Pharmacogenomics and Pharmacogenetics in Osteosarcoma: Translational Studies and Clinical Impact


Abstract
:
1. Introduction
2. Germline Pharmacogenetic Markers Associated with Risk to Develop HGOS
2.1. Common Genetic Variants
2.2. Rare Genetic Variants
2.3. Meta-Analyses
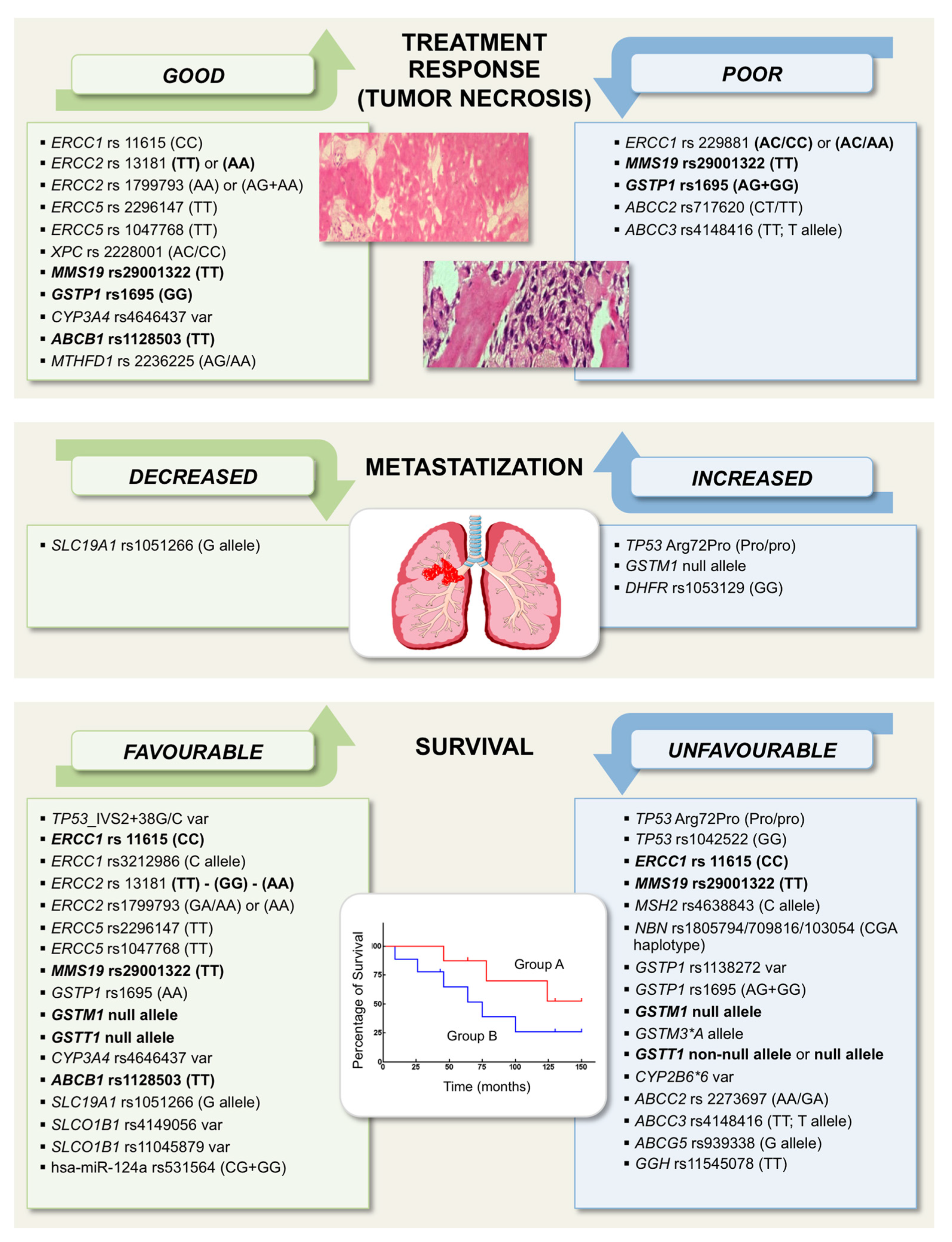
3. Pharmacogenetic and Pharmacogenomic Markers Associated with Treatment Response and/or Survival
3.1. TP53 and Its Regulators MDM2 and Mouse Double Minute 4 (MDM4)
3.2. DNA Repair-Related Genes
3.3. Genes Involved in Drug Metabolism
- The GSTM1 null allele was reported to be associated with an increased relapse rate in non-metastatic patients, and with poor survival in metastatic patients [87].
- The non-null allele of GSTT1 was correlated with poor survival in metastatic patients [87]. On the other hand, poor event-free survival was shown to be associated with the GSTT1 null allele in a study on Caucasian patients [85]. Partially in contrast with these findings, GSTM1 and/or GSTT1 null genotypes were reported to be associated with better survival rates in a Chinese study [86].
