Rationally Designed Antibodies as Research Tools to Study the Structure–Toxicity Relationship of Amyloid-β Oligomers

 , , , , , and
, , , , , and
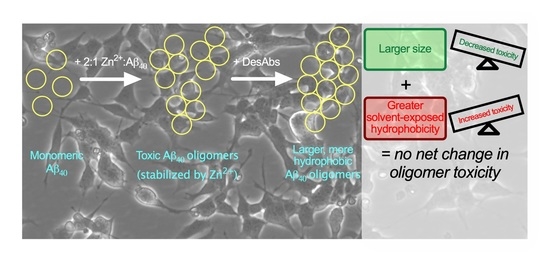
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
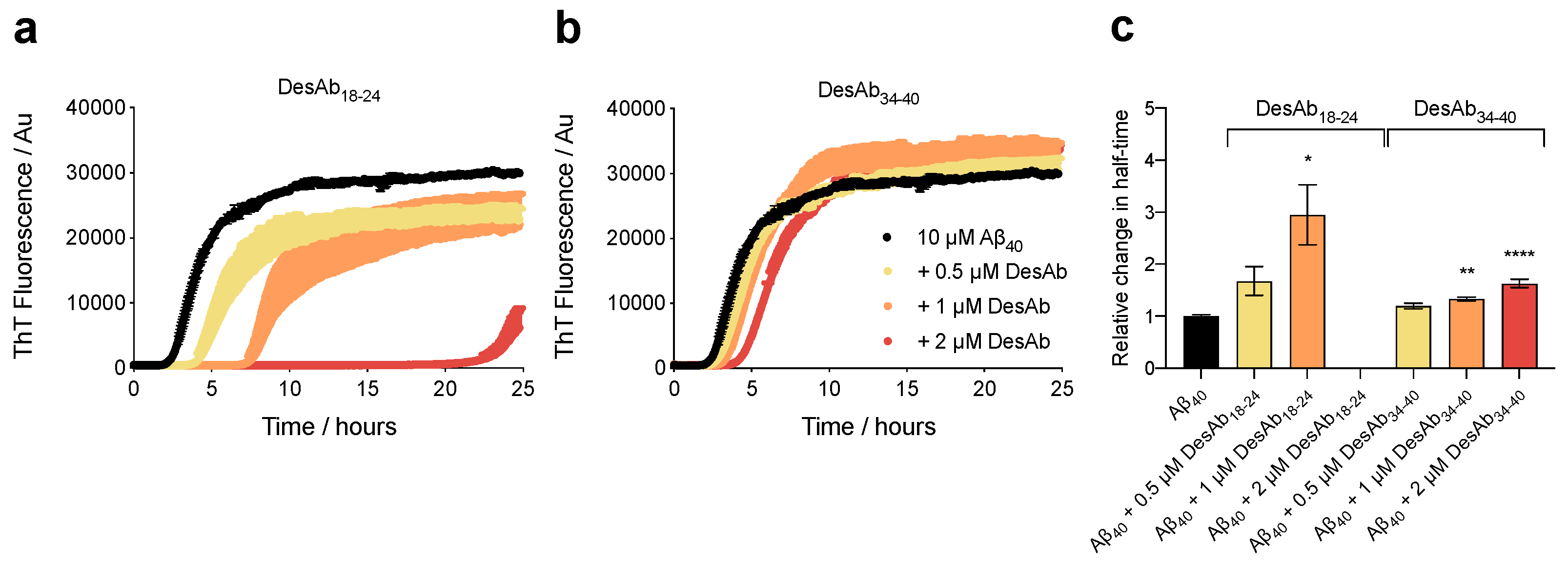
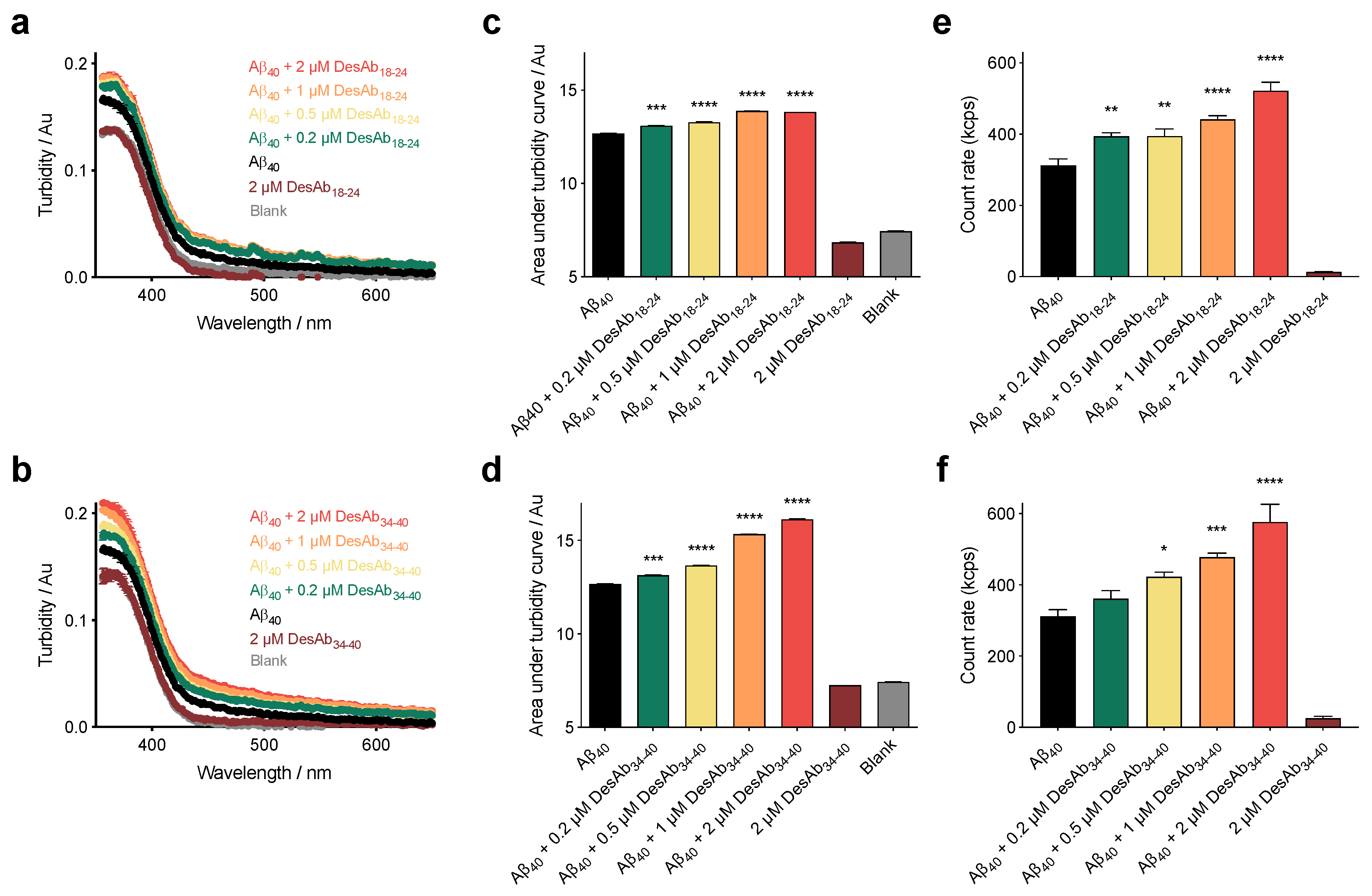
2.1. The DesAbs Inhibit Aβ40 Aggregation
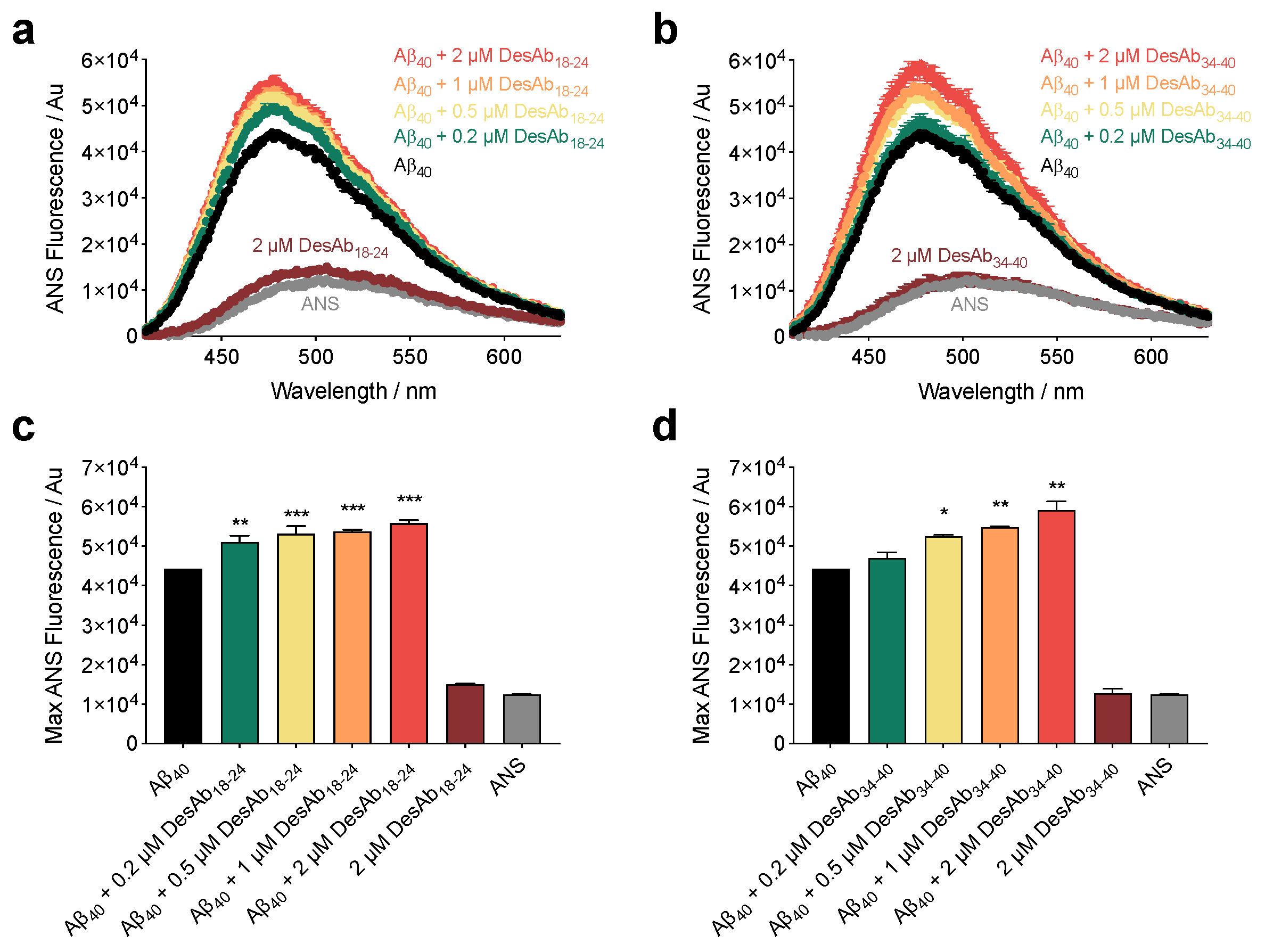
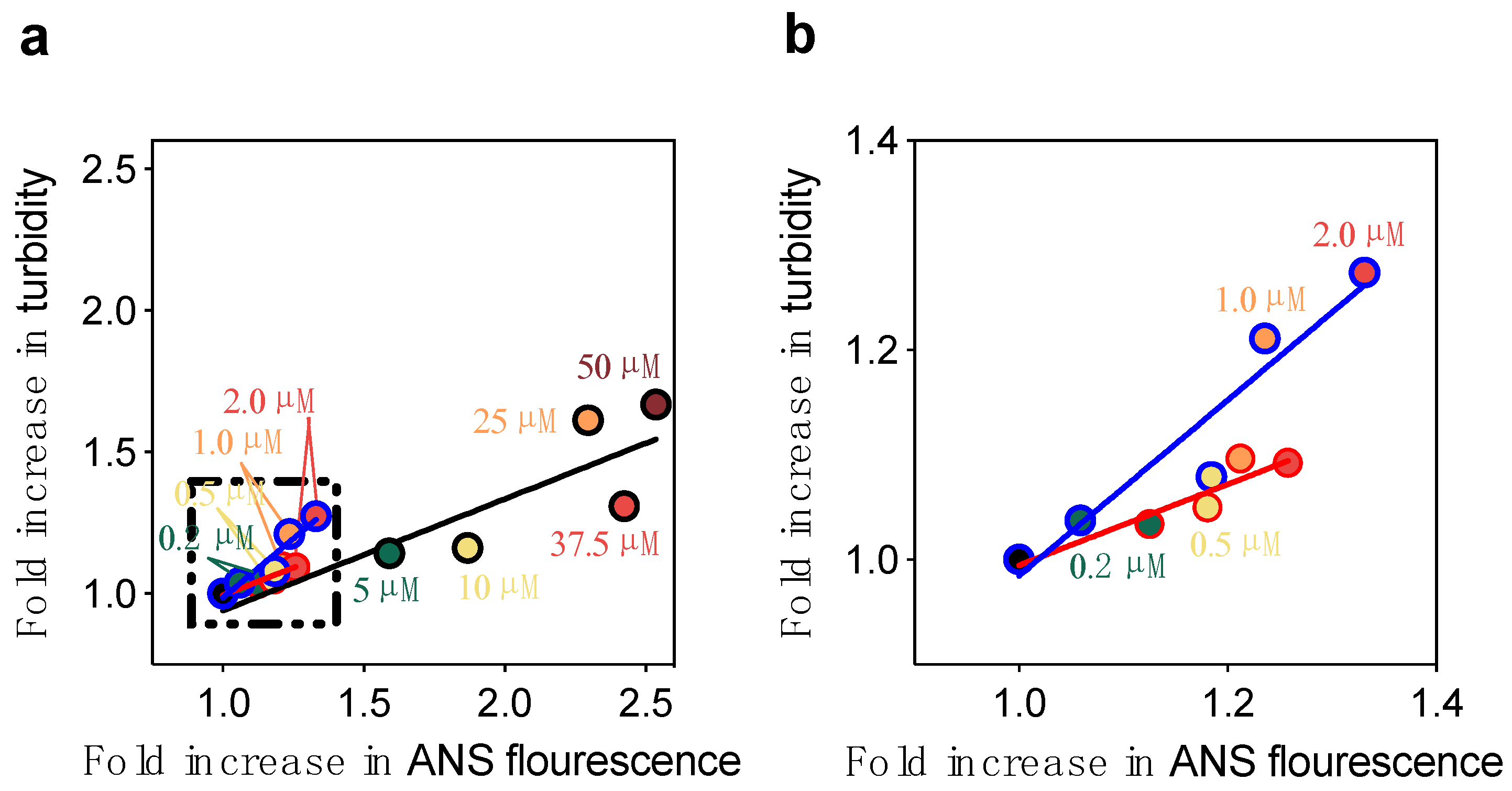
2.2. The DesAbs Bind Stabilized Aβ40 Oligomers and Increase Their Hydrophobic Surface Area
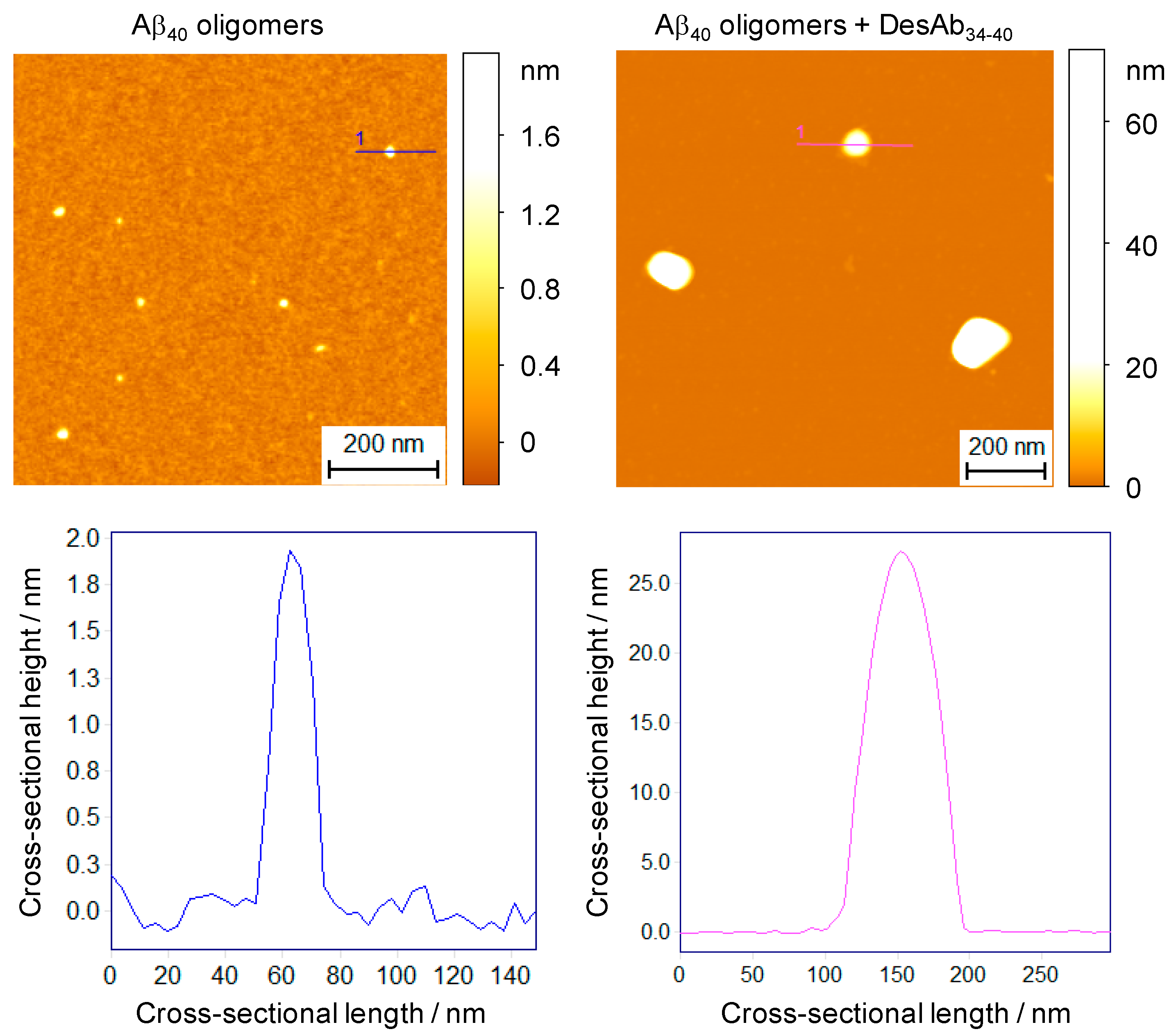
2.3. DesAbs Increase the Size of Stabilized Aβ40 Oligomers
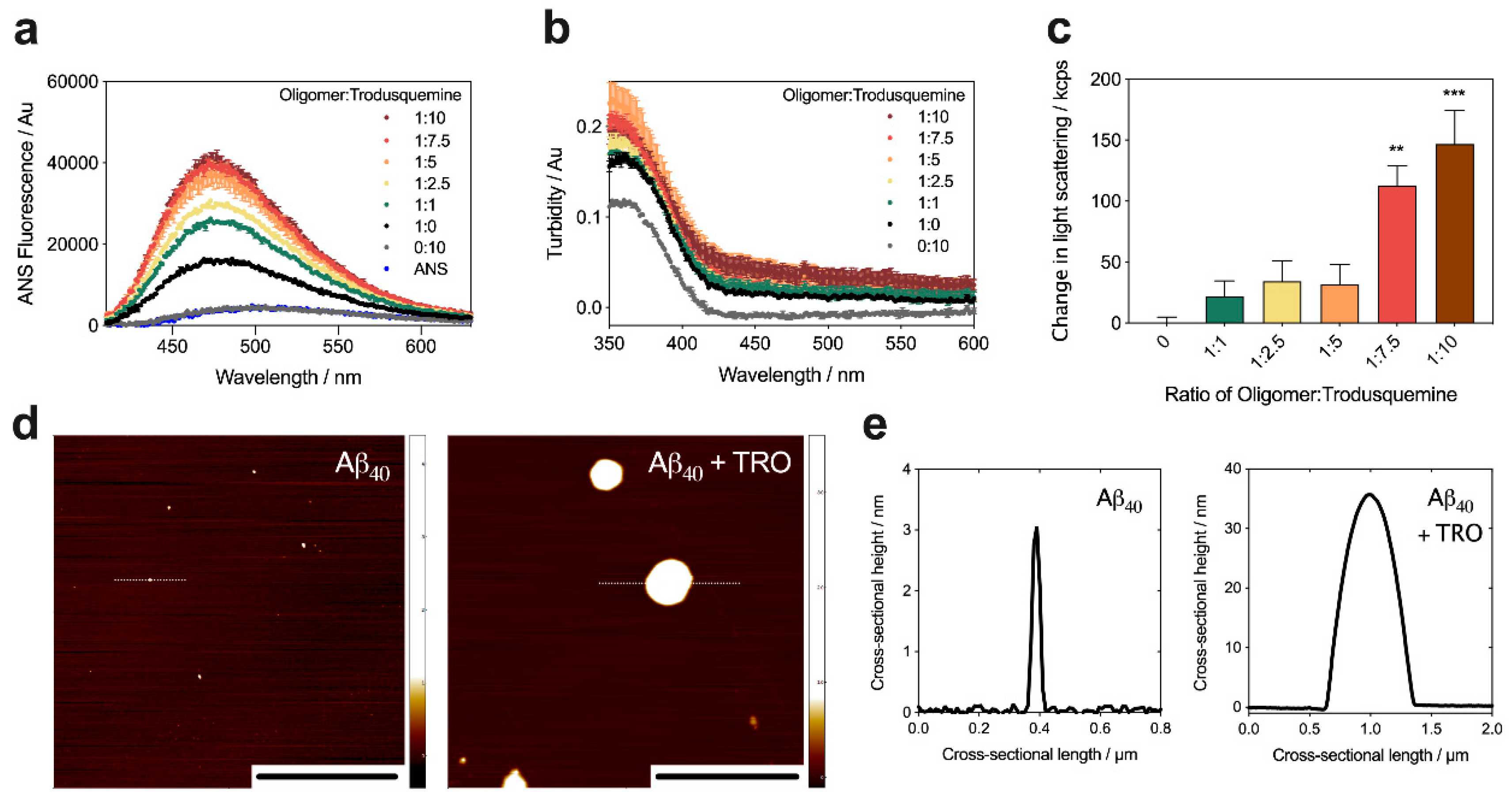
2.4. Trodusquemine Binds Stabilized Aβ40 Oligomers and Increases their Hydrophobic Surface Area
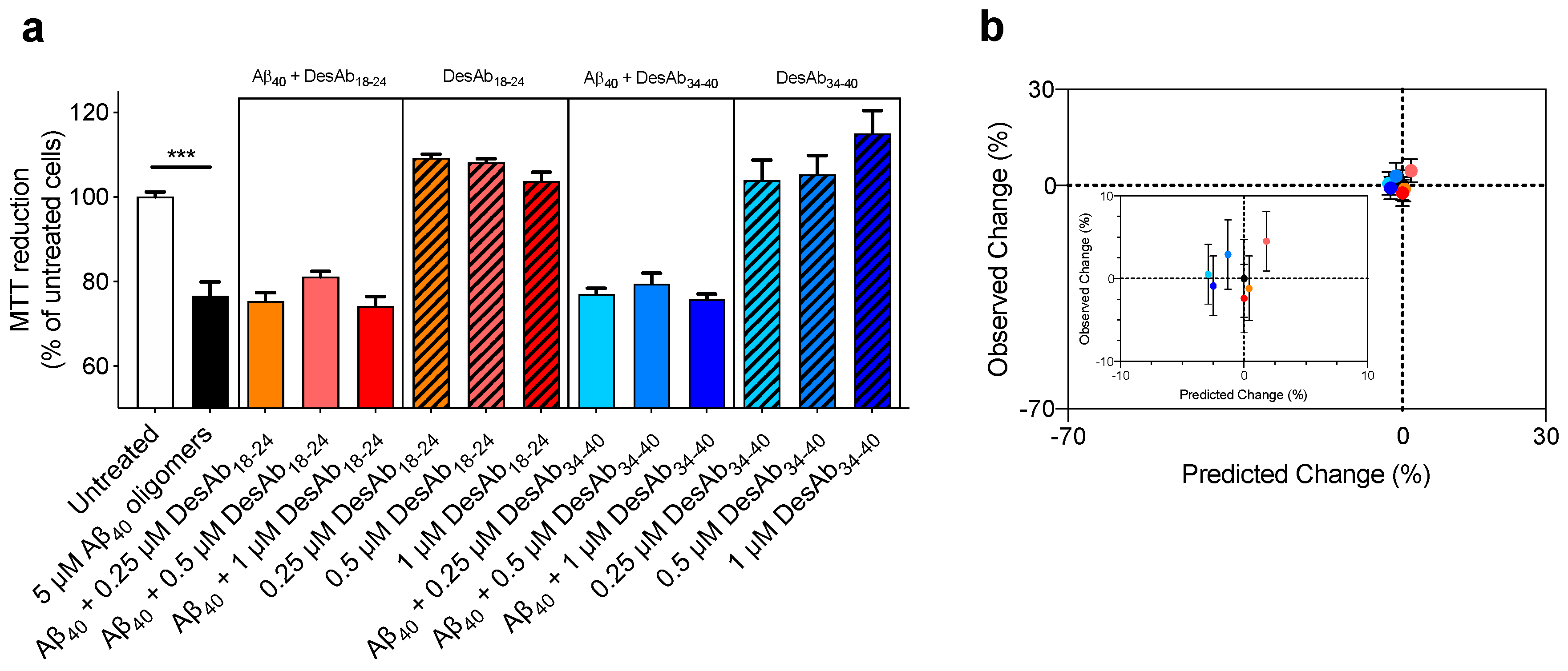
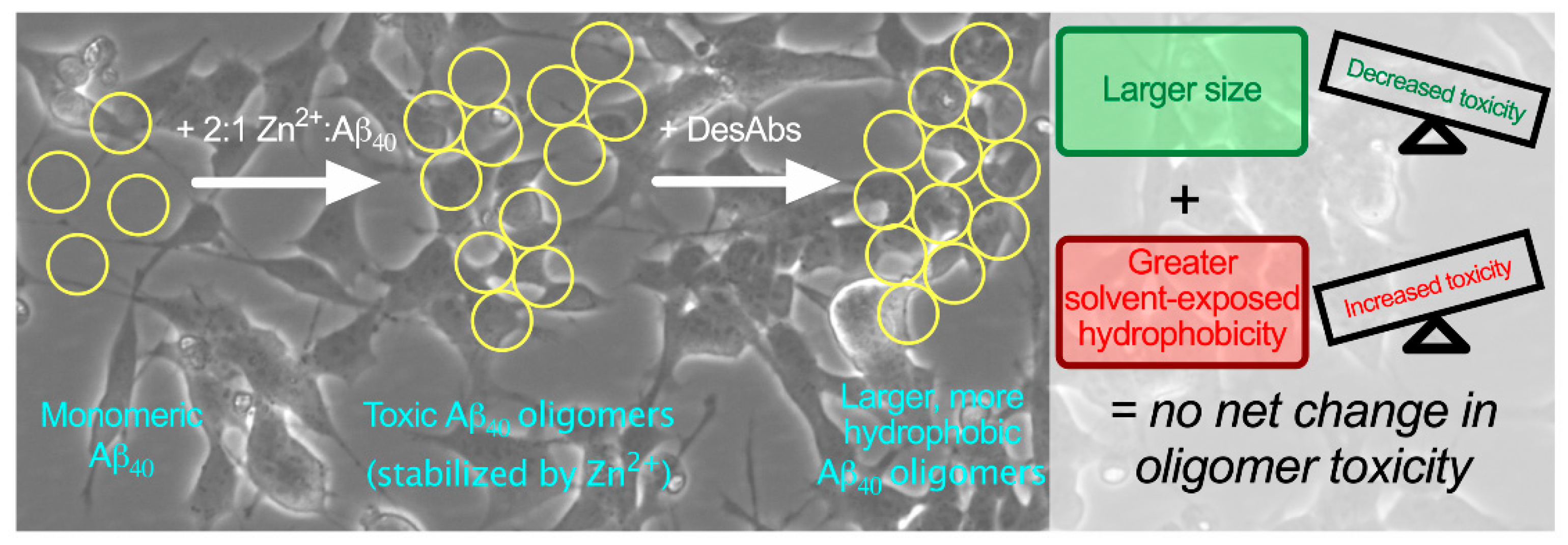
2.5. The DesAbs do not Change the Toxicity of Aβ40 Oligomers for Human Neuroblastoma Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Antibody Design, Expression and Purification
4.3. Protein Expression and Chemical Kinetics
4.4. Preparation of the Stabilized Aβ40 Oligomers
4.5. ANS Binding Measurements
4.6. Turbidimetry Measurements
4.7. SLS
4.8. Sedimentataion-Based Dot Blot Assays
4.9. Antibody Binding Dot Blot Assays
4.10. Atomic Force Microscopy
4.11. Neuroblastoma Cell Cultures
4.12. MTT Reduction Assay
4.13. Statistics and Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β peptide |
| Aβ40 | 40-residue form of the amyloid-β peptide |
| Aβ42 | 42-residue form of the amyloid-β peptide |
| AD | Alzheimer’s disease |
| AFM | atomic force microscopy |
| ANS | 8-anilinonapthalene-1-sulfonic acid |
| DesAb | designed antibody |
| HFIP | hexafluoroisopropanol |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| SEM | standard error of the mean |
| SLS | static light scattering |
