Genetic Deletion of Vasohibin-2 Exacerbates Ischemia-Reperfusion-Induced Acute Kidney Injury

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
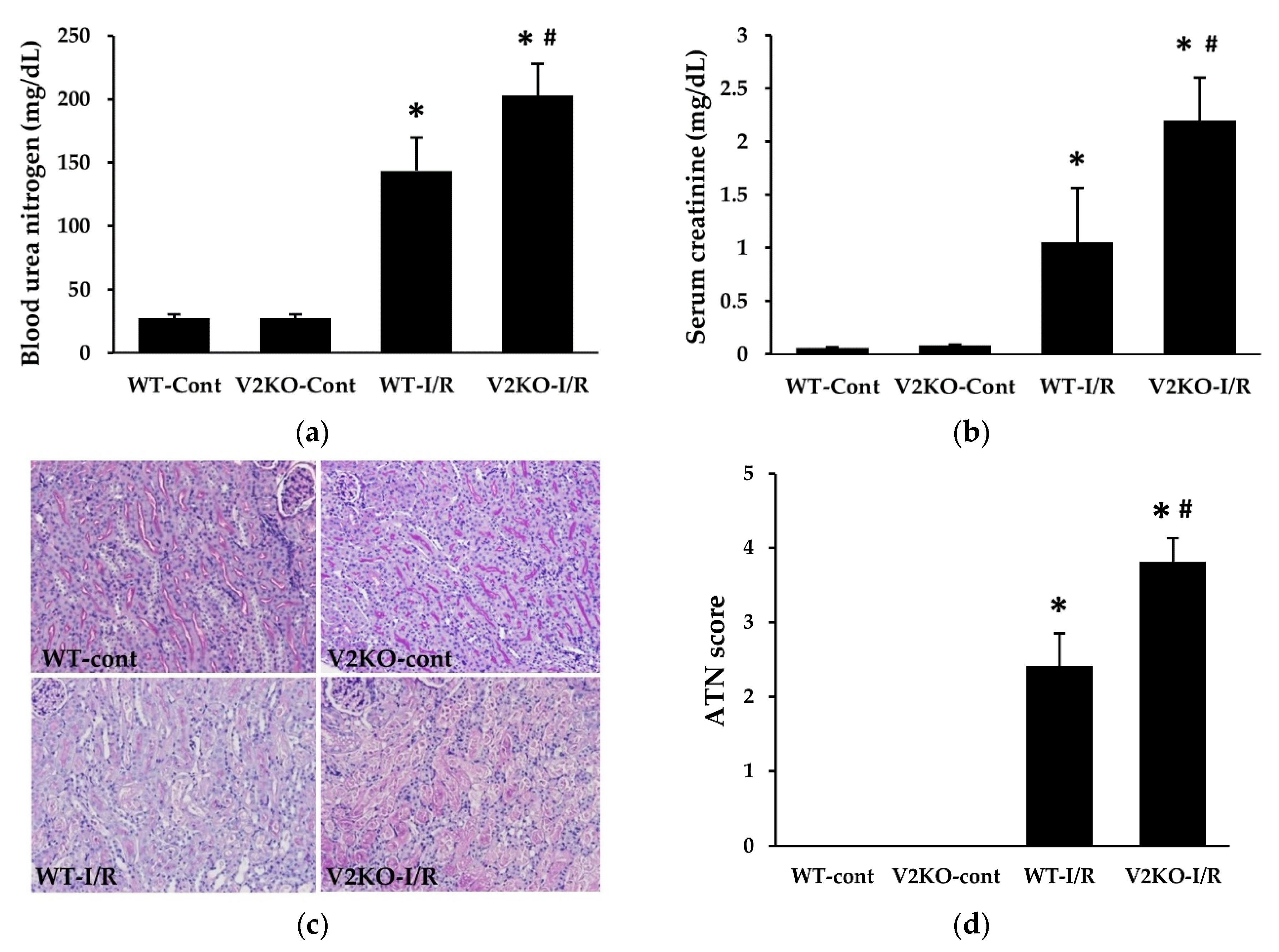
2.1. VASH2 Deficiency Accelerated I/R-Induced Renal Tubular Injury
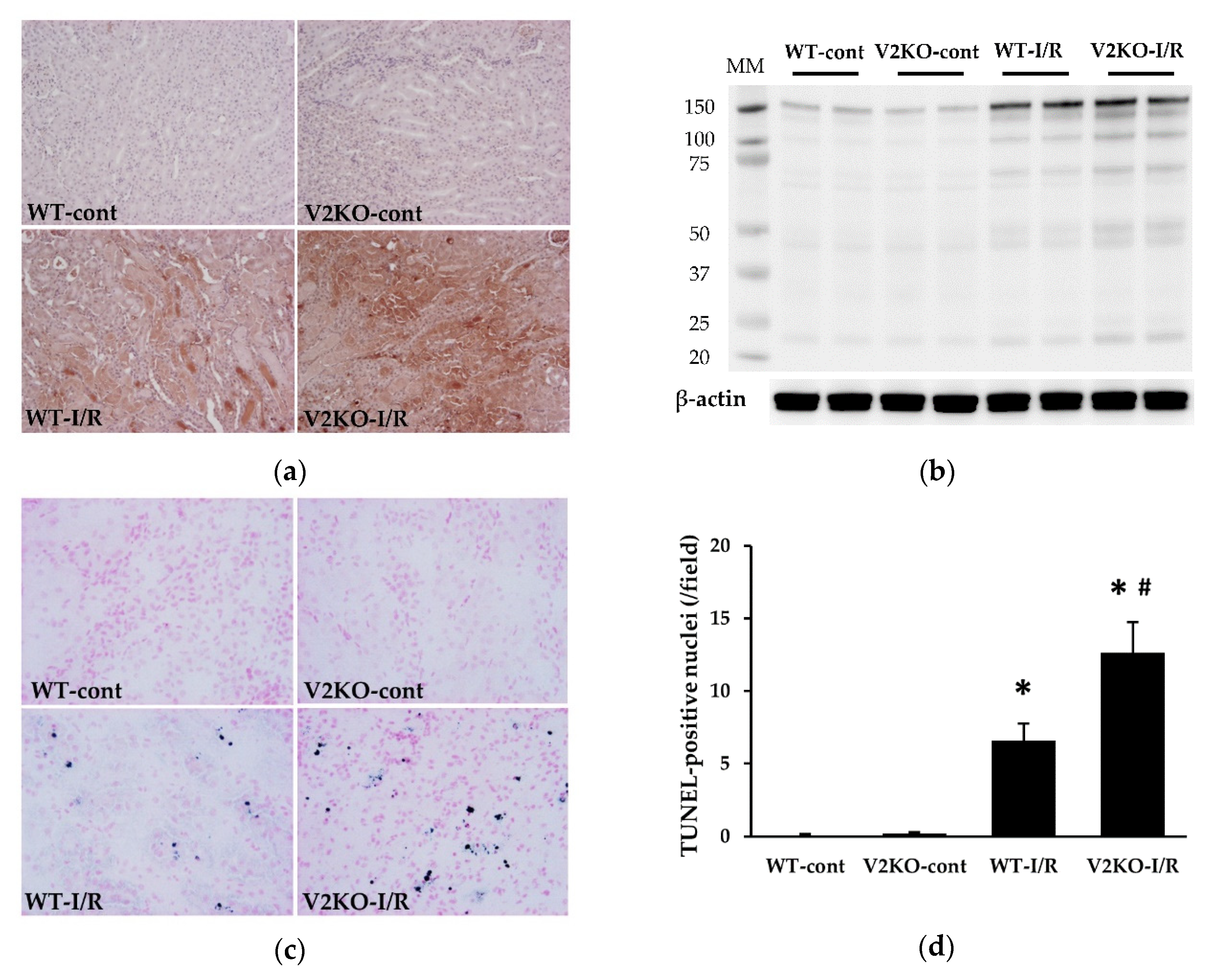
2.2. VASH2 Deficiency Promoted I/R-Induced Oxidative Stress Accumulation and Apoptosis
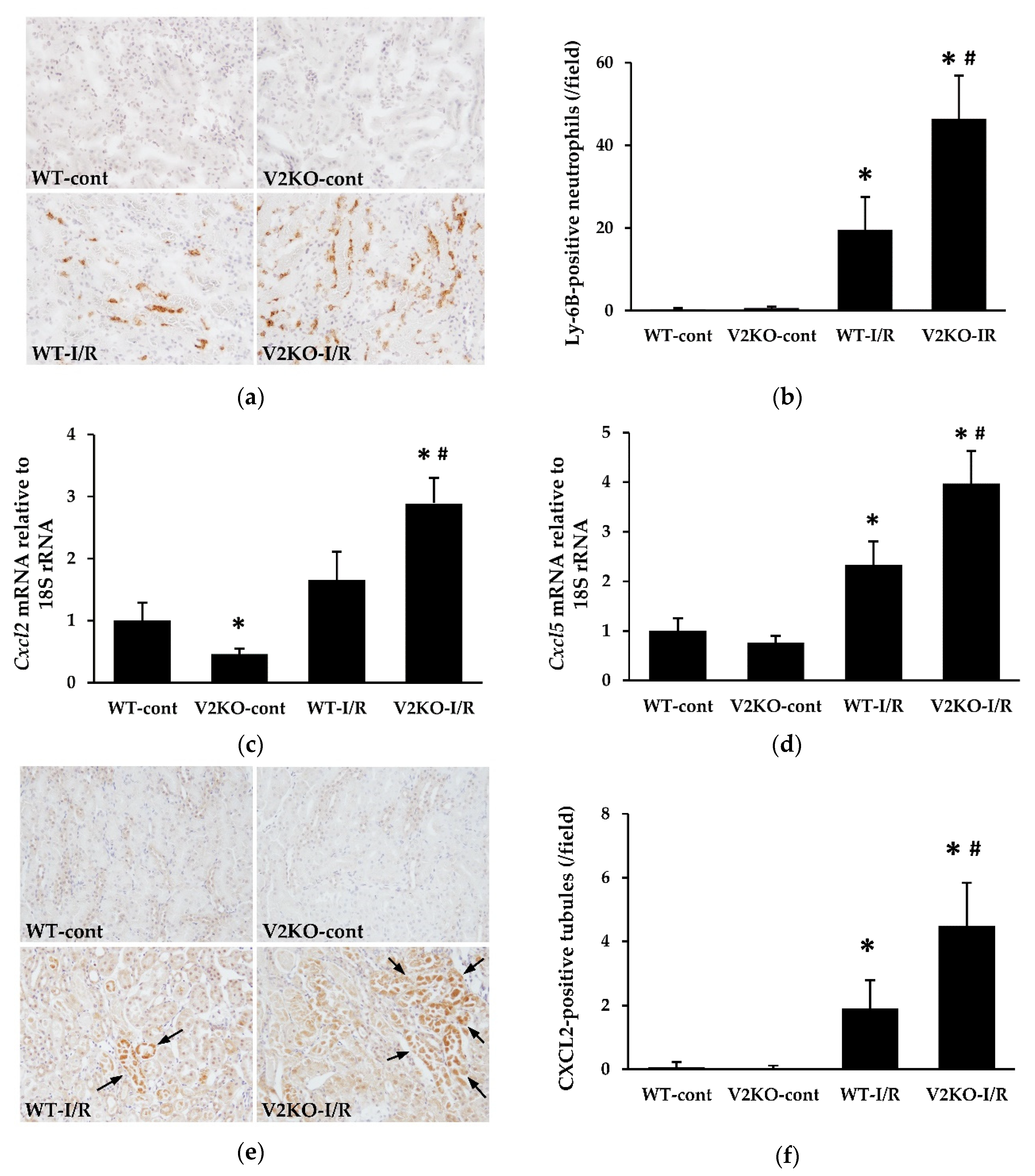
2.3. VASH2 Deficiency Enhanced Neutrophil Infiltration in I/R-Induced Renal Tubular Injury
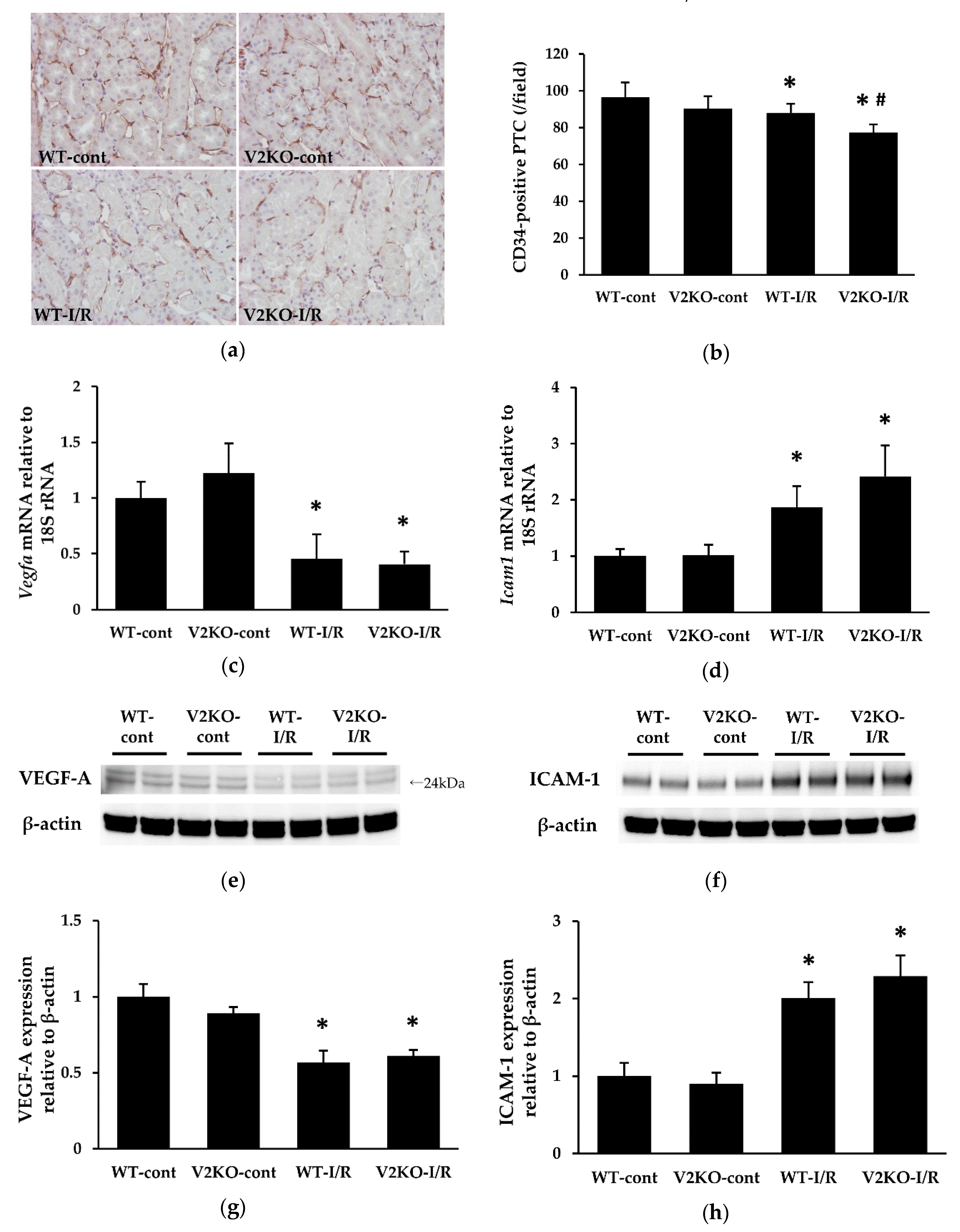
2.4. VASH2 Deficiency Accelerated PTC Loss in I/R-Induced Kidney Injury
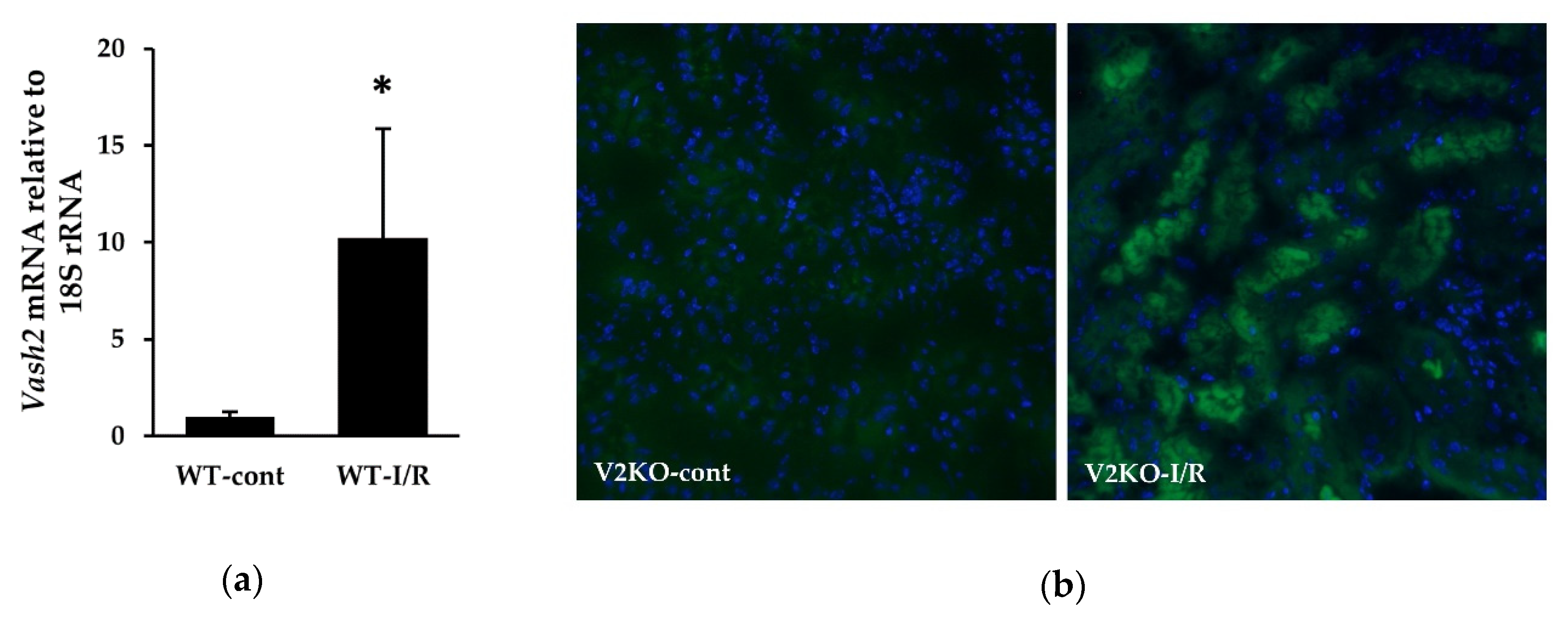
2.5. Endogenous VASH2 Expression in I/R-Induced Kidney Injury
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Protocols
4.2. Histological Analysis
4.3. Immunohistochemistry
4.4. Apoptosis Detection
4.5. Immunofluorescence
4.6. Immunoblotting
4.7. Real-Time PCR
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKI | Acute kidney injury |
| VASH2 | Vasohibin-2 |
| CKD | Chronic kidney disease |
| I/R | Ischemic reperfusion |
| NOS | Nitric oxide synthase |
| PTC | Peritubular capillary |
| VEGF | Vascular endothelial growth factor |
| WT | Wild-type |
| V2KO | VASH2 knockout |
| ATN | Acute tubular necrosis |
| MDA | Malondialdehyde |
| 4-HNE | 4-hydroxy-nonenal |
| TUNEL | Terminal uridine deoxynucleotidyl transferase-mediated dUTP nick-end labeling |
| CXCL | CXC chemokine ligand |
| ICAM-1 | Intercellular adhesion molecule-1 |
| β-gal | β-galactosidase |
| ROS | Reactive oxygen species |