3.4. Genes Involved in Drug Transport
3.5. Polymorphisms of Genes Involved in Antifolate Drugs Metabolism
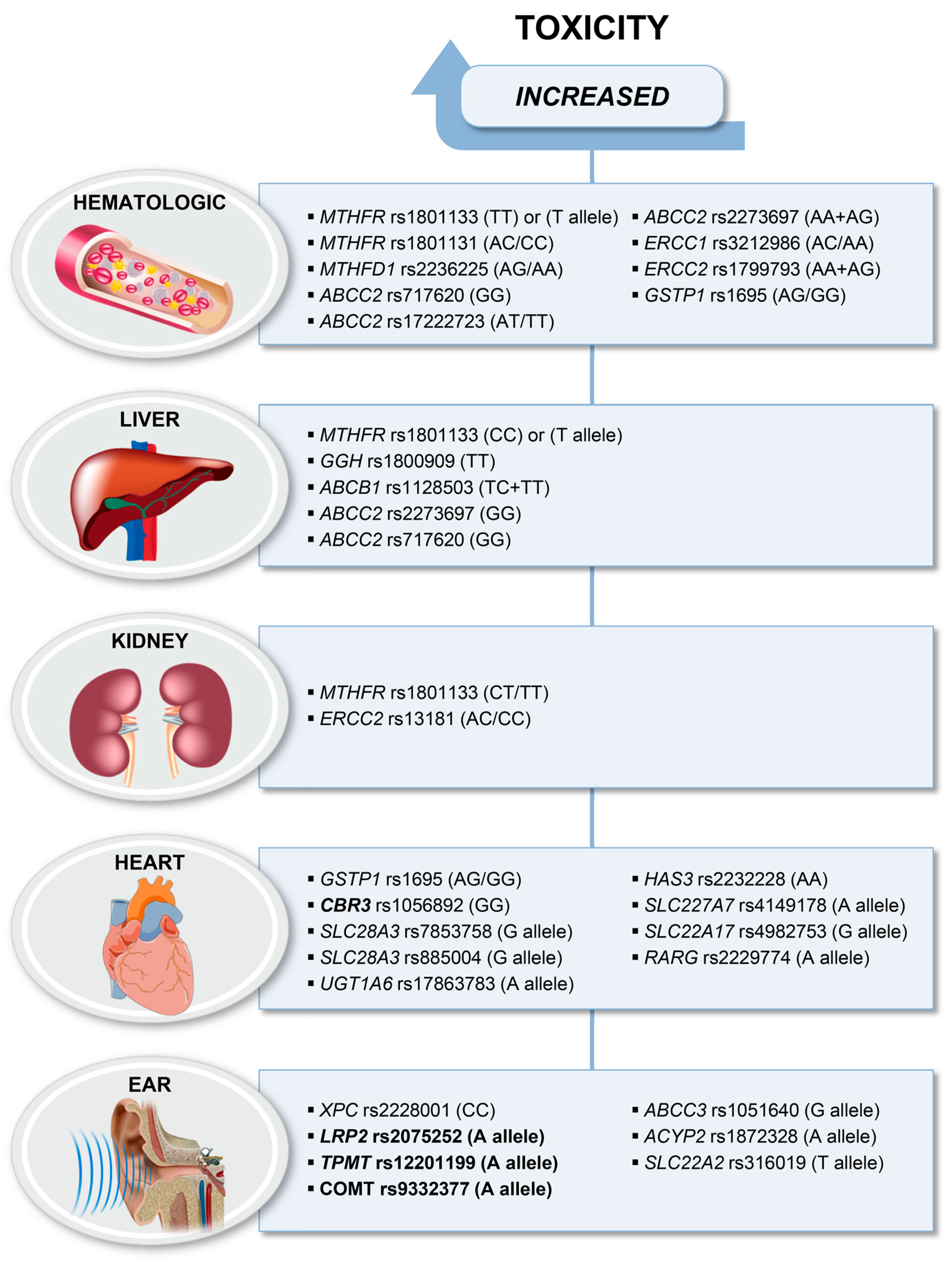
4. Gene Polymorphisms Associated with Toxicities
4.1. Haematological Toxicities
4.2. Liver Toxicity
4.3. Nephrotoxicity
4.4. Cardiotoxicity
4.5. Ototoxicity
4.6. Other Treatment-Related Toxicities
5. Polymorphisms of Non-Coding RNAs
6. Preclinical Models and Public Datasets
7. Social-Economical Aspects of Pharmacogenomics-Based Precision Medicine in HGOS
8. Future Directions
8.1. Genetic Testing and Clinical Trials
8.2. Epigenetics
- It is mandatory to determine and validate the actual value of miRNAs and lncRNAs that have been highlighted in HGOS experimental models and clinical samples as possible therapeutic targets;
- The design of non-coding RNA-based or targeted treatments with sufficient efficiency, delivery, therapeutic effects, and safety profiles to be transferred to the clinic is mostly still at a preclinical phase of development.
8.3. Immunopharmacogenomics
8.4. Paired Sequencing
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ABCB1 | ATP-binding cassette, subfamily B (MDR/TAP), member 1 |
| ABCC2 | ATP-binding cassette, subfamily C (CFTR/MRP), member 2 |
| ABCC3 | ATP-binding cassette, subfamily C (CFTR/MRP), member 3 |
| ABCC5 | ATP-binding cassette, subfamily C (CFTR/MRP), member 5 |
| ACT | anthracycline-induced cardiotoxicity |
| ACYP2 | acylphosphatase 2 |
| ADME | absorption, distribution, metabolism, excretion |
| AKT | AKT serine/threonine kinase |
| ALT | alanine transaminase |
| ATM | ataxia telangiectasia mutated) |
| ATR | ataxia telangiectasia and Rad3 related |
| ATRX | ATRX chromatin remodeler |
| AURKB | Aurora kinase B |
| B-Raf | B-Raf proto-oncogene, serine/threonine kinase |
| BRCA1 | breast related cancer antigen 1 |
| BRCA 2 | breast related cancer antigen 2 |
| Cas9 | CRISPR associated protein 9 |
| CBRs | carbonyl reductases |
| CCNE1 | cyclin E1 |
| CDK4 | cyclin-dependent kinase 4 |
| CDK6 | cyclin-dependent kinase 6 |
| CDKN2A | cyclin dependent kinase inhibitor 2A |
| CHST12 | carbohydrate sulfotransferase 12 |
| CI | confidence interval |
| CN | copy number |
| COMT | catechol O-methyltransferase |
| COSMIC | catalogue of somatic mutations in cancer |
| COX8A | cytochrome C oxidase subunit 8A |
| CRISPR | clustered regularly interspaced palindromic repeats |
| CTLA-4 | cytotoxic T-lymphocyte antigen 4 |
| CYP | cytochrome P450 |
| DHFR | dihydrofolate reductase |
| DMEs | drug metabolizing enzymes |
| ERCC 1 | excision repair cross-complementation group 1 |
| ERCC 2 | excision repair cross-complementation group 2 |
| ERCC 5 | excision repair cross-complementation group 5 |
| ERK | extracellular signal-regulated kinase |
| EZH1 | enhancer of zeste 1 polycomb repressive complex 2 subunit |
| EZH2 | enhancer of zeste 2 polycomb repressive complex 2 subunit |
| FGFR | fibroblast growth factor receptor |
| GATA1 | GATA binding protein 1 |
| GGH | gamma-glutamyl hydrolase |
| GRM4 | glutamate metabotropic receptor 4 |
| GST | glutathione S-transferase |
| GSTM1 | glutathione S-transferase M1 |
| GSTM3 | glutathione S-transferase M3 |
| GSTP1 | glutathione S-transferase P1 |
| GSTT1 | glutathione S-transferase T1 |
| GWAS | genome-wide association study |
| HAS3 | hyaluron synthase 3 |
| HGOS | high-grade osteosarcoma |
| IL-8 | interleukin-8 |
| INFORM | INdividualized therapy FOr Relapsed Malignancies |
| lncRNAs | long non-coding RNAs |
| LP | likely pathogenic |
| LRP2 | LDL receptor related protein 2, or megalin |
| MAPK | mitogen-activated protein kinase |
| MDM2 | mouse double minute 2 (also known as murine double minute 2) |
| MDM4 | mouse double minute 4 |
| MEN1 | menin 1 |
| miRNAs | microRNAs |
| MMS19 | nucleotide excision repair homolog |
| MSH2 | mutS homolog 2 |
| MTHFD1 | methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1 |
| MTHFR | methylenetetrahydrofolate reductase |
| mTOR | mammalian target of rapamycin (mechanistic target of rapamycin) |
| MTX | methotrexate |
| NBN | nibrin |
| NER | nucleotide excision repair |
| NF1 | neurofibromin 1 |
| NFE | non-Finish European |
| NGS | next generation sequencing |
| NSG | NOD Scid gamma |
| odd ratio | OR |
| P | pathogenic |
| PARP1 | poly (ADP-ribose) polymerase 1 |
| PD-1 | programmed cell death-1 |
| PD-L1 | programmed death-ligand 1 |
| PDXs or PDTXs | patient-derived tumor xenografts |
| PI3K | phosphatidylinositol 3 kinase |
| PMS2 | PMS1 homolog 2, mismatch repair system component |
| POT1 | protection of telomeres 1 |
| PRCKG | protein kinase CGMP-dependent 1 |
| PTEN | phosphatase and tensin homolog |
| RAD21 | RAD21 cohesin complex component |
| RARG | retinoic acid receptorγ |
| RB1 | RB transcriptional corepressor 1 (also known as retinoblastoma gene 1) |
| RECQL2 | RecQ like helicase 2 |
| RECQL3 | RecQ like helicase 3 |
| RECQL4 | RecQ like helicase 4 |
| RECQL5 | RecQ like helicase 5 |
| RFC | reduced folate carrier |
| RPS19 | ribosomal protein S19 |
| RPS26 | ribosomal protein S26 |
| RUNX1 | RUNX family transcription factor 1 |
| SATB2 | SATB homeobox 2 |
| SCNAs | somatic copy number aberrations |
| SLC19A1 | solute carrier family 19 (folate transporter) member 1 (also known as reduced folate carrier, RFC or reduced folate carrier 1, RFC1) |
| SLC22A17 | solute carrier family 22 member 17 |
| SLC22A2 | solute carrier family 22 member 2 |
