochizuki, A.; Sakaue-Sawano, A.; Miyawaki, A.; Yoshida, H.; et al. Local apoptosis modulates early mammalian brain development through the elimination of morphogen-producing cells. Dev. Cell 2013, 27, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botella-López, A.; Garcia-Lopez, R.; Pombero, A.; Martinez, S. Radial glia fibers translate Fgf8 morphogenetic signals to generate a thalamic nuclear complex protomap in the mantle layer. Brain Struct. Funct. 2019, 224, 661–679. [Google Scholar] [CrossRef] [Green Version]
- Marín, O. Cellular and molecular mechanisms controlling the migration of neocortical interneurons. Eur. J. Neurosci. 2013, 38, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.Y.; Sestan, N.; Anton, E.S. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 2012, 139, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucilowska, J.; Vithayathil, J.; Pagani, M.; Kelly, C.; Karlo, J.C.; Robol, C.; Morella, I.; Gozzi, A.; Brambilla, R.; Landreth, G.E. Pharmacological inhibition of ERK signaling rescues pathophysiology and behavioral phenotype associated with 16p11.2 chromosomal deletion in mice. J. Neurosci. 2018, 38, 6640–6652. [Google Scholar] [CrossRef] [PubMed]
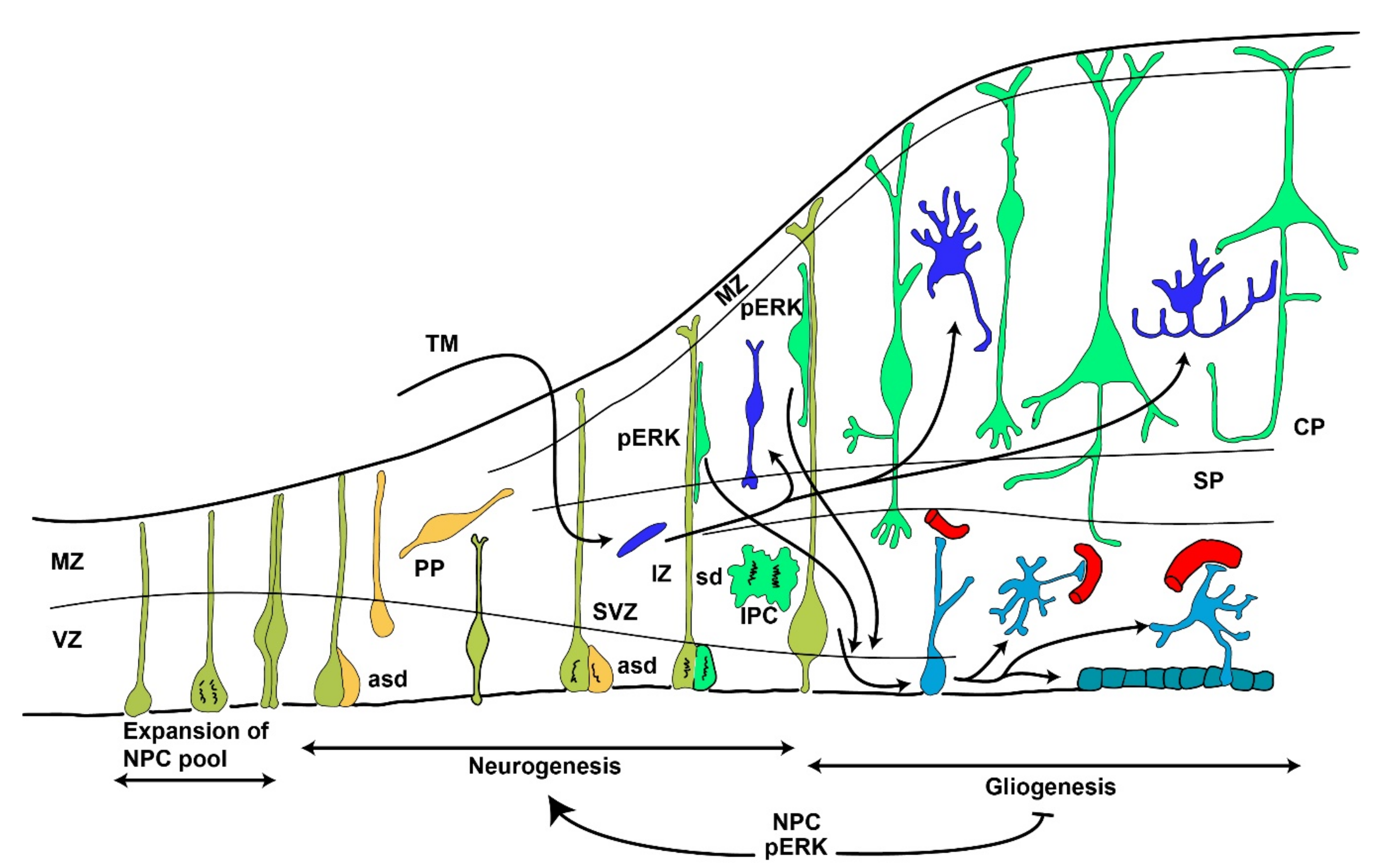
- Noctor, S.C.; Martinez-Cerdeño, V.; Ivic, L.; Kriegstein, A.R. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 2004, 7, 136–144. [Google Scholar] [CrossRef]
- Pontious, A.; Kowalczyk, T.; Englund, C.; Hevner, R.F. Role of intermediate progenitor cells in cerebral cortex development. Dev. Neurosci. 2007, 30, 24–32. [Google Scholar] [CrossRef]
- Miller, F.D.; Gauthier, A.S. Timing Is Everything: Making Neurons versus Glia in the Developing Cortex. Neuron 2007, 54, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Thomson, R.E.; Pellicano, F.; Iwata, T. Fibroblast growth factor receptor 3 kinase domain mutation increases cortical progenitor proliferation via mitogen-activated protein kinase activation. J. Neurochem. 2007, 100, 1565–1578. [Google Scholar] [CrossRef]
- Paquin, A.; Barnabé-Heider, F.; Kageyama, R.; Miller, F.D. CCAAT/enhancer-binding protein phosphorylation biases cortical precursors to generate neurons rather than astrocytes in vivo. J. Neurosci. 2005, 25, 10747–10758. [Google Scholar] [CrossRef]
- Gauthier, A.S.; Furstoss, O.; Araki, T.; Chan, R.; Neel, B.G.; Kaplan, D.R.R.; Miller, F.D. Control of CNS Cell-Fate Decisions by SHP-2 and Its Dysregulation in Noonan Syndrome. Neuron 2007, 54, 245–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, B.A.; Roberts, W.; Chung, B.; Weksberg, R.; Meyn, S.; Szatmari, P.; Joseph-George, A.M.; MacKay, S.; Whitten, K.; Noble, B.; et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J. Med. Genet. 2010, 47, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucilowska, J.; Vithayathil, J.; Tavares, E.J.; Kelly, C.; Colleen Karlo, J.; Landreth, G.E. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J. Neurosci. 2015, 35, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Stanco, A.; Pla, R.; Vogt, D.; Chen, Y.; Mandal, S.; Walker, J.; Hunt, R.F.; Lindtner, S.; Erdman, C.A.; Pieper, A.A.; et al. NPAS1 Represses the Generation of Specific Subtypes of Cortical Interneurons. Neuron 2014, 84, 940–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudy, B.; Fishell, G.; Lee, S.H.; Hjerling-Leffler, J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 2011, 71, 45–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.A.; Eisenstat, D.D.; Shi, L.; Rubenstein, J.L.R. Interneuron migration from basal forebrain to neocortex: Dependence on Dlx genes. Science 1997, 278, 474–476. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.P.; Lopes, M.W.; Rieger, D.K.; Barbosa, S.G.R.; Gonçalves, F.M.; Xikota, J.C.; Walz, R.; Leal, R.B. Differential Activation of Mitogen-Activated Protein Kinases, ERK 1/2, p38MAPK and JNK p54/p46 During Postnatal Development of Rat Hippocampus. Neurochem. Res. 2016, 41, 1160–1169. [Google Scholar] [CrossRef]