| EMT | Epithelial-to-mesenchymal transition |
| ZEB2 | zinc finger E-box-binding homeobox 2 |
References
- Khwaja, A. Kdigo clinical practice guidelines for acute kidney injury. Nephron Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef] [PubMed]
- Heung, M.; Steffick, D.E.; Zivin, K.; Gillespie, B.W.; Banerjee, T.; Hsu, C.Y.; Powe, N.R.; Pavkov, M.E.; Williams, D.E.; Saran, R.; et al. Acute kidney injury recovery pattern and subsequent risk of ckd: An analysis of veterans health administration data. Am. J. Kidney Dis. 2016, 67, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Cho, K.C.; Hsu, C.Y. Acute kidney injury in the elderly: Predisposition to chronic kidney disease and vice versa. Nephron Clin. Pract. 2011, 119 (Suppl. 1), c19–c24. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Basile, D.P.; Yoder, M.C. Renal endothelial dysfunction in acute kidney ischemia reperfusion injury. J. Clin. Investig. 2014, 14, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Mitra, A.; Poole, B.; Falk, S.; Lucia, M.S.; Tayal, S.; Schrier, R. Endothelial nitric oxide synthase-deficient mice exhibit increased susceptibility to endotoxin-induced acute renal failure. Am. J. Physiol. Renal Physiol. 2004, 287, F1044–F1048. [Google Scholar] [CrossRef]
- Cristol, J.P.; Thiemermann, C.; Mitchell, J.A.; Walder, C.; Vane, J.R. Support of renal blood flow after ischaemic-reperfusion injury by endogenous formation of nitric oxide and of cyclo-oxygenase vasodilator metabolites. Br. J. Pharmacol. 1993, 109, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Wada, J.; Sato, Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat. Rev. Nephrol. 2020, 16, 289–303. [Google Scholar] [CrossRef]