| ThT | thioflavin T |
| TRO | trodusquemine |
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Fontana, I.C.; Zimmer, A.R.; Rocha, A.S.; Gosmann, G.; Souza, D.O.; Lourenco, M.V.; Ferreira, S.T.; Zimmer, E.R. Amyloid-β oligomers in cellular models of Alzheimer’s disease. J. Neurochem. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Doody, R.; Kramer, L.; Salloway, S.; Selkoe, D.J.; Sims, J.; Sperling, R.A.; Vellas, B. The future of anti-amyloid trials. J. Prev. Alzheimers Dis. 2020, 7, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Linse, S.; Scheidt, T.; Bernfur, K.; Vendruscolo, M.; Dobson, C.M.; Cohen, S.I.A.; Sileikis, E.; Lundquist, M.; Qian, F.; O’Malley, T.; et al. Kinetic fingerprints differentiate anti-Aβ therapies. bioRxiv 2020, 815308. [Google Scholar] [CrossRef] [Green Version]
- Karran, E.; Hardy, J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol. 2014, 76, 185–205. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Holtzman, D.M.; Goate, A.; Kelly, J.; Sperling, R. Mapping the road forward in Alzheimer’s disease. Sci. Transl. Med. 2011, 3, 114ps48. [Google Scholar] [CrossRef]
- Sperling, R.A.; Jack, C.R.; Aisen, P.S. Testing the right target and the right drug at the right stage. Sci. Transl. Med. 2011, 3, 111cm33. [Google Scholar] [CrossRef] [Green Version]
- Aisen, P.S.; Vellas, B.; Hampel, H. Moving towards early clinical trials for amyloid-targeted therapy in Alzheimer’s disease. Nat. Rev. Drug Discov. 2013, 12, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, P.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 2014, 35, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Michaels, T.C.T.; Linse, S.; Månsson, C.; Emanuelsson, C.; Presto, J.; Johansson, J.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016, 7, 10948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisl, G.; Kirkegaard, J.B.; Arosio, P.; Michaels, T.C.T.; Vendruscolo, M.