- Yoshii, A.; Sheng, M.H.; Constantine-Paton, M. Eye opening induces a rapid dendritic localization of PSD-95 in central visual neurons. Proc. Natl. Acad. Sci. USA 2003, 100, 1334–1339. [Google Scholar] [CrossRef] [Green Version]
- Yufune, S.; Satoh, Y.; Takamatsu, I.; Ohta, H.; Kobayashi, Y.; Takaenoki, Y.; Pagès, G.; Pouysségur, J.; Endo, S.; Kazama, T. Transient Blockade of ERK Phosphorylation in the Critical Period Causes Autistic Phenotypes as an Adult in Mice. Sci. Rep. 2015, 5, 10252. [Google Scholar] [CrossRef] [Green Version]
- Meyza, K.Z.; Defensor, E.B.; Jensen, A.L.; Corley, M.J.; Pearson, B.L.; Pobbe, R.L.H.; Bolivar, V.J.; Blanchard, D.C.; Blanchard, R.J. The BTBR T+tf/J mouse model for autism spectrum disorders-in search of biomarkers. Behav. Brain Res. 2013, 251, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Mogha, A.; Guariglia, S.R.; Debata, P.R.; Wen, G.Y.; Banerjee, P. Serotonin 1A receptor-mediated signaling through ERK and PKCα is essential for normal synaptogenesis in neonatal mouse hippocampus. Transl. Psychiatry 2012, 2, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, S.; Moore, A.N.; Adams, F.; Dash, P.K. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J. Neurosci. 1999, 19, 3535–3544. [Google Scholar] [CrossRef] [PubMed]
- Selcher, J.C.; Atkins, C.M.; Trzaskos, J.M.; Paylor, R.; Sweatt, J.D. A necessity for MAP kinase activation in mammalian spatial learning. Learn. Mem. 1999, 6, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, A.; Kelleher, R.J.; Tonegawa, S. A clustered plasticity model of long-term memory engrams. Nat. Rev. Neurosci. 2006, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Gur, R.; Tendler, A.; Wagner, S. Long-Term Social Recognition Memory Is Mediated by Oxytocin-Dependent Synaptic Plasticity in the Medial Amygdala. Biol. Psychiatry 2014, 76, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Jurek, B.; Slattery, D.A.; Maloumby, R.; Hillerer, K.; Koszinowski, S.; Neumann, I.D.; van den Burg, E.H. Differential Contribution of Hypothalamic MAPK Activity to Anxiety-Like Behaviour in Virgin and Lactating Rats. PLoS ONE 2012, 7, e37060. [Google Scholar] [CrossRef] [PubMed]
- Blume, A.; Bosch, O.J.; Miklos, S.; Torner, L.; Wales, L.; Waldherr, M.; Neumann, I.D. Oxytocin reduces anxiety via ERK1/2 activation: local effect within the rat hypothalamic paraventricular nucleus. Eur. J. Neurosci. 2008, 27, 1947–1956. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; Bauer, E.P.; LeDoux, J.E. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J. Neurosci. 1999, 19, 10512–10519. [Google Scholar] [CrossRef]
- Schafe, G.E.; Atkins, C.M.; Swank, M.W.; Bauer, E.P.; Sweatt, J.D.; Ledoux, J.E. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J. Neurosci. 2000, 20, 8177–8187. [Google Scholar] [CrossRef]
- Duvarci, S.; Nader, K.; LeDoux, J.E. Activation of extracellular signal-regulated kinase- mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur. J. Neurosci. 2005, 21, 283–289. [Google Scholar] [CrossRef]
- Cestari, V.; Costanzi, M.; Castellano, C.; Rossi-Arnaud, C. A role for ERK2 in reconsolidation of fear memories in mice. Neurobiol. Learn. Mem. 2006, 86, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Doyère, V.; Dȩbiec, J.; Monfils, M.H.; Schafe, G.E.; LeDoux, J.E. Synapse-specific reconsolidation of distinct fear memories in the lateral amygdala. Nat. Neurosci. 2007, 10, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-R.; Kim, J.-H.; Cho, E.; Kim, M.; Park, M. Dorsal and Ventral Hippocampus Differentiate in Functional Pathways and Differentially Associate with Neurological Disease-Related Genes during Postnatal Development. Front. Mol. Neurosci. 2017, 10, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanselow, M.S.; Dong, H.-W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Burwell, R.D.; Saddoris, M.P.; Bucci, D.J.; Wiig, K.A. Corticohippocampal contributions to spatial and contextual learning. J. Neurosci. 2004, 24, 3826–3836. [Google Scholar] [CrossRef] [Green Version]