- Tanabe, K.; Sato, Y.; Wada, J. Endogenous antiangiogenic factors in chronic kidney disease: Potential biomarkers of progression. Int. J. Mol. Sci. 2018, 19, 1859. [Google Scholar] [CrossRef] [Green Version]
- Dimke, H.; Sparks, M.A.; Thomson, B.R.; Frische, S.; Coffman, T.M.; Quaggin, S.E. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J. Am. Soc. Nephrol. 2015, 26, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Fredrich, K.; Chelladurai, B.; Leonard, E.C.; Parrish, A.R. Renal ischemia reperfusion inhibits vegf expression and induces adamts-1, a novel vegf inhibitor. Am. J. Physiol. Renal Physiol. 2008, 294, F928–F936. [Google Scholar] [CrossRef] [PubMed]
- Leonard, E.C.; Friedrich, J.L.; Basile, D.P. Vegf-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am. J. Physiol. Renal Physiol. 2008, 295, F1648–F1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, T.; Watanabe, K.; Yamashita, H.; Shimizu, K.; Miyashita, H.; Abe, M.; Moriya, T.; Ohta, H.; Sonoda, H.; Shimosegawa, T.; et al. Isolation and characterization of vasohibin-2 as a homologue of vegf-inducible endothelium-derived angiogenesis inhibitor vasohibin. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1051–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Koyanagi, T.; Suzuki, Y.; Saga, Y.; Kanomata, N.; Moriya, T.; Suzuki, M.; Sato, Y. Vasohibin-2 expressed in human serous ovarian adenocarcinoma accelerates tumor growth by promoting angiogenesis. Mol. Cancer Res. 2012, 10, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Zhang, Y.; Zhi, Q.; Tu, M.; Xu, Y.; Sun, J.; Wei, J.; Lu, Z.; Miao, Y.; Gao, W. Mir200-upregulated vasohibin 2 promotes the malignant transformation of tumors by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. Cell Commun. Signal. 2014, 12, 62. [Google Scholar] [CrossRef]
- Masuda, K.; Tanabe, K.; Ujike, H.; Hinamoto, N.; Miyake, H.; Tanimura, S.; Sugiyama, H.; Sato, Y.; Maeshima, Y.; Wada, J. Deletion of pro-angiogenic factor vasohibin-2 ameliorates glomerular alterations in a mouse diabetic nephropathy model. PLoS ONE 2018, 13, e0195779. [Google Scholar] [CrossRef]
- Arata, Y.; Tanabe, K.; Hinamoto, N.; Yamasaki, H.; Sugiyama, H.; Maeshima, Y.; Kanomata, N.; Sato, Y.; Wada, J. Immunohistochemistry of vasohibin-2 in human kidney disease: Implications in impaired glucose tolerance and reduced renal function. Acta Med. Okayama 2017, 71, 369–380. [Google Scholar] [PubMed]
- Basile, D.P.; Donohoe, D.; Roethe, K.; Osborn, J.L. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am. J. Physiol. Renal Physiol. 2001, 281, F887–F899. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Miyashita, H.; Suzuki, Y.; Kobayashi, M.; Watanabe, K.; Sonoda, H.; Ohta, H.; Fujiwara, T.; Shimosegawa, T.; Sato, Y. Distinctive localization and opposed roles of vasohibin-1 and vasohibin-2 in the regulation of angiogenesis. Blood 2009, 113, 4810–4818. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, P. Update on mechanisms of ischemic acute kidney injury. J. Am. Soc. Nephrol. 