Low-Dose Ionizing Radiation Modulates Microglia Phenotypes in the Models of Alzheimer’s Disease

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
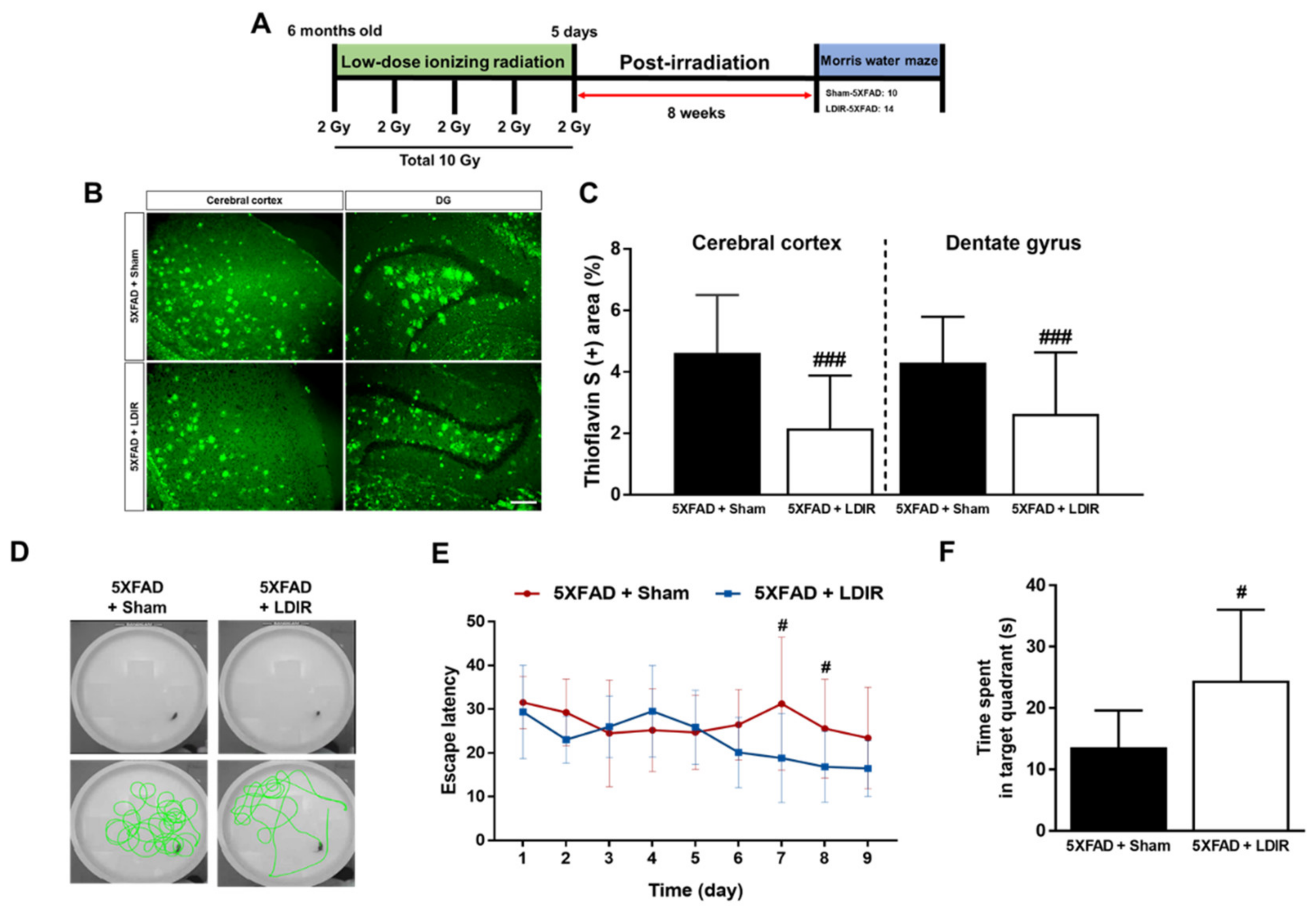
2.1. LDIR Inhibits Aβ Deposition and Improved Cognitive Deficits in 5XFAD Mice
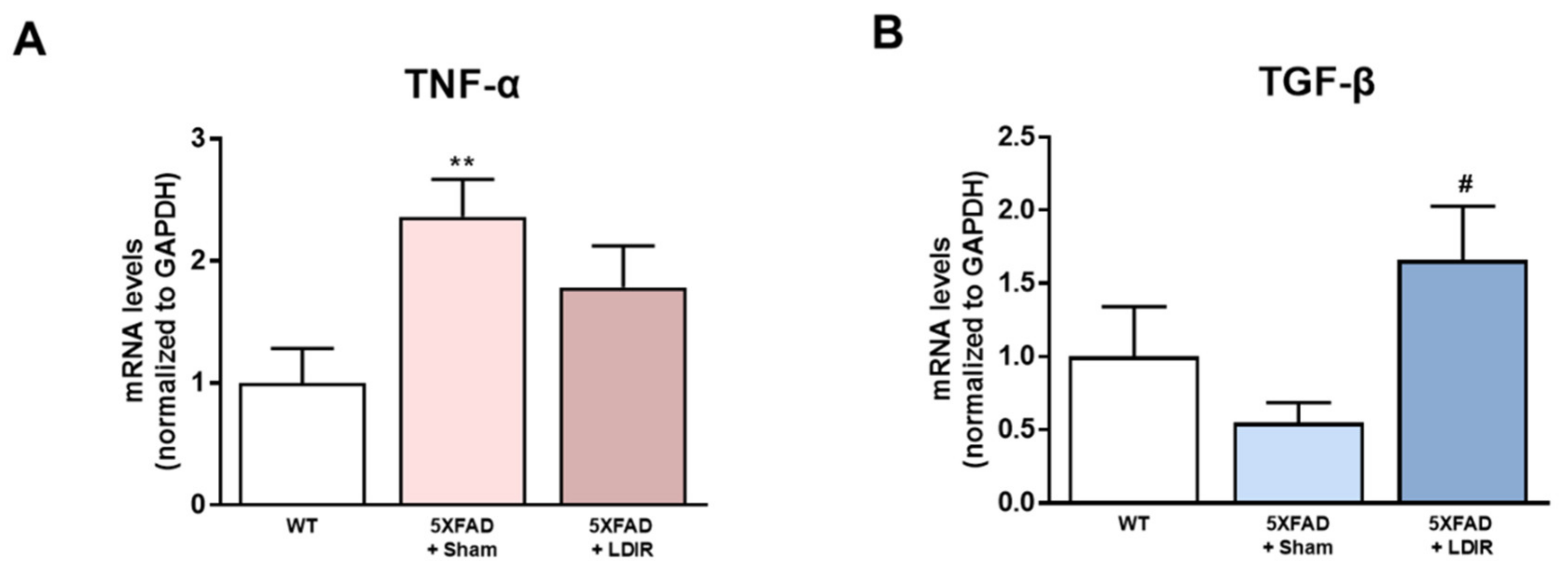
2.2. LDIR Regulates Aβ-Induced Production of Inflammatory Cytokines in the 5XFAD Mice
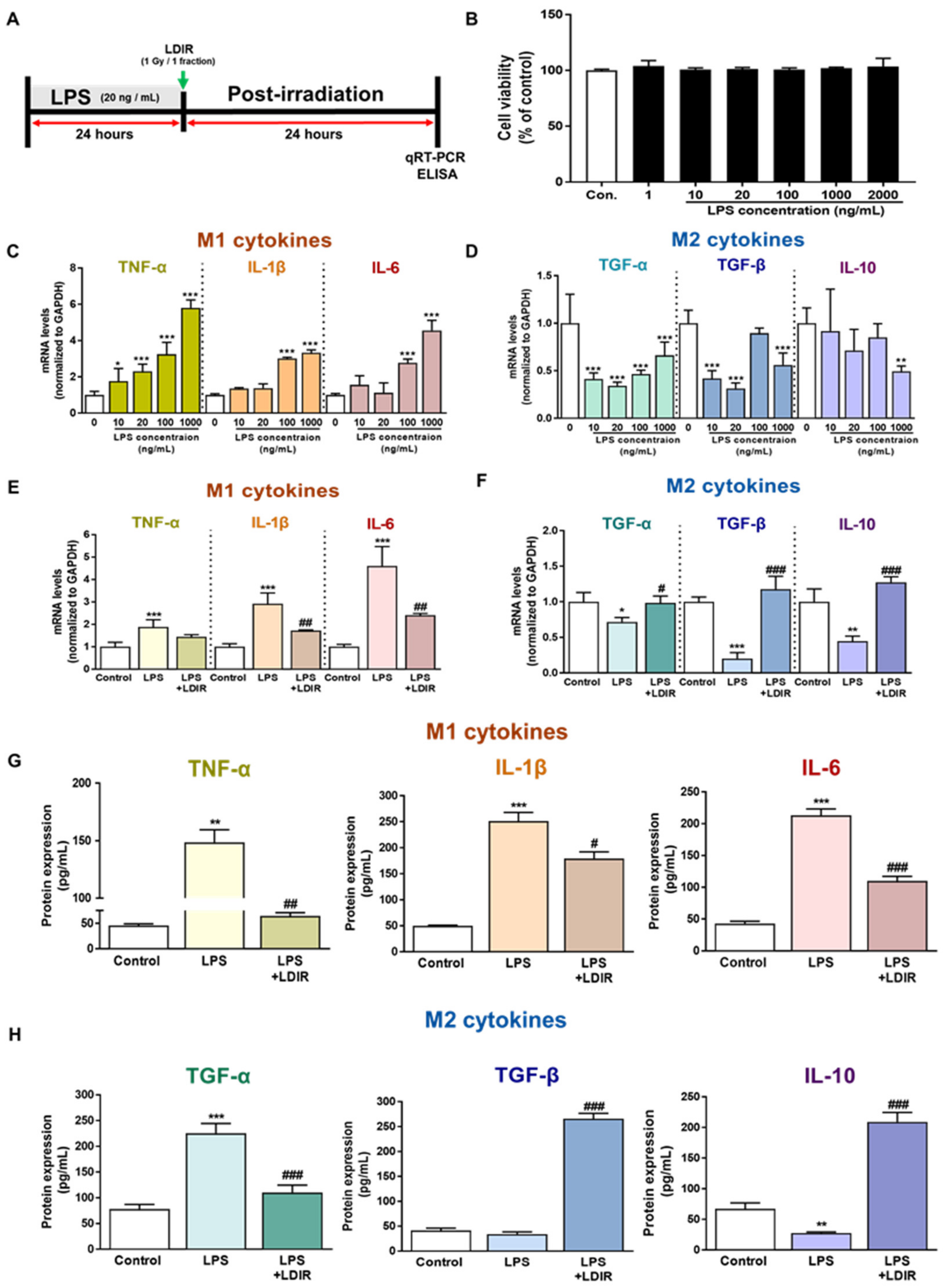
2.3. LDIR Modulates the Levels of M1/M2 Cytokines in LPS-Treated BV-2 Cells
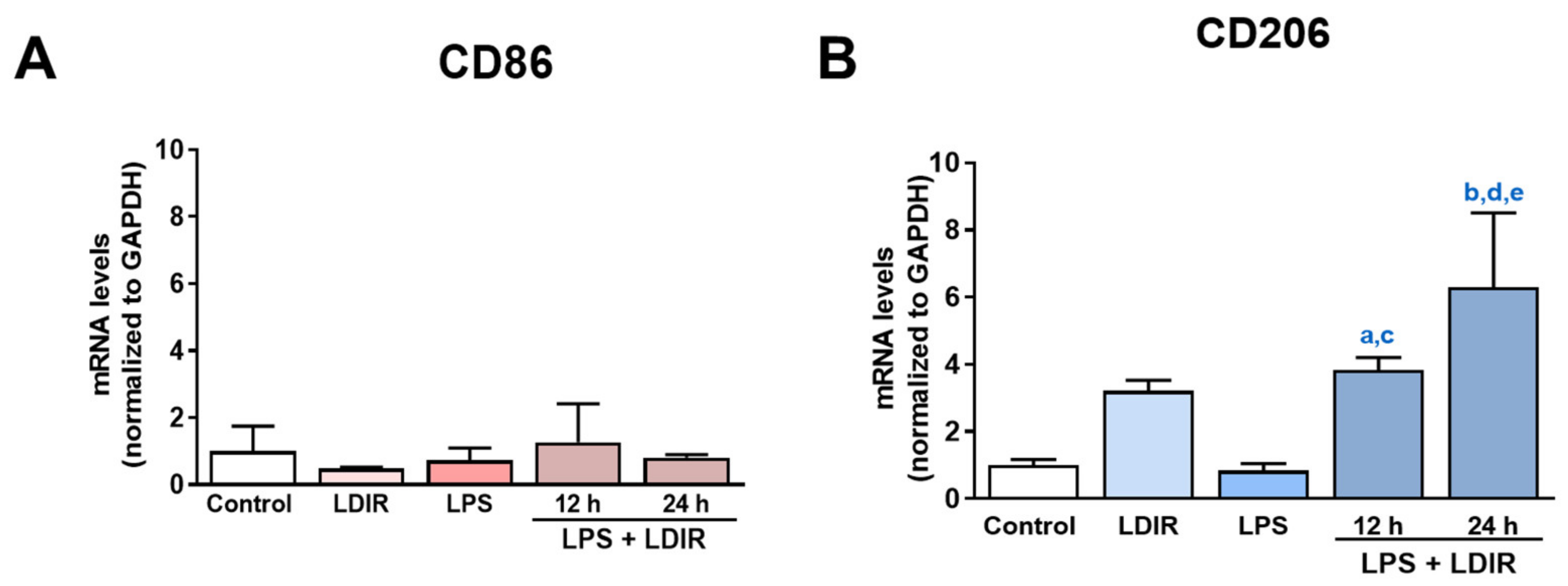
2.4. LDIR Affects Changes of M1/M2 Phenotypes of LPS-Treated BV-2 Cells
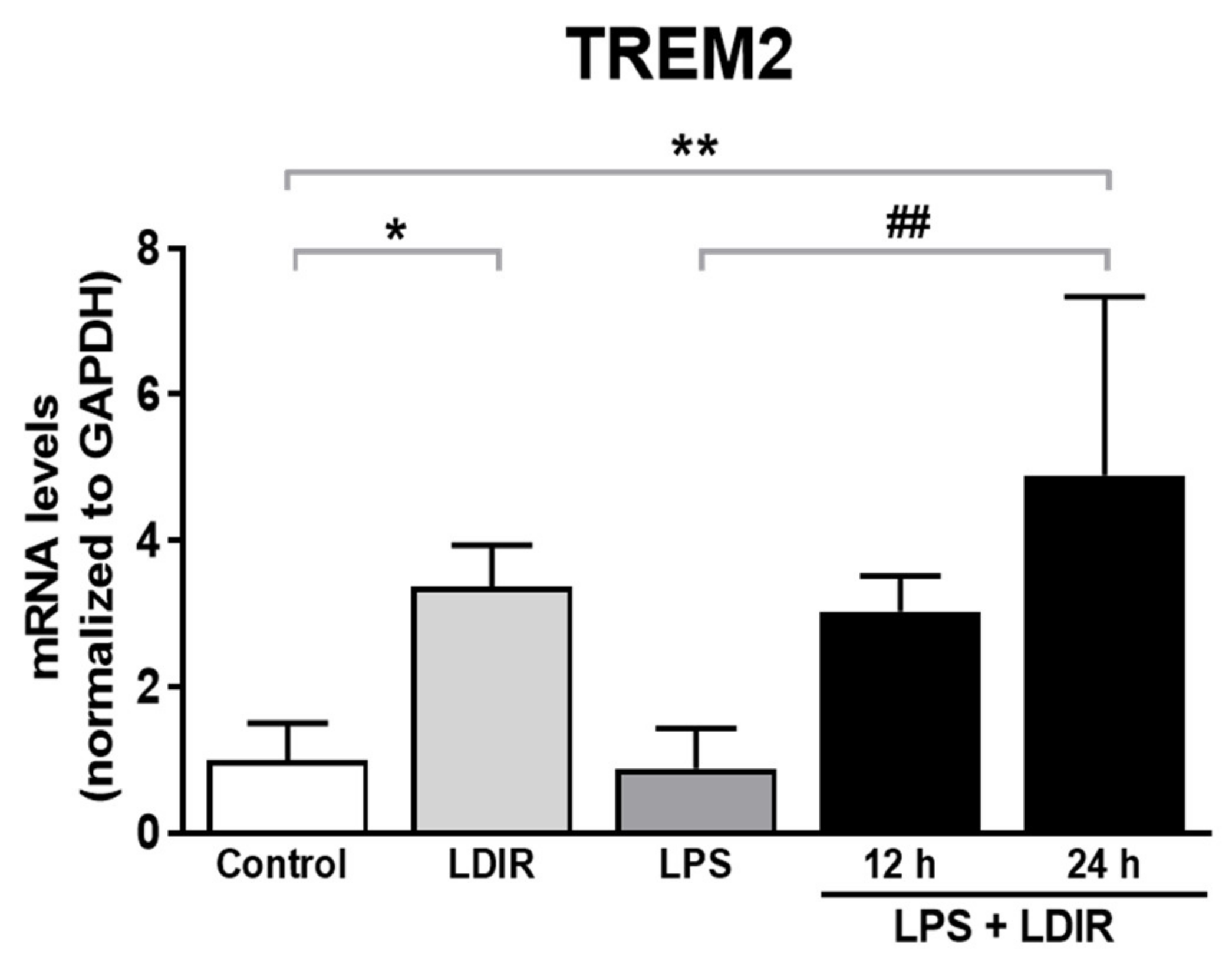
2.5. LDIR Induces the Up-Regulation of TREM2 in LPS-Treated BV-2 Cells
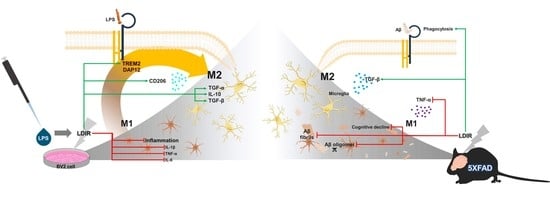
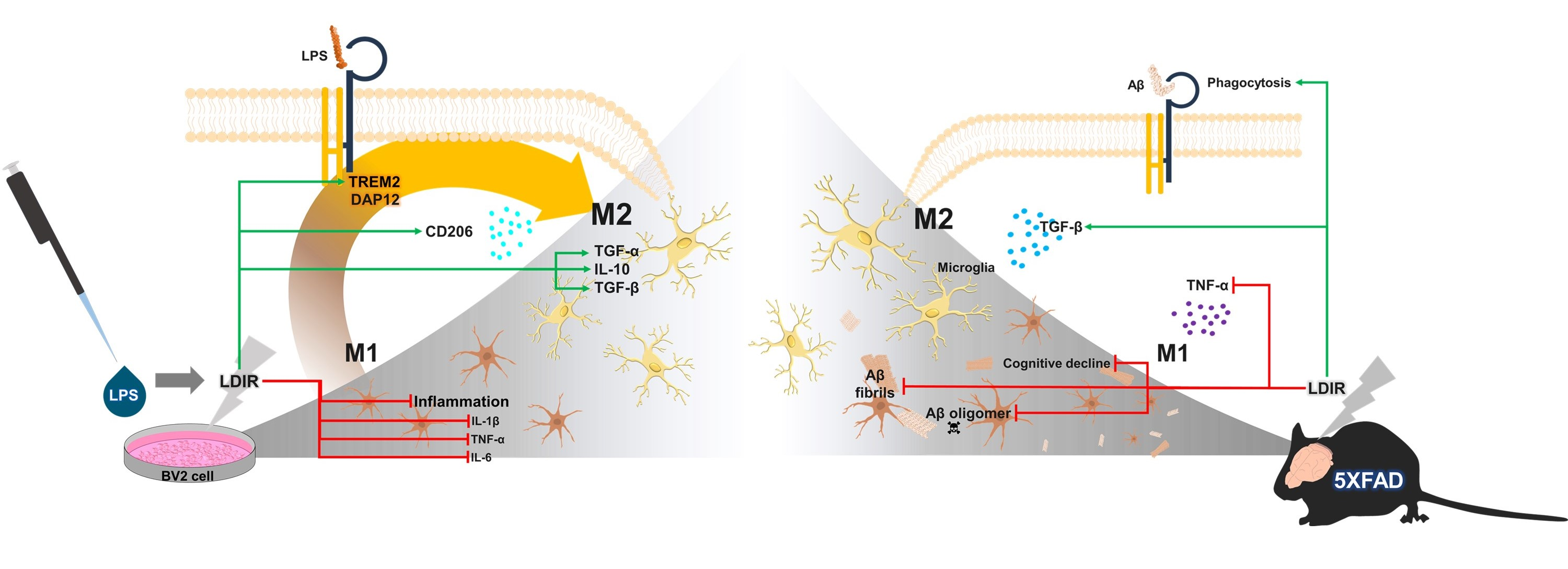
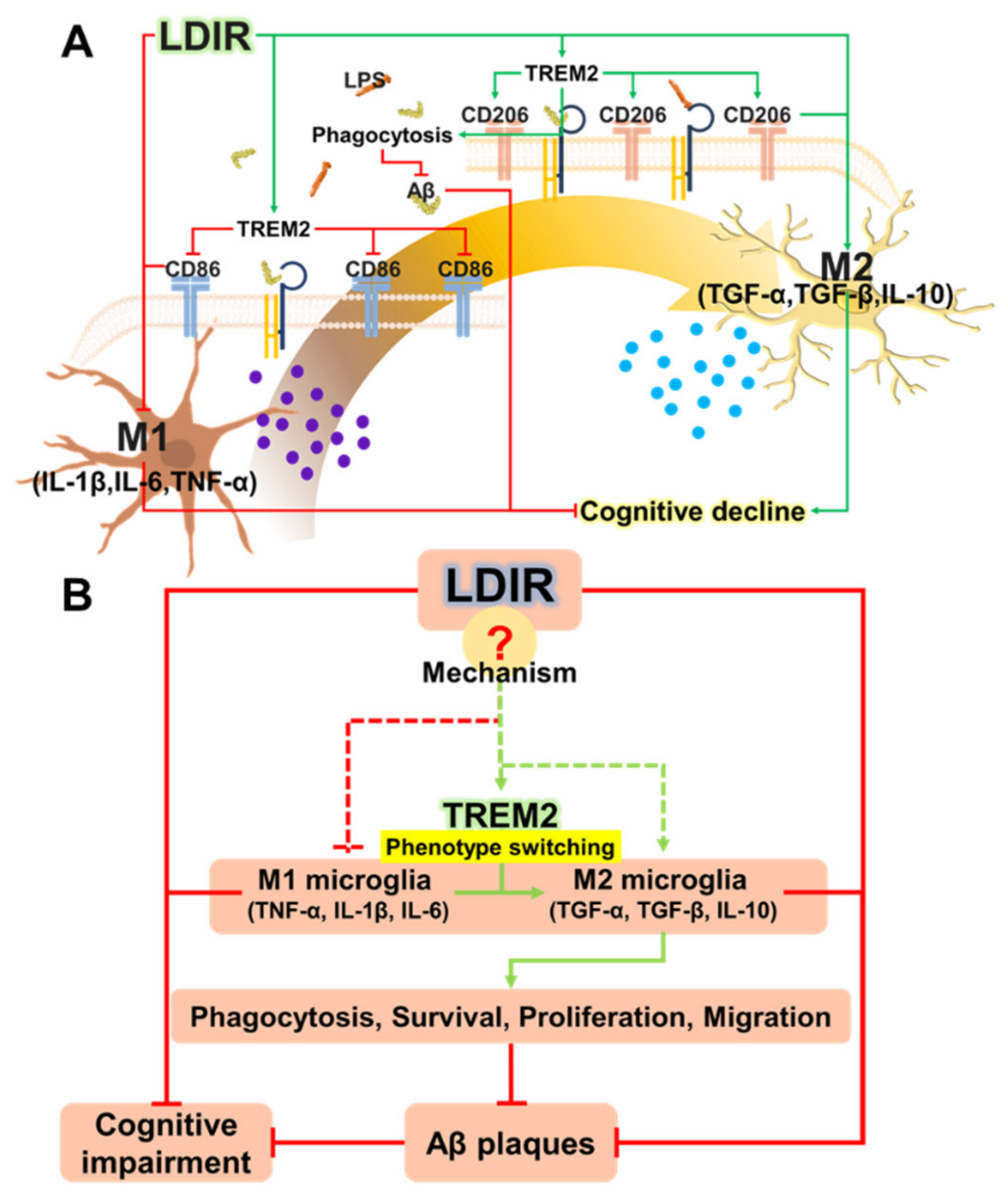
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Radiation Exposure
4.4. Memory Test
4.5. Confocal Microscopy
4.6. Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction
4.7. Enzyme-Linked Immunosorbent Assay
4.8. BV-2 Cell Culture
4.9. Cell Viability
4.10. Immunoblotting
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ANOVA | one-way analysis of variance |
| Aβ | amyloid-β |
| BBB | blood brain barrier |
| CAM | intercellular adhesion molecule |
| CCK-8 | Cell counting kit-8 |
| DMEM | Dulbecco’s Modified Eagle Medium |
| IL-1β | interleukin-1 beta |
| INF-γ | interferon-gamma |
| LDIR | Low-dose ionizing radiation |
| LPS | lipopolysaccharides |
| R47H | Heterozygous rare variants |
| TNF-α | tumor necrosis factor alpha |
| TREM2 | triggering receptor expressed on myeloid cells 2 |
| VEGF | vascular endothelial growth factors |
| WT | Wild-type |
References
- Lu, Q.; Powles, R.L.; Abdallah, S.; Ou, D.; Wang, Q.; Hu, Y.; Lu, Y.; Liu, W.; Li, B.; Mukherjee, S.; et al. Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease. Lancet 2016, 388, 078865. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Bolos, M.; Úbeda-Portugués, J.R.P.; Avila, J. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.-Y.; Han, S.-B.; Oh, K.-W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.G.; Bora, S.H.; Xu, G.; Borchelt, D.R.; Price, D.L.; Koliatsos, V.E. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 2003, 14, 133–145. [Google Scholar] [CrossRef]