- Vertes, R.P.; Fortin, W.J.; Crane, A.M. Projections of the median raphe nucleus in the rat. J. Comp. Neurol. 1999, 407, 555–582. [Google Scholar] [CrossRef]
- Steinbusch, H.W. Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience 1981, 6, 557–618. [Google Scholar] [CrossRef]
- Goto, M.; Swanson, L.W.; Canteras, N.S. Connections of the nucleus incertus. J. Comp. Neurol. 2001, 438, 86–122. [Google Scholar] [CrossRef]
- Olucha-Bordonau, F.E.; Teruel, V.; Barcia-Gonzalez, J.; Ruiz-Torner, A.; Valverde-Navarro, A.A.; Martinez-Soriano, F. Cytoarchitecture and efferent projections of the nucleus incertus of the rat. J. Comp. Neurol. 2003, 464, 62–97. [Google Scholar] [CrossRef]
- Bekiari, C.; Giannakopoulou, A.; Siskos, N.; Grivas, I.; Tsingotjidou, A.; Michaloudi, H.; Papadopoulos, G.C. Neurogenesis in the septal and temporal part of the adult rat dentate gyrus. Hippocampus 2015, 25, 511–523. [Google Scholar] [CrossRef]
- Kheirbek, M.A.; Hen, R. Dorsal vs ventral hippocampal neurogenesis: Implications for cognition and mood. Neuropsychopharmacology 2011, 36, 373–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, N.M.; Lee, B.; Banasr, M.; Elsayed, M.; Duman, R.S. Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK- and PI3K/Akt-dependent signaling. Neuropharmacology 2012, 63, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooney, M.; Messaoudi, E.; Maher, F.O.; Bramham, C.R.; Lynch, M.A. BDNF-induced LTP in dentate gyrus is impaired with age: Analysis of changes in cell signaling events. Neurobiol. Aging 2004, 25, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Richer, M.; David, M.; Villeneuve, L.R.; Trieu, P.; Ethier, N.; Pétrin, D.; Mamarbachi, A.M.; Hébert, T.E. GABA-B1 Receptors are Coupled to the ERK1/2 MAP Kinase Pathway in the Absence of GABA-B2 Subunits. J. Mol. Neurosci. 2009, 38, 67–79. [Google Scholar] [CrossRef]
- Tu, H.; Rondard, P.; Xu, C.; Bertaso, F.; Cao, F.; Zhang, X.; Pin, J.-P.; Liu, J. Dominant role of GABAB2 and Gβγ for GABAB receptor-mediated-ERK1/2/CREB pathway in cerebellar neurons. Cell. Signal. 2007, 19, 1996–2002. [Google Scholar] [CrossRef] [Green Version]
- Teles-Grilo Ruivo, L.M.; Mellor, J.R. Cholinergic modulation of hippocampal network function. Front. Synaptic Neurosci. 2013, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, J.D. The neuronal MAP kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001, 76, 1–10. [Google Scholar] [CrossRef]
- Richter, K.; Wolf, G.; Engelmann, M. Social recognition memory requires two stages of protein synthesis in mice. Learn. Mem. 2005, 12, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Trainor, B.C.; Crean, K.K.; Fry, W.H.D.; Sweeney, C. Activation of extracellular signal-regulated kinases in social behavior circuits during resident-intruder aggression tests. Neuroscience 2010, 165, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Iio, W.; Matsukawa, N.; Tsukahara, T.; Kohari, D.; Toyoda, A. Effects of chronic social defeat stress on MAP kinase cascade. Neurosci. Lett. 2011, 504, 281–284. [Google Scholar] [CrossRef]
- Ma, S.K.; Blasiak, A.; Olucha-Bordonau, F.E.; Verberne, A.J.M.; Gundlach, A.L. Heterogeneous responses of nucleus incertus neurons to corticotrophin-releasing factor and coherent activity with hippocampal theta rhythm in the rat. J. Physiol. 2013, 591, 3981–4001. [Google Scholar] [CrossRef] [PubMed]