| SLC22A7 | solute carrier family 22 member 7 |
| SLC22A8 | solute carrier family 22 member 8 |
| SLC28A3 | solute carrier family 28 member 3 |
| SLCO1B1 | solute carrier organic anion transporter family member 1B1 |
| SMARCA4 | SWI/SNF related matrix associated actin dependent regulator of chromatin, subfamily A, member 4 |
| SMARCB1 | SWI/SNF related matrix associated actin dependent regulator of chromatin, subfamily B, member 1 |
| SNPs | single nucleotide polymorphisms |
| SNV | somatic nucleotide variants |
| SV | structural variations |
| TARGET | Therapeutically Applicable Research to Generate Effective Treatments |
| TGFB1 | transforming growth factor beta 1 |
| TMB | tumor mutational burden |
| TNF | tumor necrosis factor |
| TP53 | tumor protein 53 |
| TPMT | thiopurine S-methyltransferase |
| TS | thymidylate synthase |
| TSC | TSC complex subunit |
| UGT1A6 | UDP glucuronosyltransferase 1 A6 |
| UGT2B15 | UDP glucuronosyltransferase family 2 member B15 |
| VEGF | vascular endothelial growth factor |
| VHL | Von Hippel-Lindau tumor suppressor |
| WES | whole-exome sequencing |
| WGS | whole-genome sequencing |
| XPC | xeroderma pigmentosum complementation group C |
| XRCC3 | X-ray repair cross complementing 3 |
References
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [Green Version]
- Picci, P. Osteosarcoma. In Diagnosis of Musculoskeletal Tumors and Tumor-Like Conditions, 2nd ed.; Picci, P., Manfrini, M., Donati, D., Gambarotti, M., Righi, A., Vanel, D., Dei Tos, A., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2020; pp. 185–212. [Google Scholar]
- Whelan, J.S.; Davis, L.E. Osteosarcoma, Chondrosarcoma, and Chordoma. J. Clin. Oncol. 2018, 36, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.J.; Geller, D.S.; Gill, J.; Lewis, V.O.; Gorlick, R. Current and future therapeutic approaches for osteosarcoma. Expert Rev. Anticancer. Ther. 2017, 18, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Kager, L.; Tamamyan, G.; Bielack, S. Novel insights and therapeutic interventions for pediatric osteosarcoma. Futur. Oncol. 2017, 13, 357–368. [Google Scholar] [CrossRef]
- Simpson, E.; Brown, H.L. Understanding osteosarcomas. J. Am. Acad. Physician Assist. 2018, 31, 15–19. [Google Scholar] [CrossRef]
- Ferrari, S.; Serra, M. An update on chemotherapy for osteosarcoma. Expert Opin. Pharmacother. 2015, 16, 2727–2736. [Google Scholar] [CrossRef] [PubMed]
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.C.W.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and prognosis with osteosarcoma: Outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Hattinger, C.M.; Vella, S.; Tavanti, E.; Fanelli, M.; Picci, P.; Serra, M. Pharmacogenomics of second-line drugs used for treatment of unresponsive or relapsed osteosarcoma patients. Pharmacogenomics 2016, 17, 2097–2114. [Google Scholar] [CrossRef] [Green Version]
- Roden, D.M.; McLeod, H.L.; Relling, M.V.; Williams, M.S.; Mensah, G.A.; Peterson, J.F.; Van Driest, S.L. Pharmacogenomics. Lancet 2019, 394, 521–532. [Google Scholar] [CrossRef]
- Serra, M.; Hattinger, C.M. The pharmacogenomics of osteosarcoma. Pharm. J. 2017, 17, 11–20. [Google Scholar] [CrossRef]
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating Functional Genomics into Rational Therapeutics. Science 1999, 286, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, L.; Zhang, L.; Peng, Y.; Huang, R.S. Pharmacogenetics and pharmacogenomics: A bridge to individualized cancer therapy. Pharmacogenomics 2013, 14, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianferante, D.M.; Mirabello, L.; Savage, S.A. Germline and somatic genetics of osteosarcoma—Connecting aetiology, biology and therapy. Nat. Rev. Endocrinol. 2017, 13, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Macari, E.R.; Jessop, L.; Ellis, S.R.; Myers, T.; Giri, N.; Taylor, A.M.; McGrath, K.E.; Humphries, J.M.; Ballew, B.J.; et al. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families. Blood 2014, 124, 24–32. [Google Scholar] [CrossRef]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet. 2018, 103, 930–947. [Google Scholar] [CrossRef] [Green Version]
- Rickel, K.; Fang, F.; Tao, J. Molecular genetics of osteosarcoma. Bone 2016, 102, 69–79. [Google Scholar] [CrossRef]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef] [Green Version]
- Hameed, M.; Mandelker, D. Tumor Syndromes Predisposing to Osteosarcoma. Adv. Anat. Pathol. 2018, 25, 217–222. [Google Scholar] [CrossRef]
- Wang, L.L.; Levy, M.L.; Lewis, R.A.; Chintagumpala, M.M.; Lev, R.; Rogers, M.; Plon, S.E. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am. J. Med. Genet. 2001, 102, 11–17. [Google Scholar] [CrossRef]
- Siitonen, H.A.; Sotkasiira, J.; Biervliet, M.; Benmansour, A.; Capri, Y.; Cormier-Daire, V.; Crandall, B.; Hannula-Jouppi, K.; Hennekam, R.; Herzog, D.; et al. The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 2008, 17, 151–158. [Google Scholar] [CrossRef]
- Calvert, G.T.; Randall, R.L.; Jones, K.B.; Cannon-Albright, L.A.; Lessnick, S.; Schiffman, J.D. At-Risk Populations for Osteosarcoma: The Syndromes and Beyond. Sarcoma 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.M.; Federman, N.; Khabbaze, Y.; Schwartz, C.L.; Hilliard, L.M.; Clark, J.I.; Vlachos, A. Osteogenic Sarcoma Associated with Diamond-Blackfan Anemia: A Report From the Diamond-Blackfan Anemia Registry. J. Pediatr. Hematol. 2001, 23, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachos, A.; Rosenberg, P.S.; Atsidaftos, E.; Kang, J.; Onel, K.; Sharaf, R.N.; Alter, B.P.; Lipton, J.M. Increased risk of colon cancer and osteogenic sarcoma in Diamond-Blackfan anemia. Blood 2018, 132, 2205–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, M.; Cunniff, C.M. Bloom Syndrome. In GeneReviews; University of Washington: Seattle, WA, USA, 2006; pp. 1993–2020. [Google Scholar]