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 2016, 11, 252–272. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.A.; Sormanni, P.; Perni, M.; Arosio, P.; Linse, S.; Knowles, T.P.J.; Dobson, C.M.; Vendruscolo, M. Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Sci. Adv. 2017, 3, e1700488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habchi, J.; Chia, S.; Limbocker, R.; Mannini, B.; Ahn, M.; Perni, M.; Hansson, O.; Arosio, P.; Kumita, J.R.; Challa, P.K.; et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2016, 201615613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, S.; Habchi, J.; Michaels, T.C.T.; Cohen, S.I.A.; Linse, S.; Dobson, C.M.; Knowles, T.P.J.; Vendruscolo, M. SAR by kinetics for drug discovery in protein misfolding diseases. Proc. Natl. Acad. Sci. USA 2018, 115, 10245–10250. [Google Scholar] [CrossRef] [Green Version]
- Michaels, T.C.T.; Šarić, A.; Curk, S.; Bernfur, K.; Arosio, P.; Meisl, G.; Dear, A.J.; Cohen, S.I.A.; Dobson, C.M.; Vendruscolo, M.; et al. Dynamics of oligomer populations formed during the aggregation of Alzheimer’s Aβ42 peptide. Nat. Chem. 2020, 12, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Elvin, J.G.; Couston, R.G.; van der Walle, C.F. Therapeutic antibodies: Market considerations, disease targets and bioprocessing. Int. J. Pharm. 2013, 440, 83–98. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Whiten, D.R.; Ruggeri, F.S.; Hughes, C.; Rodrigues, M.; Sideris, D.I.; Taylor, C.G.; Aprile, F.A.; Muyldermans, S.; Knowles, T.P.J.; et al. Soluble aggregates present in cerebrospinal fluid change in size and mechanism of toxicity during Alzheimer’s disease progression. Acta Neuropathol. Commun. 