- English, J.D.; David Sweatt, J. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J. Biol. Chem. 1996, 271, 24329–24332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafe, G.E.; Nadel, N.V.; Sullivan, G.M.; Harris, A.; LeDoux, J.E. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn. Mem. 1999, 6, 97–110. [Google Scholar] [PubMed]
- Schafe, G.E.; Swank, M.W.; Rodrigues, S.M.; Debiec, J.; Doyère, V. Phosphorylation of ERK/MAP kinase is required for long-term potentiation in anatomically restricted regions of the lateral amygdala in vivo. Learn. Mem. 2008, 15, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Barros, D.M.; Izquierdo, L.A.; Mello e Souza, T.; Ardenghi, P.G.; Pereira, P.; Medina, J.H.; Izquierdo, I. Molecular signalling pathways in the cerebral cortex are required for retrieval of one-trial avoidance learning in rats. Behav. Brain Res. 2000, 114, 183–192. [Google Scholar] [CrossRef]
- Szapiro, G.; Izquierdo, L.A.; Alonso, M.; Barros, D.; Paratcha, G.; Ardenghi, P.; Pereira, P.; Medina, J.H.; Izquierdo, I. Participation of hippocampal metabotropic glutamate receptors, protein kinase A and mitogen-activated protein kinases in memory retrieval. Neuroscience 2000, 99, 1–5. [Google Scholar] [CrossRef]
- Szapiro, G.; Vianna, M.R.M.; McGaugh, J.L.; Medina, J.H.; Izquierdo, I. The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus 2003, 13, 53–58. [Google Scholar] [CrossRef]
- Nader, K.; Schafe, G.E.; Le Doux, J.E. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 2000, 406, 722–726. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Schafe, G.E.; Ledoux, J.E. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron 2004, 44, 75–91. [Google Scholar] [CrossRef] [Green Version]
- Antoine, B.; Serge, L.; Jocelyne, C. Comparative dynamics of MAPK/ERK signalling components and immediate early genes in the hippocampus and amygdala following contextual fear conditioning and retrieval. Brain Struct. Funct. 2014, 219, 415–430. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat Rev Neurol 2010, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.M.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forno, L.S. The Neuropathology Of Parkinson’S Disease. In Progress in Parkinson Research; Springer: Boston, MA, USA, 1988; pp. 11–21. [Google Scholar]
- Karimi-Moghadam, A.; Charsouei, S.; Bell, B.; Jabalameli, M.R. Parkinson Disease from Mendelian Forms to Genetic Susceptibility: New Molecular Insights into the Neurodegeneration Process. Cell. Mol. Neurobiol. 2018, 38, 1153–1178. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139, 59–74. [Google Scholar] [CrossRef]
- Verma, M.; Steer, E.K.; Chu, C.T. ERKed by LRRK2: A cell biological perspective on hereditary and sporadic Parkinson’s disease. Biochim. Biophys. Acta - Mol. Basis Dis. 2014, 1842, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic Correction of a LRRK2 Mutation in Human iPSCs Links Parkinsonian Neurodegeneration to ERK-Dependent Changes in Gene Expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- White, L.R.; Toft, M.; Kvam, S.N.; Farrer, M.J.; Aasly, J.O. MAPK-pathway activity, Lrrk2 G2019S, and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 1288–1294. [Google Scholar] [CrossRef]
- Kulich, S.M.; Chu, C.T. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: Implications for Parkinson’s disease. J. Neurochem. 2001, 77, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.R.; Jayakumar, M.; Ahmed, M.S.; Zamaleeva, A.I.; Tao, J.; Li, E.H.; Job, J.K.; Pittenger, C.; Ohtsu, H.; Rajadas, J. Pharmacological antagonism of histamine H2R ameliorated L-DOPA–induced dyskinesia via normalization of GRK3 and by suppressing FosB and ERK in PD. Neurobiol. Aging 2019, 81, 177–189. [Google Scholar] [CrossRef]