- Yu, W.; Clyne, M.; Khoury, M.J.; Gwinn, M. Phenopedia and Genopedia: Disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics 2009, 26, 145–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabello, L.; Yu, K.; Berndt, S.I.; Burdette, L.; Wang, Z.; Chowdhury, S.; Teshome, K.; Uzoka, A.; Hutchinson, A.; Grotmol, T.; et al. A comprehensive candidate gene approach identifies genetic variation associated with osteosarcoma. BMC Cancer 2011, 11, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, S.A.; Mirabello, L.; Wang, Z.; Gastier-Foster, J.M.; Gorlick, R.; Khanna, C.; Flanagan, A.M.; Tirabosco, R.; Andrulis, I.L.; Wunder, J.S.; et al. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat. Genet. 2013, 45, 799–803. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Chen, H.; Shao, L.; Dong, Y. GRM4 gene polymorphism is associated with susceptibility and prognosis of osteosarcoma in a Chinese Han population. Med. Oncol. 2014, 31, 50. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhao, J.; He, M.; Fowdur, M.; Jiang, T.; Luo, S. Association of GRM4 gene polymorphisms with susceptibility and clinicopathological characteristics of osteosarcoma in Guangxi Chinese population. Tumour Biol. 2016, 37, 1105–1112. [Google Scholar] [CrossRef]
- Yang, Y.; Basu, S.; Mirabello, L.; Spector, L.; Zhang, L. A Bayesian Gene-Based Genome-Wide Association Study Analysis of Osteosarcoma Trio Data Using a Hierarchically Structured Prior. Cancer Inform. 2018, 17, 1176935118775103. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Mai, P.L.; Gastier-Foster, J.M.; Gorlick, R.; Khanna, C.; Patino-Garcia, A.; Sierrasesumaga, L.; Lecanda, F.; Andrulis, I.L.; et al. Germline TP53 variants and susceptibility to osteosarcoma. J. Natl. Cancer Inst. 2015, 107, djv101. [Google Scholar] [CrossRef]
- Ballinger, M.L.; Goode, D.L.; Ray-Coquard, I.; James, P.A.; Mitchell, G.; Niedermayr, E.; Puri, A.; Schiffman, J.D.; Dite, G.S.; Cipponi, A.; et al. Monogenic and polygenic determinants of sarcoma risk: An international genetic study. Lancet Oncol. 2016, 17, 1261–1271. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, F.; Zhang, Z.; Shao, Z. Association between TP53 rs1042522 gene polymorphism and the risk of malignant bone tumors: A meta-analysis. Biosci. Rep. 2019, 39, BSR20181832. [Google Scholar] [CrossRef]
- Moghimi, M.; Sobhan, M.R.; Jarahzadeh, M.H.; Morovati-Sharifabad, M.; Aghili, K.; Ahrar, H.; Zare-Shehneh, M.; Neamatzadeh, H. Association of GSTM1, GSTT1, GSTM3, and GSTP1 Genes Polymorphisms with Susceptibility to Osteosarcoma: A Case-Control Study and Meta-Analysis. Asian Pac. J. Cancer Prev. 2019, 20, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Barys, L.; O‘Reilly, T.; Young, S.; Gorbatcheva, B.; Monahan, J.; Zumstein-Mecker, S.; Choong, P.F.; Dickinson, I.; Crowe, P.; et al. Comprehensive mapping of p53 pathway alterations reveals an apparent role for both SNP309 and MDM2 amplification in sarcomagenesis. Clin. Cancer Res. 2011, 17, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Toffoli, G.; Biason, P.; Russo, A.; De Mattia, E.; Cecchin, E.; Hattinger, C.M.; Pasello, M.; Alberghini, M.; Ferrari, C.; Scotlandi, K.; et al. Effect of TP53 Arg72Pro and MDM2 SNP309 polymorphisms on the risk of high-grade osteosarcoma development and survival. Clin. Cancer Res. 2009, 15, 3550–3556. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, Z.; Jing, P.; Shao, L.; Chen, L.; He, X.; Gong, W. Effects of murine double minute 2 polymorphisms on the risk and survival of osteosarcoma: A systemic review and meta-analysis. Tumour Biol. 2014, 35, 1649–1652. [Google Scholar] [CrossRef]
- Bilbao-Aldaiturriaga, N.; Askaiturrieta, Z.; Granado-Tajada, I.; Goricar, K.; Dolzan, V.; For The Slovenian Osteosarcoma Study, G.; Garcia-Miguel, P.; Garcia de Andoin, N.; Martin-Guerrero, I.; Garcia-Orad, A. A systematic review and meta-analysis of MDM2 polymorphisms in osteosarcoma susceptibility. Pediatr. Res. 2016, 80, 472–479. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z. Systematic meta-analysis of genetic variants associated with osteosarcoma susceptibility. Medicine (Baltimore) 2018, 97, e12525. [Google Scholar] [CrossRef] [PubMed]
- Ognjanovic, S.; Olivier, M.; Bergemann, T.L.; Hainaut, P. Sarcomas in TP53 germline mutation carriers: A review of the IARC TP53 database. Cancer 2012, 118, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Pakos, E.E.; Kyzas, P.A.; Ioannidis, J.P. Prognostic significance of TP53 tumor suppressor gene expression and mutations in human osteosarcoma: A meta-analysis. Clin. Cancer Res. 2004, 10, 6208–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoenen, E.; Curl, A.; Iwakuma, T. TP53 in bone and soft tissue sarcomas. Pharmacol. Ther. 2019, 202, 149–164. [Google Scholar] [CrossRef]