2006, 17, 1503–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.K.; Bajwa, A.; Ye, H.; Vergis, A.L.; Awad, A.S.; Kharel, Y.; Lynch, K.R.; Okusa, M.D. Divergent roles of sphingosine kinases in kidney ischemia-reperfusion injury. Kidney Int. 2009, 75, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, H.; Dragun, D.; Miethke, A.; Park, J.K.; Weis, A.; Lippoldt, A.; Gross, V.; Luft, F.C. Antisense oligonucleotides for icam-1 attenuate reperfusion injury and renal failure in the rat. Kidney Int. 1996, 50, 473–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyanagi, T.; Saga, Y.; Takahashi, Y.; Suzuki, Y.; Suzuki, M.; Sato, Y. Downregulation of vasohibin-2, a novel angiogenesis regulator, suppresses tumor growth by inhibiting angiogenesis in endometrial cancer cells. Oncol. Lett. 2013, 5, 1058–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmer, R.; Haase, A.; Merkert, S.; Cui, W.; Palecek, J.; Ran, C.; Kirschning, A.; Scheper, T.; Glage, S.; Miller, K.; et al. Long term expansion of undifferentiated human ips and es cells in suspension culture using a defined medium. Stem Cell Res. 2010, 5, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, H.; Watanabe, T.; Hayashi, H.; Suzuki, Y.; Nakamura, T.; Ito, S.; Ono, M.; Hoshikawa, Y.; Okada, Y.; Kondo, T.; et al. Angiogenesis inhibitor vasohibin-1 enhances stress resistance of endothelial cells via induction of sod2 and sirt1. PLoS ONE 2012, 7, e46459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimura, S.; Tanabe, K.; Miyake, H.; Masuda, K.; Tsushida, K.; Morioka, T.; Sugiyama, H.; Sato, Y.; Wada, J. Renal tubular injury exacerbated by vasohibin-1 deficiency in a murine cisplatin-induced acute kidney injury model. Am. J. Physiol. Cell Physiol. 2019, 317, F264–F274. [Google Scholar] [CrossRef]