- Zhu, J.H.; Gusdon, A.M.; Cimen, H.; Van Houten, B.; Koc, E.; Chu, C.T. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: Dual roles for ERK1/2. Cell Death Dis. 2012, 3, e312. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, N.; Wu, C.; Lu, Y.; Gao, G.; Duan, C.; Yang, H.; Lu, L. Galectin-1 attenuates neurodegeneration in Parkinson’s disease model by modulating microglial MAPK/IκB/NFκB axis through its Carbohydrate-recognition domain. Brain. Behav. Immun. 2019, 83, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-H.; Kulich, S.M.; Oury, T.D.; Chu, C.T. Cytoplasmic Aggregates of Phosphorylated Extracellular Signal-Regulated Protein Kinases in Lewy Body Diseases. Am. J. Pathol. 2002, 161, 2087–2098. [Google Scholar] [CrossRef] [Green Version]
- Dineley, K.T.; Westerman, M.; Bui, D.; Bell, K.; Ashe, K.H.; Sweatt, J.D. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. 2001, 21, 4125–4133. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.A.; O’Riordan, K.J.; Sweatt, J.D.; Dineley, K.T. MAPK recruitment by beta-amyloid in organotypic hippocampal slice cultures depends on physical state and exposure time. J. Neurochem. 2004, 91, 349–361. [Google Scholar] [CrossRef]
- Russo, C.; Dolcini, V.; Salis, S.; Venezia, V.; Zambrano, N.; Russo, T.; Schettini, G. Signal Transduction through Tyrosine-phosphorylated C-terminal Fragments of Amyloid Precursor Protein via an Enhanced Interaction with Shc/Grb2 Adaptor Proteins in Reactive Astrocytes of Alzheimer’s Disease Brain. J. Biol. Chem. 2002, 277, 35282–35288. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.N.; Ma, K.G.; Chen, X.L.; Shi, L.L.; Bu, G.; Hu, X.D.; Han, H.; Liu, Y.; Qian, Y.H. Mitogen-activated protein kinase signaling pathways are involved in regulating α7 nicotinic acetylcholine receptor-mediated amyloid-β uptake in SH-SY5Y cells. Neuroscience 2014, 278, 276–290. [Google Scholar] [CrossRef]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-β1-42-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.E.; Thomas, R.S.; Freeman, T.J.; Hvoslef-Eide, M.; Good, M.A.; Kidd, E.J. Selective reduction of APP-BACE1 activity improves memory via NMDA-NR2B receptor-mediated mechanisms in aged PDAPP mice. Neurobiol. Aging 2019, 75, 136–149. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Ivanov, A.; Tyzio, R.; Pellegrino, C.; Ben-Ari, Y.; Clapham, D.E.; Medina, I. The NMDA Receptor Is Coupled to the ERK Pathway by a Direct Interaction between NR2B and RasGRF1. Neuron 2003, 40, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.E.; Miners, J.S.; Piva, G.; Willis, C.L.; Heard, D.M.; Kidd, E.J.; Good, M.A.; Kehoe, P.G. ACE2 activation protects against cognitive decline and reduces amyloid pathology in the Tg2576 mouse model of Alzheimer’s disease. Acta Neuropathol. 2020, 139, 485–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlson, E.; Jeong, G.-B.; Ross, J.L.; Dixit, R.; Wallace, K.E.; Kalb, R.G.; Holzbaur, E.L.F. A Switch in Retrograde Signaling from Survival to Stress in Rapid-Onset Neurodegeneration. J. Neurosci. 2009, 29, 9903–9917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodai, L.; Marsh, J.L. A novel target for Huntington’s disease: ERK at the crossroads of signaling. The ERK signaling pathway is implicated in Huntington’s disease and its upregulation ameliorates pathology. Bioessays 2012, 34, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Roze, E.; Betuing, S.; Deyts, C.; Marcon, E.; Brami-Cherrier, K.; Pagès, C.; Humbert, S.; Mérienne, K.; Caboche, J. Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 2008, 22, 1083–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppington, K.M.; Brown, D.R. Resistance of cell lines to prion toxicity aided by phospho-ERK expression. J. Neurochem. 2008, 105, 842–852. [Google Scholar] [CrossRef]