- Hauss-Wegrzyniak, B.; Wenk, G. Beta-amyloid deposition in the brains of rats chronically infused with thiorphan or lipopolysaccharide: The role of ascorbic acid in the vehicle. Neurosci. Lett. 2002, 322, 75–78. [Google Scholar] [CrossRef]
- Pretorius, E.; Bester, J.; Kell, D.B. A Bacterial Component to Alzheimer’s-Type Dementia Seen via a Systems Biology Approach that Links Iron Dysregulation and Inflammagen Shedding to Disease. J. Alzheimer’s Dis. 2016, 53, 1237–1256. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-J.; Choi, N.-Y.; Yun, Y.-P.; Han, S.-B.; Oh, K.-W.; Hong, J.T. Epigallocatechin-3-gallate prevents systemic inflammation-induced memory deficiency and amyloidogenesis via its anti-neuroinflammatory properties. J. Nutr. Biochem. 2013, 24, 298–310. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2015, 136, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.M.; Salinas-Navarro, M.; Cordeiro, M.F.; Moons, L.; De Groef, L. Characterizing microglia activation: A spatial statistics approach to maximize information extraction. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, J.; You, Z. Switching of the Microglial Activation Phenotype Is a Possible Treatment for Depression Disorder. Front. Cell. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Chiba, K. Role of microglial m1/m2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals (Basel) 2014, 7, 1028–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathipati, P.; Müller, S.; Jiang, X.; Ferriero, N. Phenotype and Secretory Responses to Oxidative Stress in Microglia. Dev. Neurosci. 2013, 35, 241–254. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- A Wynn, T.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. et Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Wang, X.; Liu, C.; Zhang, H.-L. Pharmacological Targeting of Microglial Activation: New Therapeutic Approach. Front. Cell. Neurosci. 2019, 13, 514. [Google Scholar] [CrossRef] [Green Version]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, S.; Nie, K.; Li, Y.; Gao, Y.; Gan, R.; Wang, L.; Li, B.; Sun, X.; Wang, L.; et al. TREM2 modulates microglia phenotypes in the neuroinflammation of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2018, 499, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Ma, Y.; Huang, W.; Cheng, X.; Gao, N.; Li, G.; Tian, S. Up-regulation of TREM2 accelerates the reduction of amyloid deposits and promotes neuronal regeneration in the hippocampus of amyloid beta1-42 injected mice. J. Chem. Neuroanat. 2019, 97, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y.