- Overholtzer, M.; Rao, P.H.; Favis, R.; Lu, X.Y.; Elowitz, M.B.; Barany, F.; Ladanyi, M.; Gorlick, R.; Levine, A.J. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc. Natl. Acad. Sci. USA 2003, 100, 11547–11552. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; Mejia-Hernandez, J.O.; Vijayakumaran, R.; Keam, S.P.; Haupt, Y. The long and the short of it: The MDM4 tail so far. J. Mol. Cell Biol. 2019, 11, 231–244. [Google Scholar] [CrossRef]
- Duhamel, L.A.; Ye, H.; Halai, D.; Idowu, B.D.; Presneau, N.; Tirabosco, R.; Flanagan, A.M. Frequency of Mouse Double Minute 2 (MDM2) and Mouse Double Minute 4 (MDM4) amplification in parosteal and conventional osteosarcoma subtypes. Histopathology 2012, 60, 357–359. [Google Scholar] [CrossRef]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef]
- Lenos, K.; Grawenda, A.M.; Lodder, K.; Kuijjer, M.L.; Teunisse, A.F.; Repapi, E.; Grochola, L.F.; Bartel, F.; Hogendoorn, P.C.; Wuerl, P.; et al. Alternate splicing of the p53 inhibitor HDMX offers a superior prognostic biomarker than p53 mutation in human cancer. Cancer Res. 2012, 72, 4074–4084. [Google Scholar] [CrossRef] [Green Version]
- Lenos, K.; Jochemsen, A.G. Functions of MDMX in the modulation of the p53-response. J. Biomed. Biotechnol. 2011, 2011, 876173. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Yang, M. The functional MDM2 T309G genetic variant but not P53 Arg72Pro polymorphism is associated with risk of sarcomas: A meta-analysis. J. Cancer Res. Clin. Oncol. 2012, 138, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M.; Biason, P.; Iacoboni, E.; Gagno, S.; Fanelli, M.; Tavanti, E.; Vella, S.; Ferrari, S.; Roli, A.; Roncato, R.; et al. Candidate germline polymorphisms of genes belonging to the pathways of four drugs used in osteosarcoma standard chemotherapy associated with risk, survival and toxicity in non-metastatic high-grade osteosarcoma. Oncotarget 2016, 7, 61970–61987. [Google Scholar] [CrossRef] [Green Version]
- Savage, S.A.; Burdett, L.; Troisi, R.; Douglass, C.; Hoover, R.N.; Chanock, S.J. Germ-line genetic variation of TP53 in osteosarcoma. Pediatr. Blood Cancer 2007, 49, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Ru, J.Y.; Cong, Y.; Kang, W.B.; Yu, L.; Guo, T.; Zhao, J.N. Polymorphisms in TP53 are associated with risk and survival of osteosarcoma in a Chinese population. Int. J. Clin. Exp. Pathol. 2015, 8, 3198–3203. [Google Scholar] [PubMed]
- Madhusudan, S.; Middleton, M.R. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat. Rev. 2005, 31, 603–617. [Google Scholar] [CrossRef]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, H.B. DNA damage repair and response proteins as targets for cancer therapy. Curr. Med. Chem. 2008, 15, 360–367. [Google Scholar] [CrossRef]
- Cao, Z.H.; Yin, H.P.; Jiang, N.; Yu, B. Association between ERCC1 and ERCC2 gene polymorphisms and chemotherapy response and overall survival in osteosarcoma. Genet. Mol. Res. 2015, 14, 10145–10151. [Google Scholar] [CrossRef]
- Ji, W.P.; He, N.B. Investigation on the DNA repaired gene polymorphisms and response to chemotherapy and overall survival of osteosarcoma. Int. J. Clin. Exp. Pathol. 2015, 8, 894–899. [Google Scholar]
- Sun, Y.; Wu, Y.; Li, W.; Kong, Z.; Zou, X. Genetic polymorphisms in nucleotide excision repair pathway influences response to chemotherapy and overall survival in osteosarcoma. Int. J. Clin. Exp. Pathol. 2015, 8, 7905–7912. [Google Scholar]
- Zhang, H.; Ge, J.; Hong, H.; Bi, L.; Sun, Z. Genetic polymorphisms in ERCC1 and ERCC2 genes are associated with response to chemotherapy in osteosarcoma patients among Chinese population: A meta-analysis. World J. Surg. Oncol. 2017, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lv, L.Y.; Li, B.J.; Zhang, J.; Wei, F. Investigation of ERCC1 and ERCC2 gene polymorphisms and response to chemotherapy and overall survival in osteosarcoma. Genet. Mol. Res. 2015, 14, 11235–11241. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Feng, W.; Zhang, J.; Sun, Y.J.; Wang, G. Association of four ERCC1 and ERCC2 SNPs with survival of bone tumour patients. Asian Pac. J. Cancer Prev. 2012, 13, 3821–3824. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.J.; Zhu, Y.; Guo, X.J.; Tian, Z.Z. Genetic variability of genes involved in DNA repair influence treatment outcome in osteosarcoma. Genet. Mol. Res. 2015, 14, 11652–11657. [Google Scholar] [CrossRef] [PubMed]
- Obiedat, H.; Alrabadi, N.; Sultan, E.; Al Shatti, M.; Zihlif, M. The effect of ERCC1 and ERCC2 gene polymorphysims on response to cisplatin based therapy in osteosarcoma patients. BMC Med. Genet. 2018, 19, 112. [Google Scholar] [CrossRef] [PubMed]
- Caronia, D.; Patino-Garcia, A.; Milne, R.L.; Zalacain-Diez, M.; Pita, G.; Alonso, M.R.; Moreno, L.T.; Sierrasesumaga-Ariznabarreta, L.; Benitez, J.; Gonzalez-Neira, A. Common variations in ERCC2 are associated with response to cisplatin chemotherapy and clinical outcome in osteosarcoma patients. Pharmacogn J. 2009, 9, 347–353. [Google Scholar] [CrossRef]
- Biason, P.; Hattinger, C.M.; Innocenti, F.; Talamini, R.; Alberghini, M.; Scotlandi, K.; Zanusso, C.; Serra, M.; Toffoli, G. Nucleotide excision repair gene variants and association with survival in osteosarcoma patients treated with neoadjuvant chemotherapy. Pharmacogn. J. 2012, 12, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, S.; Wang, W.; Zhang, K.; Liu, Z.; Zhang, C.; Chen, S.; Wu, S. ERCC polymorphisms and prognosis of patients with osteosarcoma. Tumour Biol. 2014, 35, 10129–10136. [Google Scholar] [CrossRef]