- LaCasse, R.; Striebel, J.; Favara, C.; Kercher, L.; Chesebro, B. Role of Erk1/2 activation in prion disease pathogenesis: Absence of CCR1 leads to increased Erk1/2 activation and accelerated disease progression. J. Neuroimmunol. 2008, 196, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Seese, R.R.; Maske, A.R.; Lynch, G.; Gall, C.M. Long-term memory deficits are associated with elevated synaptic ERK1/2 activation and reversed by mGluR5 antagonism in an animal model of autism. Neuropsychopharmacology 2014, 39, 1664–1673. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, M.; Timmerman, C.K.; Schwartz, J.L.; Pham, D.L.; Meffert, M.K. Characterizing autism spectrum disorders by key biochemical pathways. Front. Neurosci. 2015, 9, 313. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef] [Green Version]
- Plowey, E.D.; Cherra 3rd, S.J.; Liu, Y.J.; Chu, C.T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008, 105, 1048–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Santos, C.; Ferrer, I.; Reiriz, J.; Viñals, F.; Barrachina, M.; Ambrosio, S. MPP+ increases α-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblastoma SH-SY5Y cells. Brain Res. 2002, 935, 32–39. [Google Scholar] [CrossRef]
- Sarkar, S.; Lu, E.; Raymick, J.; Hanig, J.; Gu, Q. ERK/MAP Kinase Activation is Evident in Activated Microglia of the Striatum and Substantia Nigra in an Acute and Chronically-Induced Mouse Model of Parkinson’s Disease. Curr. Neurovasc. Res. 2019, 15, 336–344. [Google Scholar]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Pozueta, J.; Lefort, R.; Shelanski, M.L. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 2013, 251, 51–65. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E. Genetically altered transgenic models of Alzheimer’s disease. J. Neural Transm. Suppl. 2000, 59, 175–183. [Google Scholar]
- Decker, M.W. The effects of aging on hippocampal and cortical projections of the forebrain cholinergic system. Brain Res. 1987, 434, 423–438. [Google Scholar]
- Bruel-Jungerman, E.; Lucassen, P.J.; Francis, F. Cholinergic influences on cortical development and adult neurogenesis. Behav. Brain Res. 2011, 221, 379–388. [Google Scholar] [CrossRef]
- VanGuilder, H.D.; Yan, H.; Farley, J.A.; Sonntag, W.E.; Freeman, W.M. Aging alters the expression of neurotransmission-regulating proteins in the hippocampal synaptoproteome. J. Neurochem. 2010, 113, 1577–1588. [Google Scholar]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Castellani, R.J.; Takeda, A.; Nunomura, A.; Atwood, C.S.; Perry, G.; Smith, M.A. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: The ‘two hit’ hypothesis. Mech. Ageing Dev. 2001, 123, 39–46. [Google Scholar] [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieślik, M.; Czapski, G.A.; Strosznajder, J.B. The Molecular Mechanism of Amyloid β42 Peptide Toxicity: The Role of Sphingosine Kinase-1 and Mitochondrial Sirtuins. PLoS ONE 2015, 10, e0137193. [Google Scholar] [CrossRef]
- Wang, H.-Q.; Xu, Y.-X.; Zhu, C.-Q. Upregulation of Heme Oxygenase-1 by Acteoside Through ERK and PI3 K/Akt Pathway Confer Neuroprotection Against Beta-Amyloid-Induced Neurotoxicity. Neurotox. Res. 2012, 21, 368–378. [Google Scholar] [CrossRef]
- Rowland, L.P. How amyotrophic lateral sclerosis got its name: The clinical-pathologic genius of Jean-Martin Charcot. Arch. Neurol. 2001, 58, 512–515. [Google Scholar] [CrossRef]
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef]
- Brooks, B.R. El Escorial World Federation of Neurology Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical Limits of Amyotrophic Lateral Sclerosis” Workshop Contributors. J. Neurol. Sci. 1994, 124, S96–S107. [Google Scholar]
- De Carvalho, M.; Swash, M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph. Lateral Scler. 2009, 10, 53–57. [Google Scholar] [CrossRef]
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, K.L.; Kalmar, B.; Rhymes, E.R.; Fellows, A.D.; Ahmed, M.; Whiting, P.; Davies, C.H.; Greensmith, L.; Schiavo, G. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 2018, 9, 596. [Google Scholar] [CrossRef] [PubMed]