- Suzuki, Y.; Kitahara, S.; Suematsu, T.; Oshima, M.; Sato, Y. Requisite role of vasohibin-2 in spontaneous gastric cancer formation and accumulation of cancer-associated fibroblasts. Cancer Sci. 2017, 108, 2342–2351. [Google Scholar] [CrossRef]
- Kim, J.C.; Kim, K.T.; Park, J.T.; Kim, H.J.; Sato, Y.; Kim, H.S. Expression of vasohibin-2 in pancreatic ductal adenocarcinoma promotes tumor progression and is associated with a poor clinical outcome. Hepatogastroenterology 2015, 62, 251–256. [Google Scholar]
- Ninomiya, Y.; Ozawa, S.; Oguma, J.; Kazuno, A.; Nitta, M.; Kajiwara, H.; Sato, Y. Expression of vasohibin-1 and -2 predicts poor prognosis among patients with squamous cell carcinoma of the esophagus. Oncol. Lett. 2018, 16, 5265–5274. [Google Scholar] [CrossRef]
- Kozako, T.; Matsumoto, N.; Kuramoto, Y.; Sakata, A.; Motonagare, R.; Aikawa, A.; Imoto, M.; Toda, A.; Honda, S.; Shimeno, H.; et al. Vasohibin induces prolyl hydroxylase-mediated degradation of hypoxia-inducible factor-1α in human umbilical vein endothelial cells. FEBS Lett. 2012, 586, 1067–1072. [Google Scholar] [CrossRef] [Green Version]
- Choudhry, H.; Harris, A.L. Advances in hypoxia-inducible factor biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Maeshima, Y.; Sato, Y.; Wada, J. Antiangiogenic therapy for diabetic nephropathy. Biomed. Res. Int. 2017, 2017, 5724069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Kauttu, T.; Seppanen, H.; Vainionpaa, S.; Ye, Y.; Wang, S.; Mustonen, H.; Puolakkainen, P. Vasohibin-1 and vasohibin-2 expression in gastric cancer cells and tams. Med. Oncol. 2012, 29, 2718–2726. [Google Scholar] [CrossRef]