- Yang, L.M.; Li, X.H.; Bao, C.F. Glutathione S-transferase P1 and DNA polymorphisms influence response to chemotherapy and prognosis of bone tumors. Asian Pac. J. Cancer Prev. 2012, 13, 5883–5886. [Google Scholar] [CrossRef]
- Goricar, K.; Kovac, V.; Jazbec, J.; Zakotnik, B.; Lamovec, J.; Dolzan, V. Genetic variability of DNA repair mechanisms and glutathione-S-transferase genes influences treatment outcome in osteosarcoma. Cancer Epidemiol. 2015, 39, 182–188. [Google Scholar] [CrossRef]
- Liu, Z.F.; Asila, A.L.; Aikenmu, K.; Zhao, J.; Meng, Q.C.; Fang, R. Influence of ERCC2 gene polymorphisms on the treatment outcome of osteosarcoma. Genet. Mol. Res. 2015, 14, 12967–12972. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.B.; Chen, H.X.; Bao, Y.X.; Luo, X.; Zhong, J.J. Predictive impact of common variations in DNA repair genes on clinical outcome of osteosarcoma. Asian Pac. J. Cancer Prev. 2013, 14, 3677–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.H.; Hou, W.G.; Zhao, H.X.; Zhao, Y.L.; Ma, C.; Liu, Y. Single nucleotide polymorphisms in the NER pathway and clinical outcome of patients with bone malignant tumors. Asian Pac. J. Cancer Prev. 2013, 14, 2049–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.L.; Yang, L.B.; Geng, X.L.; Zhou, Q.L.; Qin, H.; Yang, L.; Dong, Y.Z.; Zhong, J.J. The association of XPG and MMS19L polymorphisms response to chemotherapy in osteosarcoma. Pak. J. Med. Sci. 2013, 29, 1225–1229. [Google Scholar] [CrossRef]
- Hagleitner, M.M.; Coenen, M.J.; Gelderblom, H.; Makkinje, R.R.; Vos, H.I.; de Bont, E.S.; van der Graaf, W.T.; Schreuder, H.B.; Flucke, U.E.; van Leeuwen, F.N.; et al. A first step towards personalized medicine in osteosarcoma: Pharmacogenetics as predictive marker of outcome after chemotherapy based treatment. Clin. Cancer Res. 2015, 21, 3436–3441. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Z.; Deng, C.; Tian, Y.; Ma, X. Meta-analysis showing that ERCC1 polymorphism is predictive of osteosarcoma prognosis. Oncotarget 2017, 8, 62769–62779. [Google Scholar] [CrossRef] [Green Version]
- Michael, M.; Doherty, M.M. Tumoral drug metabolism: Overview and its implications for cancer therapy. J. Clin. Oncol. 2005, 23, 205–229. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Lo, H.W.; Ali-Osman, F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr. Opin. Pharmacol. 2007, 7, 367–374. [Google Scholar] [CrossRef]
- Li, J.Z.; Tian, Z.Q.; Jiang, S.N.; Feng, T. Effect of variation of ABCB1 and GSTP1 on osteosarcoma survival after chemotherapy. Genet. Mol. Res. 2014, 13, 3186–3192. [Google Scholar] [CrossRef]
- Liu, S.; Yi, Z.; Ling, M.; Shi, J.; Qiu, Y.; Yang, S. Predictive potential of ABCB1, ABCC3, and GSTP1 gene polymorphisms on osteosarcoma survival after chemotherapy. Tumour Biol. 2014, 35, 9897–9904. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.W.; Yang, Z.M.; Li, J.; Xu, B. Predictive role of Glutathione S-transferases (GSTs) on the prognosis of osteosarcoma patients treated with chemotherapy. Pak. J. Med. Sci. 2013, 29, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Windsor, R.E.; Strauss, S.J.; Kallis, C.; Wood, N.E.; Whelan, J.S. Germline genetic polymorphisms may influence chemotherapy response and disease outcome in osteosarcoma: A pilot study. Cancer 2012, 118, 1856–1867. [Google Scholar] [CrossRef]
- Zhang, S.L.; Mao, N.F.; Sun, J.Y.; Shi, Z.C.; Wang, B.; Sun, Y.J. Predictive potential of glutathione S-transferase polymorphisms for prognosis of osteosarcoma patients on chemotherapy. Asian Pac. J. Cancer Prev. 2012, 13, 2705–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinas-Souza, C.; Petrilli, A.S.; de Toledo, S.R. Glutathione S-transferase polymorphisms in osteosarcoma patients. Pharm. Genom. 2010, 20, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Barnette, P.; Scholl, R.; Blandford, M.; Ballard, L.; Tsodikov, A.; Magee, J.; Williams, S.; Robertson, M.; Ali-Osman, F.; Lemons, R.; et al. High-throughput detection of glutathione s-transferase polymorphic alleles in a pediatric cancer population. Cancer Epidemiol. Biomark. Prev. 2004, 13, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Pu, F.; Chen, F.; Chen, S.; Wang, B.; Liu, J.; Shao, Z. Association between GSTP1 polymorphisms and prognosis of osteosarcoma in patients treated with chemotherapy: A meta-analysis. Onco Targets Ther. 2015, 8, 1835–1842. [Google Scholar]
- Wang, Z.; Xu, H.; He, M.; Wu, H.; Zhu, Y.; Su, Z. The association of glutathione S-transferase polymorphisms in patients with osteosarcoma: Evidence from a meta-analysis. Eur. J. Cancer Care (Engl.) 2015, 24, 417–424. [Google Scholar] [CrossRef]
- Lan, J.; Yang, Q.; Zhou, M.; Xu, R.; Zhou, C.; Wang, J.; Zheng, H. A meta-analysis of association between glutathione S-transferase gene polymorphism and osteosarcoma chemosensitivity in Chinese population. J. Cancer Res. Ther. 2016, 12, 64–67. [Google Scholar]
- McFadyen, M.C.; Melvin, W.T.; Murray, G.I. Cytochrome P450 enzymes: Novel options for cancer therapeutics. Mol. Cancer Ther. 2004, 3, 363–371. [Google Scholar]