- Sawa, A.; Nagata, E.; Sutcliffe, S.; Dulloor, P.; Cascio, M.B.; Ozeki, Y.; Roy, S.; Ross, C.A.; Snyder, S.H. Huntingtin is cleaved by caspases in the cytoplasm and translocated to the nucleus via perinuclear sites in Huntington’s disease patient lymphoblasts. Neurobiol. Dis. 2005, 20, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Lakkaraju, A.K.K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 2015, 26, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Invest. 2017, 127, 3230–3239. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Min, J.-S.; Kim, B.; Chae, U.-B.; Yun, J.W.; Choi, M.-S.; Kong, I.-K.; Chang, K.-T.; Lee, D.-S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef]
- Lee, H.-P.; Jun, Y.-C.; Choi, J.-K.; Kim, J.-I.; Carp, R.I.; Kim, Y.-S. Activation of mitogen-activated protein kinases in hamster brains infected with 263K scrapie agent. J. Neurochem. 2005, 95, 584–593. [Google Scholar] [CrossRef]
- Kanner, L. Autistic disturbances of affective contact. Nerv Child 1943, 2, 217–250. [Google Scholar]
- Asperger, H. Die „Autistischen Psychopathen” im Kindesalter. Arch. Psychiatr. Nervenkr. 1944, 117, 76–136. [Google Scholar] [CrossRef]
- Gillberg, C. Outcome in Autism and Autistic-like Conditions. J. Am. Acad. Child Adolesc. Psychiatry 1991, 30, 375–382. [Google Scholar] [CrossRef]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. MAPK signalling in cellular metabolism: Stress or wellness? EMBO Rep. 2010, 11, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Bilder, D.A.; Esplin, M.S.; Coon, H.; Burghardt, P.; Clark, E.A.S.; Fraser, A.; Smith, K.R.; Worsham, W.; Chappelle, K.; Rayner, T.; et al. Early Second Trimester Maternal Serum Steroid-Related Biomarkers Associated with Autism Spectrum Disorder. J. Autism Dev. Disord. 2019, 49, 4572–4583. [Google Scholar] [CrossRef] [PubMed]
- Kohane, I.S.; McMurry, A.; Weber, G.; MacFadden, D.; Rappaport, L.; Kunkel, L.; Bickel, J.; Wattanasin, N.; Spence, S.; Murphy, S.; et al. The co-morbidity burden of children and young adults with autism spectrum disorders. PLoS ONE 2012, 7, e33224. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakar, A.L.; Dölen, G.; Bear, M.F. The Pathophysiology of Fragile X (and What It Teaches Us about Synapses). Annu. Rev. Neurosci. 2012, 35, 417–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of Tsc1 In Vivo Impairs Hippocampal mGluR-LTD and Increases Excitatory Synaptic Function. J. Neurosci. 2011, 31, 8862–8869. [Google Scholar] [CrossRef] [Green Version]
- Zoghbi, H.Y.; Bear, M.F. Synaptic Dysfunction in Neurodevelopmental Disorders Associated with Autism and Intellectual Disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; DeFranco, D.B. Opposing Roles for ERK1/2 in Neuronal Oxidative Toxicity. J. Biol. Chem. 2006, 281, 16436–16442. [Google Scholar] [CrossRef] [Green Version]
- Colucci-D’Amato, L.; Perrone-Capano, C.; Di Porzio, U. Chronic activation of ERK and neurodegenerative diseases. BioEssays 2003, 25, 1085–1095. [Google Scholar] [CrossRef]




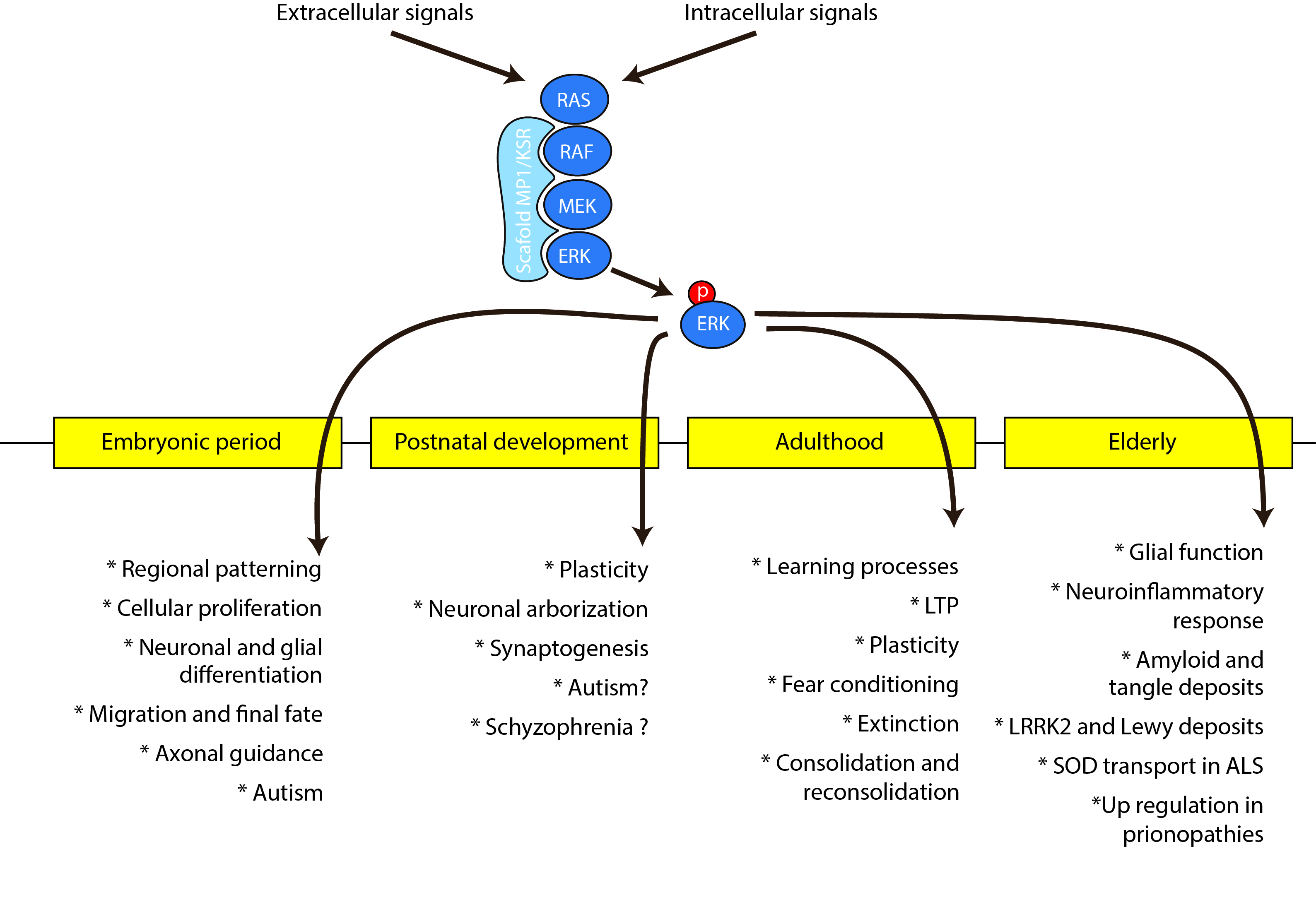{kind=link}
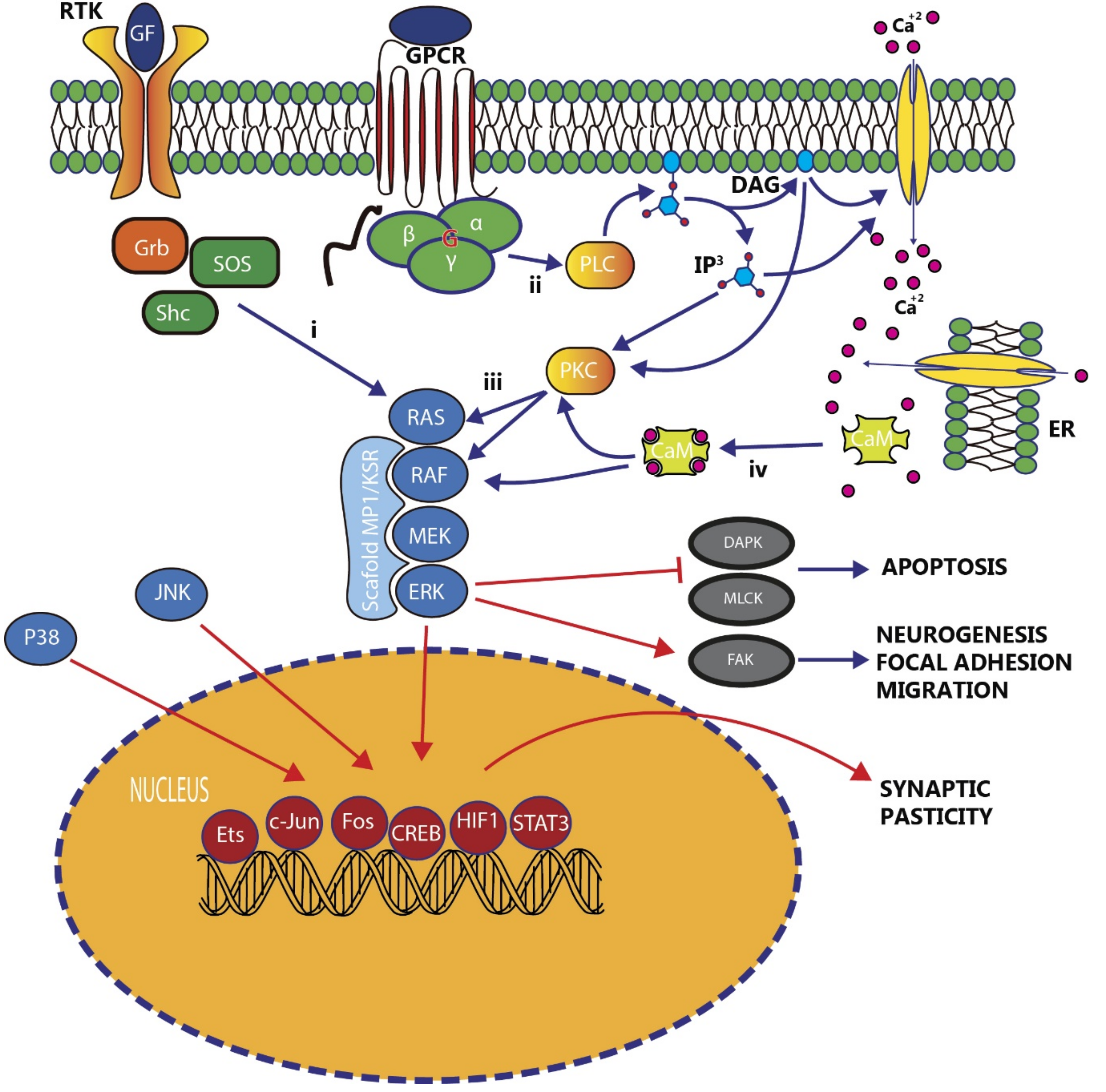{kind=link}
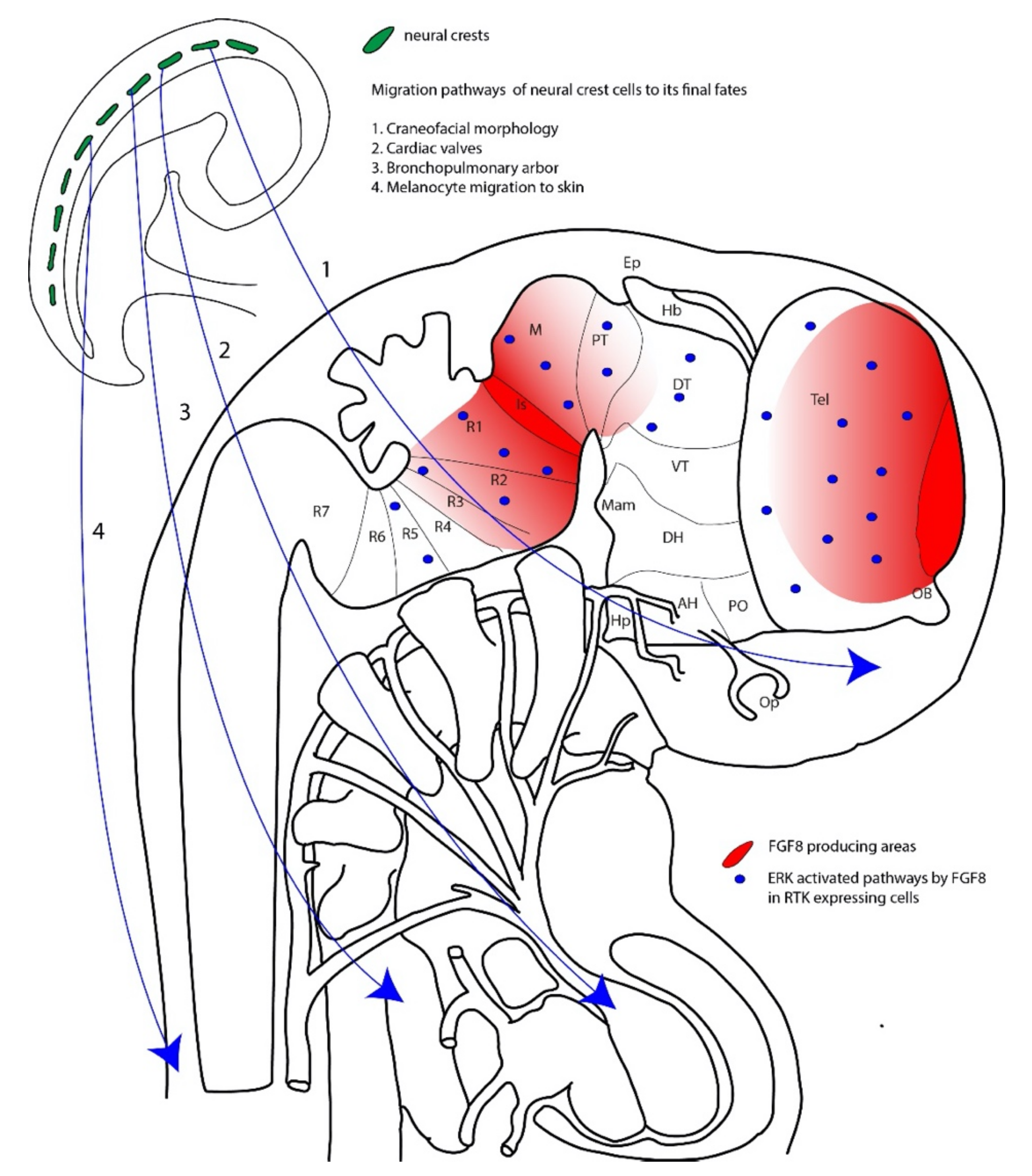{kind=link}
{kind=link}
| Ligand | Receptor/Type | Process | Reference |
| GABA | GABAB-R/GPCR | Long-term inhibition of synaptic transmission | [6,7,8] |
| Association with trafficking | |||
| Developing brain | |||
| Glu | m-Glu/GPCR | Fine control over glutamate activity | [8,9] |
| Synaptic plasticity | |||
| Developing brain | |||
| CRH | CRH-R/GPCR | Response to stress | [10,11] |
| OXT | OXTR/GPCR | Promotes social recognition memory | [12,13] |
| Anxiolytic | |||
| RLN3 | RXFP3/GPCR | Inhibits social recognition memory, and spatial short-term memory | [14,15,16,17,18,19,20,21] |
| Stress response | |||
| Anxiolytic | |||
| Food intake | |||