- Norita, R.; Suzuki, Y.; Furutani, Y.; Takahashi, K.; Yoshimatsu, Y.; Podyma-Inoue, K.A.; Watabe, T.; Sato, Y. Vasohibin-2 is required for epithelial-mesenchymal transition of ovarian cancer cells by modulating transforming growth factor-beta signaling. Cancer Sci. 2017, 108, 419–426. [Google Scholar] [CrossRef]
- Sato, Y. The vasohibin family: A novel family for angiogenesis regulation. J. Biochem. 2013, 153, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Tamura, Y.; Lanaspa, M.A.; Miyazaki, M.; Suzuki, N.; Sato, W.; Maeshima, Y.; Schreiner, G.F.; Villarreal, F.J.; Johnson, R.J.; et al. Epicatechin limits renal injury by mitochondrial protection in cisplatin nephropathy. Am. J. Physiol. Renal Physiol. 2012, 303, F1264–F1274. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Maeshima, Y.; Ichinose, K.; Kitayama, H.; Takazawa, Y.; Hirokoshi, K.; Kinomura, M.; Sugiyama, H.; Makino, H. Endostatin peptide, an inhibitor of angiogenesis, prevents the progression of peritoneal sclerosis in a mouse experimental model. Kidney Int. 2007, 71, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Lanaspa, M.A.; Kitagawa, W.; Rivard, C.J.; Miyazaki, M.; Klawitter, J.; Schreiner, G.F.; Saleem, M.A.; Mathieson, P.W.; Makino, H.; et al. Nicorandil as a novel therapy for advanced diabetic nephropathy in the enos-deficient mouse. Am. J. Physiol. Renal Physiol. 2012, 302, F1151–F1160. [Google Scholar] [CrossRef]