- Caronia, D.; Patino-Garcia, A.; Perez-Martinez, A.; Pita, G.; Moreno, L.T.; Zalacain-Diez, M.; Molina, B.; Colmenero, I.; Sierrasesumaga, L.; Benitez, J.; et al. Effect of ABCB1 and ABCC3 polymorphisms on osteosarcoma survival after chemotherapy: A pharmacogenetic study. PLoS ONE 2011, 6, e26091. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Li, A.; Geng, X.; Liu, Y.; Zhao, H.; Zhao, Y. Effect of ABCB1 polymorphism on the clinical outcome of osteosarcoma patients after receiving chemotherapy. Pak. J. Med. Sci. 2014, 30, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, Z.G.; Cai, H.Q.; Li, Y.C.; Xu, Y.L. Effect of variation of ABCB1 and ABCC3 genotypes on the survival of bone tumor cases after chemotherapy. Asian Pac. J. Cancer Prev. 2013, 14, 4595–4598. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Jiang, M.; Zhao, R.K.; Gu, G.H. Quantitative Assessment of the Association between ABC Polymorphisms and Osteosarcoma Response: A Meta-analysis. Asian Pac. J. Cancer Prev 2015, 16, 4659–4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffaud, F.; Egerer, G.; Ferrari, S.; Rassam, H.; Boecker, U.; Bui-Nguyen, B. A phase II trial of second-line pemetrexed in adults with advanced/metastatic osteosarcoma. Eur. J. Cancer 2012, 48, 564–570. [Google Scholar] [CrossRef] [PubMed]
- O’Day, K.; Gorlick, R. Novel therapeutic agents for osteosarcoma. Expert Rev. Anticancer Ther. 2009, 9, 511–523. [Google Scholar] [CrossRef]
- Warwick, A.B.; Malempati, S.; Krailo, M.; Melemed, A.; Gorlick, R.; Ames, M.M.; Safgren, S.L.; Adamson, P.C.; Blaney, S.M. Phase 2 trial of pemetrexed in children and adolescents with refractory solid tumors: A Children’s Oncology Group study. Pediatr. Blood Cancer 2013, 60, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Jabeen, S.; Holmboe, L.; Alnaes, G.I.; Andersen, A.M.; Hall, K.S.; Kristensen, V.N. Impact of genetic variants of RFC1, DHFR and MTHFR in osteosarcoma patients treated with high-dose methotrexate. Pharmacogn. J. 2015, 15, 385–390. [Google Scholar] [CrossRef]
- Goricar, K.; Kovac, V.; Jazbec, J.; Zakotnik, B.; Lamovec, J.; Dolzan, V. Influence of the folate pathway and transporter polymorphisms on methotrexate treatment outcome in osteosarcoma. Pharmacogn. Genom. 2014, 24, 514–521. [Google Scholar] [CrossRef]
- Janeway, K.A.; Grier, H.E. Sequelae of osteosarcoma medical therapy: A review of rare acute toxicities and late effects. Lancet Oncol. 2010, 11, 670–678. [Google Scholar] [CrossRef]
- Vos, H.I.; Coenen, M.J.; Guchelaar, H.J.; Te Loo, D.M. The role of pharmacogenetics in the treatment of osteosarcoma. Drug Discov. Today 2016, 21, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Aminkeng, F.; Ross, C.J.; Rassekh, S.R.; Hwang, S.; Rieder, M.J.; Bhavsar, A.P.; Smith, A.; Sanatani, S.; Gelmon, K.A.; Bernstein, D.; et al. Recommendations for genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity. Br. J. Clin. Pharmacol. 2016, 82, 683–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Pussegoda, K.; Rassekh, S.R.; Monzon, J.G.; Liu, G.; Hwang, S.; Bhavsar, A.P.; Pritchard, S.; Ross, C.J.; Amstutz, U.; et al. Clinical Practice Recommendations for the Management and Prevention of Cisplatin-Induced Hearing Loss Using Pharmacogenetic Markers. Ther. Drug Monit. 2016, 38, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattinger, C.M.; Tavanti, E.; Fanelli, M.; Vella, S.; Picci, P.; Serra, M. Pharmacogenomics of genes involved in antifolate drug response and toxicity in osteosarcoma. Expert Opin. Drug Metab. Toxicol. 2017, 13, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Patino-Garcia, A.; Zalacain, M.; Marrodan, L.; San-Julian, M.; Sierrasesumaga, L. Methotrexate in pediatric osteosarcoma: Response and toxicity in relation to genetic polymorphisms and dihydrofolate reductase and reduced folate carrier 1 expression. J. Pediatr. 2009, 154, 688–693. [Google Scholar] [CrossRef]
- Xie, L.; Guo, W.; Yang, Y.; Ji, T.; Xu, J. More severe toxicity of genetic polymorphisms on MTHFR activity in osteosarcoma patients treated with high-dose methotrexate. Oncotarget 2018, 9, 11465–11476. [Google Scholar] [CrossRef] [Green Version]
- Hegyi, M.; Arany, A.; Semsei, A.F.; Csordas, K.; Eipel, O.; Gezsi, A.; Kutszegi, N.; Csoka, M.; Muller, J.; Erdelyi, D.J.; et al. Pharmacogenetic analysis of high-dose methotrexate treatment in children with osteosarcoma. Oncotarget 2017, 8, 9388–9398. [Google Scholar] [CrossRef] [Green Version]
- Te Loo, D.M.W.; Hagleitner, M.M.; Coenen, M.J. Is there a role for the MTHFR 677C>T and 1298A>C polymorphisms in methotrexate-induced liver toxicity? Pharmacogenomics 2014, 15, 1401–1403. [Google Scholar] [CrossRef]
- Hagleitner, M.M.; Coenen, M.J.; Aplenc, R.; Patino-Garcia, A.; Chiusolo, P.; Gemmati, D.; De Mattei, M.; Ongaro, A.; Krajinovic, M.; Hoogerbrugge, P.M.; et al. The role of the MTHFR 677C>T polymorphism in methotrexate-induced liver toxicity: A meta-analysis in patients with cancer. Pharmacogn. J. 2014, 14, 115–119. [Google Scholar] [CrossRef]
- Dogan, M.; Karabulut, H.G.; Tukun, A.; Demirkazik, A.; Utkan, G.; Yalcin, B.; Dincol, D.; Akbulut, H.; Icli, F. Relationship between antimetabolite toxicity and pharmacogenetics in Turkish cancer patients. Asian Pac. J. Cancer Prev. 2012, 13, 1553–1556. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Kralovanszky, J.; Adleff, V.; Pap, E.; Nemeth, K.; Komlosi, V.; Kovacs, G. Toxic encephalopathy and delayed MTX clearance after high-dose methotrexate therapy in a child homozygous for the MTHFR C677T polymorphism. Anticancer Res. 2008, 28, 3051–3054. [Google Scholar]