-W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395. [Google Scholar] [CrossRef] [Green Version]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A Dap12-Mediated Pathway Regulates Expression of Cc Chemokine Receptor 7 and Maturation of Human Dendritic Cells. J. Exp. Med. 2001, 194, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Zhao, Y.; Dua, P.; Rogaev, E.I.; Lukiw, W.J. microRNA-34a-Mediated Down-Regulation of the Microglial-Enriched Triggering Receptor and Phagocytosis-Sensor TREM2 in Age-Related Macular Degeneration. PLoS ONE 2016, 11, e0150211. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, C.-C.; Atagi, Y.; Chen, X.-F.; Jia, L.; Yang, L.; He, W.; Zhang, X.; Kang, S.S.; Rosenberry, T.L.; et al. Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging 2016, 42, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.D.; Daggett, A.; Gu, X.; Jiang, L.-L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Zhang, Y.; Chen, Q.; Gao, Q.; Zhu, X.-C.; Zhou, J.; Shi, J.-Q.; Lu, H.; Tan, L.; Yu, J. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 2016, 105, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervois, P.; Lambrichts, I. The Emerging Role of Triggering Receptor Expressed on Myeloid Cells 2 as a Target for Immunomodulation in Ischemic Stroke. Front. Immunol. 2019, 10, 1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Monje, M.L.; Mizumatsu, S.; Fike, J.R.; Palmer, T.D. Irradiation induces neural precursor-cell dysfunction. Nat. Med. 2002, 8, 955–962. [Google Scholar] [CrossRef]
- Chakraborti, A.; Allen, A.; Allen, B.; Rosi, S.; Fike, J.R. Cranial Irradiation Alters Dendritic Spine Density and Morphology in the Hippocampus. PLoS ONE 2012, 7, e40844. [Google Scholar] [CrossRef]
- Shirai, K.; Mizui, T.; Suzuki, Y.; Okamoto, M.; Hanamura, K.; Yoshida, Y.; Hino, M.; Noda, S.-E.; Al-Jahdari, W.S.; Chakravarti, A.; et al. X Irradiation Changes Dendritic Spine Morphology and Density through Reduction of Cytoskeletal Proteins in Mature Neurons. Radiat. Res. 2013, 179, 630–636. [Google Scholar] [CrossRef]
- Wilson, G.D.; Marples, B. A New Use for an Old Treatment: Radiation Therapy and Alzheimer’s Disease. Radiat. Res. 2016, 185, 443–448. [Google Scholar] [CrossRef]