2019, 7, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, S.; Wirthensohn, D.C.; Flagmeier, P.; Hughes, C.; Aprile, F.A.; Ruggeri, F.S.; Whiten, D.R.; Emin, D.; Xia, Z.; Varela, J.A.; et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 2019, 10, 1541. [Google Scholar] [CrossRef] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Aprile, F.A.; Sormanni, P.; Podpolny, M.; Chhangur, S.; Needham, L.-M.; Ruggeri, F.S.; Perni, M.; Limbocker, R.; Heller, G.T.; Sneideris, T.; et al. Rational design of a conformation-specific antibody for the quantification of Aβ oligomers. Proc. Natl. Acad. Sci. USA 2020, 201919464. [Google Scholar] [CrossRef]
- Yang, T.; O’Malley, T.T.; Kanmert, D.; Jerecic, J.; Zieske, L.R.; Zetterberg, H.; Hyman, B.T.; Walsh, D.M.; Selkoe, D.J. A highly sensitive novel immunoassay specifically detects low levels of soluble Aβ oligomers in human cerebrospinal fluid. Alzheimers Res. Ther. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine enhances Aβ42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 2019, 10, 225. [Google Scholar] [CrossRef] [Green Version]
- Mannini, B.; Mulvihill, E.; Sgromo, C.; Cascella, R.; Khodarahmi, R.; Ramazzotti, M.; Dobson, C.M.; Cecchi, C.; Chiti, F. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 2014, 9, 2309–2317. [Google Scholar] [CrossRef]
- Ojha, J.; Masilamoni, G.; Dunlap, D.; Udoff, R.A.; Cashikar, A.G. Sequestration of toxic oligomers by HspB1 as a cytoprotective mechanism. Mol. Cell. Biol. 2011, 31, 3146–3157. [Google Scholar] [CrossRef] [Green Version]
- Mannini, B.; Chiti, F. Chaperones as suppressors of protein misfolded oligomer toxicity. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Mannini, B.; Cascella, R.; Zampagni, M.; van Waarde-Verhagen, M.; Meehan, S.; Roodveldt, C.; Campioni, S.; Boninsegna, M.; Penco, A.; Relini, A.; et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, S.; Penco, A.; Mannini, B.; Cascella, R.; Wilson, M.R.; Ecroyd, H.; Li, X.; Buxbaum, J.N.; Dobson, C.M.; Cecchi, C.; et al. Effect of molecular chaperones on aberrant protein oligomers in vitro: Super-versus sub-stoichiometric chaperone concentrations. Biol. Chem. 2016, 397, 401–415. [Google Scholar] [CrossRef]