| Cannabinoid | CB1-GPCR | Addiction | [22] |
| INS/IGF1 | INSR-IGF1R /RTK | Mitogenesis, glucose uptake, and metabolism | [23] |
| Neuronal survival, neuronal and glia development, and synaptic plasticity | |||
| Reproductive endocrinology | |||
| FGF8 | FGFR1-3 TrkA-B/RTK | Axonal growth and regeneration | [24,25,26,27,28,29,30] |
| Neural and neural crest differentiation, migration, and final fate | |||
| NGF | Trk-A/RTK | Neuronal survival | [31,32] |
| BDNF | Trk-B/RTK | Neuronal and glia developmentand synaptic plasticity | [33] |
| Memory | [34] | ||
| Addiction | [35] | ||
| Addiction-extinction | [36] | ||
| EGF | EGFR/RTK | Development and maintenance | [37] |
| Appetite suppression and neuroendocrine alterations | |||
| Acute and chronic pathological processes |
| Behavior | ERK (+) Activation (−) Inhibition | Reference |
|---|---|---|
| Spatial memory | (+) LTP generation, spatial learning (−) impairs effects | [85,86] |
| (−) PD098059 infusion inhibits long-term memory formation | [87] | |
| Social recognition memory | (+) promotes social recognition memory | [88] |
| Elevated plus maze | (+) activation at PVN causes an anxiolytic effect (−) eliminates the anxiolytic effect | [89,90] |
| Fear conditioning | (−) disrupts LTP at the amygdalo-thalamic pathway | [91,92] |
| Consolidation of fear memory | (−) infusion of U0126 prior to the retrieval of memory impair reconsolidation | [93,94,95] |
| Parkinson’s disease | References | |
| LRRK2 | p-ERK present in Lewy bodies | [130,131,132] |
| 6-OHDA model | 6-OHDA elicits sustained ERK phosphorylation related to LID | [133,134] |
| MPTP model | ERK phosphorylation is implicated in neuroinflammation | [135,136] |
| PD patients | ERK phosphorylated deposits close to Lewy bodies | [137] |
| Alzheimer’s disease | ||
| AD patients | Aβ dysregulates hippocampal ERK | [138,139,140] |
| SH-SY5Y cells | α7nACh induce tau phosphorylation and neurofibrillary tangle formation after binding to soluble Aβ | [141] |
| PC12 cells | HO1 protects against Aβ-induced oxidative stress | [142] |
| Transgenic mice | ERK-signaling induces Aβ-associated behavioral deficits | [143,144,145,146] |
| ALS and HD | ||
| SOD1 transgenic mice | ERK is down regulated, which induces a dysregulation in axonal transport | [147] |
| Mutant Htt model | ERK deficiency triggers striatal degeneration and increases glutamate susceptibility | [148,149] |
| Prion diseases | ||
| Prion infected mice | ERK is neuroprotective following prion infection | [150,151] |
| Autism | ||
| BTBR mice | p-ERK immunolabeling negatively correlates with cognitive function in the prefrontal cortex | [152] |
| ERK 1 /2 KO mice | Critical window between 6th and 14th postnatal day when an ERK blockade promotes autistic behaviors | [82,153] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albert-Gascó, H.; Ros-Bernal, F.; Castillo-Gómez, E.; Olucha-Bordonau, F.E. MAP/ERK Signaling in Developing Cognitive and Emotional Function and Its Effect on Pathological and Neurodegenerative Processes. Int. J. Mol. Sci. 2020, 21, 4471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124471
Albert-Gascó H, Ros-Bernal F, Castillo-Gómez E, Olucha-Bordonau FE. MAP/ERK Signaling in Developing Cognitive and Emotional Function and Its Effect on Pathological and Neurodegenerative Processes. International Journal of Molecular Sciences. 2020; 21(12):4471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124471
Chicago/Turabian StyleAlbert-Gascó, Héctor, Francisco Ros-Bernal, Esther Castillo-Gómez, and Francisco E. Olucha-Bordonau. 2020. "MAP/ERK Signaling in Developing Cognitive and Emotional Function and Its Effect on Pathological and Neurodegenerative Processes" International Journal of Molecular Sciences 21, no. 12: 4471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124471