- Michael, D.B.; Wilson, G.D.; Hanna, A.; Wilson, T.; Martinez, A.A.; Chinnaiyan, P.; Maddens, M.E.; Fontanesi, J. Radiation therapy for the treatment of Alzheimer’s disease. Neurol. Neurosurg. 2019, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Ceyzériat, K.; Tournier, B.B.; Millet, P.; Frisoni, G.B.; Garibotto, V.; Zilli, T. Low-Dose Radiation Therapy: A New Treatment Strategy for Alzheimer’s Disease? J. Alzheimer’s Dis. 2020, 74, 411–419. [Google Scholar] [CrossRef]
- Kim, S.; Nam, Y.; Kim, C.; Lee, H.; Hong, S.; Kim, H.S.; Shin, S.J.; Park, Y.H.; Mai, H.N.; Oh, S.-M.; et al. Neuroprotective and Anti-Inflammatory Effects of Low–Moderate Dose Ionizing Radiation in Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3678. [Google Scholar] [CrossRef] [PubMed]
- Cuttler, J.S.W.J.M.; Moore, E.R.; Hosfeld, V.D.; Nadolski, D.L. Treatment of Alzheimer Disease With CT Scans. Dose Response 2016, 14, 1559325816640073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Hinshaw, R.G.; Le, K.X.; Park, M.-A.; Wang, S.; Belanger, A.P.; Dubey, S.; Frost, J.L.; Shi, Q.; Holton, P.; et al. Space-like 56Fe irradiation manifests mild, early sex-specific behavioral and neuropathological changes in wildtype and Alzheimer’s-like transgenic mice. Sci. Rep. 2019, 9, 12118. [Google Scholar] [CrossRef]
- Wei, L.-C.; Ding, Y.-X.; Liu, Y.; Duan, L.; Bai, Y.; Shi, M.; Chen, L.-W. Low-dose radiation stimulates Wnt/?-catenin signaling, neural stem cell proliferation and neurogenesis of the mouse hippocampus in vitro and in vivo. Curr. Alzheimer Res. 2012, 9, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Marples, B.; McGee, M.; Callan, S.; Bowen, S.; Thibodeau, B.J.; Michael, D.B.; Wilson, G.D.; Maddens, M.E.; Fontanesi, J.; Martinez, A.A. Cranial irradiation significantly reduces beta amyloid plaques in the brain and improves cognition in a murine model of Alzheimer’s Disease (AD). Radiother. Oncol. 2016, 118, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Kempf, S.J.; Janik, D.; Barjaktarovic, Z.; Braga-Tanaka, I., 3rd; Tanaka, S.; Neff, F.; Saran, A.; Larsen, M.R.; Tapio, S. Chronic low-dose-rate ionising radiation affects the hippocampal phosphoproteome in the ApoE-/- Alzheimer’s mouse model. Oncotarget 2016, 7(44), 71817–71832. [Google Scholar] [CrossRef] [PubMed]
- Otani, A.; Kojima, H.; Guo, C.; Oishi, A.; Yoshimura, N. Low-Dose-Rate, Low-Dose Irradiation Delays Neurodegeneration in a Model of Retinitis Pigmentosa. Am. J. Pathol. 2012, 180, 328–336. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Norris, C.M.; Sompol, P.; Wilcock, D.M.; Goulding, D.; Neltner, J.H.; Clair, D.S.; Watterson, D.M.; Van Eldik, L.J. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer’s disease-related pathology. J. Neurosci. 2012, 32, 10201–10210. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2015, 53, 1181–1194. [Google Scholar] [CrossRef]