- Bolognesi, B.; Kumita, J.R.; Barros, T.P.; Esbjorner, E.K.; Luheshi, L.M.; Crowther, D.C.; Wilson, M.R.; Dobson, C.M.; Favrin, G.; Yerbury, J.J. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 2010, 5, 735–740. [Google Scholar] [CrossRef]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 9902–9907. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, A.; Luheshi, L.M.; Söllvander, S.; de Barros, T.P.; Macao, B.; Knowles, T.P.J.; Biverstål, H.; Lendel, C.; Ekholm-Petterson, F.; Dubnovitsky, A.; et al. Stabilization of neurotoxic Alzheimer amyloid-β oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [Google Scholar] [CrossRef] [Green Version]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Klein, W.L. Aβ toxicity in Alzheimer’s disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 2002, 41, 345–352. [Google Scholar] [CrossRef]
- Mannini, B.; Habchi, J.; Chia, S.; Ruggeri, F.S.; Perni, M.; Knowles, T.P.J.; Dobson, C.M.; Vendruscolo, M. Stabilization and characterization of cytotoxic Aβ40 oligomers isolated from an aggregation reaction in the presence of zinc ions. ACS Chem. Neurosci. 2018, 9, 2959–2971. [Google Scholar] [CrossRef] [PubMed]
- Stryer, L. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin: A fluorescent probe of non-polar binding sites. J. Mol. Biol. 1965, 13, 482–495. [Google Scholar] [CrossRef]
- Vivoli Vega, M.; Cascella, R.; Chen, S.W.; Fusco, G.; De Simone, A.; Dobson, C.M.; Cecchi, C.; Chiti, F. The toxicity of misfolded protein oligomers is independent of their secondary structure. ACS Chem. Biol. 2019, 14, 1593–1600. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Vieweg, S.; Cendrowska, U.; Longo, G.; Chiki, A.; Lashuel, H.A.; Dietler, G. Nanoscale studies link amyloid maturity with polyglutamine diseases onset. Sci. Rep. 2016, 6, 31155. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, F.S.; Šneideris, T.; Vendruscolo, M.; Knowles, T.P.J. Atomic force microscopy for single molecule characterisation of protein aggregation. Arch. Biochem. Biophys. 2019, 664, 134–148. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Habchi, J.; Cerreta, A.; Dietler, G. AFM-based single molecule techniques: Unraveling the amyloid pathogenic species. Curr. Pharm. Des. 2016, 22, 3950–3970. [Google Scholar] [CrossRef] [Green Version]
- Adamcik, J.; Jung, J.-M.; Flakowski, J.; De Los Rios, P.; Dietler, G.; Mezzenga, R. Understanding amyloid aggregation by statistical analysis of atomic force microscopy images. Nat. Nanotechnol. 2010, 5, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, F.S.; Charmet, J.; Kartanas, T.; Peter, Q.; Chia, S.; Habchi, J.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J. Microfluidic deposition for resolving single-molecule protein architecture and heterogeneity. Nat. Commun. 2018, 9, 3890. [Google Scholar] [CrossRef] [Green Version]
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2018, 119, 57–70. [Google Scholar] [CrossRef]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure–neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Condron, M.M.; Teplow, D.B. Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 2010, 285, 23186–23197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Kedia, N.; Illes-Toth, E.; Haralampiev, I.; Prisner, S.; Herrmann, A.; Wanker, E.E.; Bieschke, J. Amyloid-β(1–42) aggregation initiates its cellular uptake and cytotoxicity. J. Biol. Chem. 2016, 291, 19590–19606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, S.; Foley, A.R.; Warner, C.J.A.; Zhang, X.; Rolandi, M.; Abrams, B.; Raskatov, J.A. Suppression of oligomer formation and formation of non-toxic fibrils upon addition of mirror-image Aβ42 to the natural L-enantiomer. Angew. Chem. Int. Ed. 2017, 56, 11506–11510. [Google Scholar] [CrossRef]