- Tsushida, K.; Tanabe, K.; Masuda, K.; Tanimura, S.; Miyake, H.; Arata, Y.; Sugiyama, H.; Wada, J. Estrogen-related receptor alpha is essential for maintaining mitochondrial integrity in cisplatin-induced acute kidney injury. Biochem. Biophys. Res. Commun. 2018, 498, 918–924. [Google Scholar] [CrossRef]
- Hinamoto, N.; Maeshima, Y.; Yamasaki, H.; Nasu, T.; Saito, D.; Watatani, H.; Ujike, H.; Tanabe, K.; Masuda, K.; Arata, Y.; et al. Exacerbation of diabetic renal alterations in mice lacking vasohibin-1. PLoS ONE 2014, 9, e107934. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyake, H.; Tanabe, K.; Tanimura, S.; Nakashima, Y.; Morioka, T.; Masuda, K.; Sugiyama, H.; Sato, Y.; Wada, J. Genetic Deletion of Vasohibin-2 Exacerbates Ischemia-Reperfusion-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2020, 21, 4545. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124545
Miyake H, Tanabe K, Tanimura S, Nakashima Y, Morioka T, Masuda K, Sugiyama H, Sato Y, Wada J. Genetic Deletion of Vasohibin-2 Exacerbates Ischemia-Reperfusion-Induced Acute Kidney Injury. International Journal of Molecular Sciences. 2020; 21(12):4545. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124545
Chicago/Turabian StyleMiyake, Hiromasa, Katsuyuki Tanabe, Satoshi Tanimura, Yuri Nakashima, Tomoyo Morioka, Kana Masuda, Hitoshi Sugiyama, Yasufumi Sato, and Jun Wada. 2020. "Genetic Deletion of Vasohibin-2 Exacerbates Ischemia-Reperfusion-Induced Acute Kidney Injury" International Journal of Molecular Sciences 21, no. 12: 4545. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124545