- Kacimi, R.; Giffard, R.G.; Yenari, M.A. Endotoxin-activated microglia injure brain derived endothelial cells via NF-kappaB, JAK-STAT and JNK stress kinase pathways. J. Inflamm. (Lond.) 2011, 8. [Google Scholar] [CrossRef] [Green Version]
- Park, B.-K.; Kim, Y.H.; Kim, Y.R.; Choi, J.J.; Yang, C.; Jang, I.S.; Lee, M.Y. Antineuroinflammatory and Neuroprotective Effects of Gyejibokryeong-Hwan in Lipopolysaccharide-Stimulated BV2 Microglia. Evid. Based Complement. Altern. Med. 2019, 2019, 7585896. [Google Scholar] [CrossRef] [PubMed]
- Lively, S.; Schlichter, L.C. The microglial activation state regulates migration and roles of matrix-dissolving enzymes for invasion. J. Neuroinflamm. 2013, 10, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Zhong, R.; Li, S.; Fu, Z.; Cheng, C.; Cai, H.; Le, W. Acute Hypoxia Induced an Imbalanced M1/M2 Activation of Microglia through NF-kappaB Signaling in Alzheimer’s Disease Mice and Wild-Type Littermates. Front. Aging Neurosci. 2017, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Nickell, C.R.G.; Chen, K.Y.; McClain, J.A.; Nixon, K. Increased expression of M1 and M2 phenotypic markers in isolated microglia after four-day binge alcohol exposure in male rats. Alcohol 2017, 62, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkenstam, N.H.; Smith, P.L.P.; Fleiss, B.; Nair, S.; Svedin, P.; Wang, W.; Boström, M.; Gressens, P.; Hagberg, H.; Brown, K.L.; et al. Temporal Characterization of Microglia/Macrophage Phenotypes in a Mouse Model of Neonatal Hypoxic-Ischemic Brain Injury. Front. Cell. Neurosci. 2016, 10, 176. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2008, 206, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Hu, Q.; Wu, J.; Mu, C.; Ren, H.-G.; Liu, C.-F.; Wang, G. P7C3 Inhibits LPS-Induced Microglial Activation to Protect Dopaminergic Neurons Against Inflammatory Factor-Induced Cell Death in vitro and in vivo. Front. Cell. Neurosci. 2018, 12, 400. [Google Scholar] [CrossRef] [Green Version]
- Katafuchi, T.; Ifuku, M.; Mawatari, S.; Noda, M.; Miake, K.; Sugiyama, M.; Fujino, T. Effects of plasmalogens on systemic lipopolysaccharide-induced glial activation and beta-amyloid accumulation in adult mice. Ann. N. Y. Acad. Sci. 2012, 1262, 85–92. [Google Scholar] [CrossRef]