- Dutta, S.; Finn, T.S.; Kuhn, A.J.; Abrams, B.; Raskatov, J.A. Chirality dependence of amyloid β cellular uptake and a new mechanistic perspective. ChemBioChem 2019, 20, 1023–1026. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [Green Version]
- Perni, M.; Flagmeier, P.; Limbocker, R.; Cascella, R.; Aprile, F.A.; Galvagnion, C.; Heller, G.T.; Meisl, G.; Chen, S.W.; Kumita, J.R.; et al. Multistep inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem. Biol. 2018, 13, 2308–2319. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, M.N.; Jones, S.R.; Shao, B.; Feibush, P.; McGuigan, M.; Tzodikov, N.; Feibush, B.; Sharkansky, I.; Snyder, B.; et al. Synthesis of Squalamine utilizing a readily accessible Spermidine equivalent. J. Org. Chem. 1998, 63, 8599–8603. [Google Scholar] [CrossRef]
- Rao, M.; Shinnar, A.; Noecker, L.; Chao, T.; Feibush, B.; Snyder, B.; Sharkansky, I.; Sarkahian, A.; Zhang, X.; Jones, S.; et al. Aminosterols from the Dogfish Shark Squalus acanthias. J. Nat. Prod. 2000, 63, 631–635. [Google Scholar] [CrossRef]
- Habchi, J.; Arosio, P.; Perni, M.; Costa, A.R.; Yagi-Utsumi, M.; Joshi, P.; Chia, S.; Cohen, S.I.A.; Müller, M.B.D.; Linse, S.; et al. An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic Aβ42 aggregates linked with Alzheimer’s disease. Sci. Adv. 2016, 2, e1501244. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, F.S.; Adamcik, J.; Jeong, J.S.; Lashuel, H.A.; Mezzenga, R.; Dietler, G. Influence of the β-sheet content on the mechanical properties of aggregates during amyloid fibrillization. Angew. Chem. Int. Ed. 2015, 54, 2462–2466. [Google Scholar] [CrossRef]