- Sorrenti, V.; Contarini, G.; Sut, S.; Dall’Acqua, S.; Confortin, F.; Pagetta, A.; Giusti, P.; Zusso, M. Curcumin Prevents Acute Neuroinflammation and Long-Term Memory Impairment Induced by Systemic Lipopolysaccharide in Mice. Front. Pharmacol. 2018, 9, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.-V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, X.L.; Quan, H.F.; Yan, L.; Pei, X.Y.; Wang, R.; Peng, X.D. Effects of Betaine on LPS-Stimulated Activation of Microglial M1/M2 Phenotypes by Suppressing TLR4/NF-kappaB Pathways in N9 Cells. Molecules 2019, 24, 367. [Google Scholar] [CrossRef] [Green Version]
- Vay, S.U.; Flitsch, L.J.; Rabenstein, M.; Rogall, R.; Blaschke, S.; Kleinhaus, J.; Reinert, N.; Bach, A.; Fink, G.R.; Schroeter, M.; et al. The plasticity of primary microglia and their multifaceted effects on endogenous neural stem cells in vitro and in vivo. J. Neuroinflamm. 2018, 15, 226. [Google Scholar] [CrossRef]
- Cianciulli, A.; Salvatore, R.; Porro, C.; Trotta, T.; Panaro, M.A. Folic Acid Is Able to Polarize the Inflammatory Response in LPS Activated Microglia by Regulating Multiple Signaling Pathways. Mediat. Inflamm. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Neumann, H.; Daly, M.J. Variant TREM2 as risk factor for Alzheimer’s disease. N. Engl. J. Med. 2012, 368, 182–184. [Google Scholar] [CrossRef]
- Owens, R.; Grabert, K.; Davies, C.L.; Alfieri, A.; Antel, J.P.; Healy, L.M.; McColl, B.W. Divergent Neuroinflammatory Regulation of Microglial TREM Expression and Involvement of NF-kappaB. Front. Cell Neurosci. 2017, 11, 56. [Google Scholar] [CrossRef]
- Jiang, T.; Tan, L.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Tan, M.-S.; Gu, L.-Z.; Wang, H.-F.; Ding, Z.-Z.; Zhang, Y.-D.; et al. Upregulation of TREM2 Ameliorates Neuropathology and Rescues Spatial Cognitive Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide Associates with Amyloid Plaques, Neurons and Oligodendrocytes in Alzheimer’s Disease Brain: A Review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.-C.; Guo, C.; Yalamanchili, H.K.; Abreha, M.; Al-Ouran, R.; Li, Y.; Dammer, E.B.; Lah, J.J.; Levey, A.I.; Bennett, D.A.; et al. Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA-Splicing and Neurodegeneration in Alzheimer’s Disease. SSRN Electron. J. 2019, 29(2), 301. [Google Scholar] [CrossRef]