- Capitini, C.; Conti, S.; Perni, M.; Guidi, F.; Cascella, R.; De Poli, A.; Penco, A.; Relini, A.; Cecchi, C.; Chiti, F. TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS ONE 2014, 9, e86720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelisti, E.; Cascella, R.; Becatti, M.; Marrazza, G.; Dobson, C.M.; Chiti, F.; Stefani, M.; Cecchi, C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 2016, 6, 32721. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, E.; Cecchi, C.; Cascella, R.; Sgromo, C.; Becatti, M.; Dobson, C.M.; Chiti, F.; Stefani, M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci. 2012, 125, 2416–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limbocker, R.; Mannini, B.; Cataldi, R.; Chhangur, S.; Wright, A.K.; Kreiser, R.P.; Albright, J.A.; Chia, S.; Habchi, J.; Sormanni, P.; et al. Rationally Designed Antibodies as Research Tools to Study the Structure–Toxicity Relationship of Amyloid-β Oligomers. Int. J. Mol. Sci. 2020, 21, 4542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124542
Limbocker R, Mannini B, Cataldi R, Chhangur S, Wright AK, Kreiser RP, Albright JA, Chia S, Habchi J, Sormanni P, et al. Rationally Designed Antibodies as Research Tools to Study the Structure–Toxicity Relationship of Amyloid-β Oligomers. International Journal of Molecular Sciences. 2020; 21(12):4542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124542
Chicago/Turabian StyleLimbocker, Ryan, Benedetta Mannini, Rodrigo Cataldi, Shianne Chhangur, Aidan K. Wright, Ryan P. Kreiser, J. Alex Albright, Sean Chia, Johnny Habchi, Pietro Sormanni, and et al. 2020. "Rationally Designed Antibodies as Research Tools to Study the Structure–Toxicity Relationship of Amyloid-β Oligomers" International Journal of Molecular Sciences 21, no. 12: 4542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124542