- Schaue, D.; Jahns, J.; Hildebrandt, G.; Trott, K.-R. Radiation treatment of acute inflammation in mice. Int. J. Radiat. Boil. 2005, 81, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2012, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jay, T.; Miller, C.M.; Cheng-Hathaway, P.; Graham, L.C.; BeMiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.; Zhu, X.-C.; Tan, L. TREM2 in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 180–185. [Google Scholar] [CrossRef]
- Shen, Z.; Bao, X.; Wang, R. Clinical PET Imaging of Microglial Activation: Implications for Microglial Therapeutics in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 314. [Google Scholar] [CrossRef]
- Jin, X.; Liu, M.-Y.; Zhang, D.-F.; Zhong, X.; Du, K.; Qian, P.; Gao, H.; Wei, Q. Natural products as a potential modulator of microglial polarization in neurodegenerative diseases. Pharmacol. Res. 2019, 145, 104253. [Google Scholar] [CrossRef]
- Marples, B.; McGee, M.; Callan, S.; Bowen, S.; Thibodeau, B.J.; Michael, D.B.; Wilson, G.D.; Maddens, M.E.; Fontanesi, J.; Martinez, A.A.; et al. Cranial irradiation significantly reduces beta amyloid plaques in the brain and improves cognition in a murine model of Alzheimer’s Disease (AD). Radiother. Oncol. 2016, 118, 43–51. [Google Scholar] [CrossRef]
- Kurrus, J.A.; Hayes, J.K.; Hoidal, J.R.; Menendez, M.M.; Elstad, M.R. Radiation therapy for tracheobronchial amyloidosis. Chest 1998, 114, 1489–1492. [Google Scholar] [CrossRef]
- Kang, J.H.; Jung, M.Y.; Yin, X.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Cell-penetrating peptides selectively targeting SMAD3 inhibit profibrotic TGF-beta signaling. J. Clin. Investig. 2017, 127, 2541–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolowski, J.D.; Mandell, J.W. Phagocytic Clearance in Neurodegeneration. Am. J. Pathol. 2011, 178, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Moravan, M.J.; Olschowka, J.A.; Williams, J.P.; O’Banion, M.K. Cranial irradiation leads to acute and persistent neuroinflammation with delayed increases in T-cell infiltration and CD11c expression in C57BL/6 mouse brain. Radiat. Res. 2011, 176, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Olschowka, J.A.; Kyrkanides, S.; Harvey, B.K.; O’Banion, M.K.; Williams, J.P.; Rubin, P.; Hansen, J.T. ICAM-1 Induction in the Mouse CNS Following Irradiation. Brain Behav. Immun. 1997, 11, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-I.; Jeon, S.G.; Kim, K.A.; Kim, J.-J.; Song, E.J.; Jeon, Y.; Kim, E.; Lee, K.B.; Kwak, J.H.; Moon, M. Platycodon grandiflorus Root Extract Improves Learning and Memory by Enhancing Synaptogenesis in Mice Hippocampus. Nutrients 2017, 9, 794. [Google Scholar] [CrossRef] [Green Version]
- Rivera, I.; Capone, R.; Cauvi, D.M.; Arispe, N.; De Maio, A. Modulation of Alzheimer’s amyloid beta peptide oligomerization and toxicity by extracellular Hsp70. Cell Stress Chaperones 2018, 23, 269–279. [Google Scholar] [CrossRef]
- Heissig, B.; Rafii, S.; Akiyama, H.; Ohki, Y.; Sato, Y.; Rafael, T.; Zhu, Z.; Hicklin, D.J.; Okumura, K.; Ogawa, H.; et al. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9–mediated progenitor cell mobilization. J. Exp. Med. 2005, 202, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Jansen, H.; Meffert, R.; Birkenfeld, F.; Petersen, W.; Pufe, T. Detection of vascular endothelial growth factor (VEGF) in moderate osteoarthritis in a rabbit model. Ann. Anat. Anat. Anz. 2012, 194, 452–456. [Google Scholar] [CrossRef]
- Sanchez, A.; Wadhwani, S.; Grammas, P. Multiple neurotrophic effects of VEGF on cultured neurons. Neuropeptides 2010, 44, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, S.D.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Stott, D.J.; Rodondi, N.; Kearney, P.M.; Ford, I.; Westendorp, R.G.J.; Mooijaart, S.P.; Sattar, N.; Aubert, C.E.; Aujesky, D.; Bauer, D.C.; et al. Thyroid Hormone Therapy for Older Adults with Subclinical Hypothyroidism. N. Engl. J. Med. 2017, 376, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Chung, H.; Ngoc Mai, H.; Nam, Y.; Shin, S.J.; Park, Y.H.; Chung, M.J.; Lee, J.K.; Rhee, H.Y.; Jahng, G.-H.; et al. Low-Dose Ionizing Radiation Modulates Microglia Phenotypes in the Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 4532. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124532
Kim S, Chung H, Ngoc Mai H, Nam Y, Shin SJ, Park YH, Chung MJ, Lee JK, Rhee HY, Jahng G-H, et al. Low-Dose Ionizing Radiation Modulates Microglia Phenotypes in the Models of Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(12):4532. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124532
Chicago/Turabian StyleKim, Sujin, Hyunju Chung, Han Ngoc Mai, Yunkwon Nam, Soo Jung Shin, Yong Ho Park, Mi Joo Chung, Jong Kil Lee, Hak Young Rhee, Geon-Ho Jahng, and et al. 2020. "Low-Dose Ionizing Radiation Modulates Microglia Phenotypes in the Models of Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 12: 4532. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124532