- Park, J.A.; Shin, H.Y. Influence of genetic polymorphisms in the folate pathway on toxicity after high-dose methotrexate treatment in pediatric osteosarcoma. Blood Res. 2016, 51, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, J.G.; Sun, C.L.; Landier, W.; Chen, L.; Esparza-Duran, D.; Leisenring, W.; Mays, A.; Friedman, D.L.; Ginsberg, J.P.; Hudson, M.M.; et al. Anthracycline-related cardiomyopathy after childhood cancer: Role of polymorphisms in carbonyl reductase genes—A report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 1415–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visscher, H.; Ross, C.J.; Rassekh, S.R.; Barhdadi, A.; Dube, M.P.; Al-Saloos, H.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; et al. Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J. Clin. Oncol. 2012, 30, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Visscher, H.; Ross, C.J.; Rassekh, S.R.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; et al. Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr. Blood Cancer 2013, 60, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, W.; Sun, C.L.; Armenian, S.H.; Hakonarson, H.; Hageman, L.; Ding, Y.; Landier, W.; Blanco, J.G.; Chen, L.; et al. Hyaluronan synthase 3 variant and anthracycline-related cardiomyopathy: A report from the children’s oncology group. J. Clin. Oncol. 2014, 32, 647–653. [Google Scholar] [CrossRef]
- Visscher, H.; Rassekh, S.R.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; Carleton, B.C.; et al. Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics 2015, 16, 1065–1076. [Google Scholar] [CrossRef]
- Aminkeng, F.; Bhavsar, A.P.; Visscher, H.; Rassekh, S.R.; Li, Y.; Lee, J.W.; Brunham, L.R.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; et al. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat. Genet. 2015, 47, 1079–1084. [Google Scholar] [CrossRef]
- Singh, P.; Wang, X.; Hageman, L.; Chen, Y.; Magdy, T.; Landier, W.; Ginsberg, J.P.; Neglia, J.P.; Sklar, C.A.; Castellino, S.M.; et al. Association of GSTM1 null variant with anthracycline-related cardiomyopathy after childhood cancer-A Children’s Oncology Group ALTE03N1 report. Cancer 2020. [Google Scholar] [CrossRef]
- Riedemann, L.; Lanvers, C.; Deuster, D.; Peters, U.; Boos, J.; Jurgens, H.; am Zehnhoff-Dinnesen, A. Megalin genetic polymorphisms and individual sensitivity to the ototoxic effect of cisplatin. Pharmacogn. J. 2008, 8, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.J.; Katzov-Eckert, H.; Dube, M.P.; Brooks, B.; Rassekh, S.R.; Barhdadi, A.; Feroz-Zada, Y.; Visscher, H.; Brown, A.M.; Rieder, M.J.; et al. Genetic variants in TPMT and COMT are associated with hearing loss in children receiving cisplatin chemotherapy. Nat. Genet. 2009, 41, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Lanvers-Kaminsky, C.; Sprowl, J.A.; Malath, I.; Deuster, D.; Eveslage, M.; Schlatter, E.; Mathijssen, R.H.; Boos, J.; Jurgens, H.; Am Zehnhoff-Dinnesen, A.G.; et al. Human OCT2 variant c.808G>T confers protection effect against cisplatin-induced ototoxicity. Pharmacogenomics 2015, 16, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pussegoda, K.; Ross, C.J.; Visscher, H.; Yazdanpanah, M.; Brooks, B.; Rassekh, S.R.; Zada, Y.F.; Dube, M.P.; Carleton, B.C.; Hayden, M.R. Replication of TPMT and ABCC3 genetic variants highly associated with cisplatin-induced hearing loss in children. Clin. Pharmacol. Ther. 2013, 94, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Hagleitner, M.M.; Coenen, M.J.; Patino-Garcia, A.; de Bont, E.S.; Gonzalez-Neira, A.; Vos, H.I.; van Leeuwen, F.N.; Gelderblom, H.; Hoogerbrugge, P.M.; Guchelaar, H.J.; et al. Influence of genetic variants in TPMT and COMT associated with cisplatin induced hearing loss in patients with cancer: Two new cohorts and a meta-analysis reveal significant heterogeneity between cohorts. PLoS ONE 2014, 9, e115869. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Robinson, G.W.; Huang, J.; Lim, J.Y.; Zhang, H.; Bass, J.K.; Broniscer, A.; Chintagumpala, M.; Bartels, U.; Gururangan, S.; et al. Common variants in ACYP2 influence susceptibility to cisplatin-induced hearing loss. Nat. Genet. 2015, 47, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Vos, H.I.; Guchelaar, H.J.; Gelderblom, H.; de Bont, E.S.; Kremer, L.C.; Naber, A.M.; Hakobjan, M.H.; van der Graaf, W.T.; Coenen, M.J.; te Loo, D.M. Replication of a genetic variant in ACYP2 associated with cisplatin-induced hearing loss in patients with osteosarcoma. Pharm. Genom. 2016, 26, 243–247. [Google Scholar] [CrossRef]
- Peters, U.; Preisler-Adams, S.; Hebeisen, A.; Hahn, M.; Seifert, E.; Lanvers, C.; Heinecke, A.; Horst, J.; Jurgens, H.; Lamprecht-Dinnesen, A. Glutathione S-transferase genetic polymorphisms and individual sensitivity to the ototoxic effect of cisplatin. Anticancer Drugs 2000, 11, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Spracklen, T.F.; Whitehorn, H.; Vorster, A.A.; Ramma, L.; Dalvie, S.; Ramesar, R.S. Genetic variation in Otos is associated with cisplatin-induced ototoxicity. Pharmacogenomics 2014, 15, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Rowley, R.; Williams, M.S.; Roden, D.; Ginsburg, G.S.; Bult, C.; Chisholm, R.L.; Deverka, P.A.; McLeod, H.L.; Mensah, G.A.; et al. Opportunities, resources, and techniques for implementing genomics in clinical care. Lancet 2019, 394, 511–520. [Google Scholar] [CrossRef]
- Bilbao-Aldaiturriaga, N.; Gutierrez-Camino, A.; Martin-Guerrero, I.; Pombar-Gomez, M.; Zalacain-Diez, M.; Patino-Garcia, A.; Lopez-Lopez, E.; Garcia-Orad, A. Polymorphisms in miRNA processing genes and their role in osteosarcoma risk. Pediatr. Blood Cancer 2015, 62, 766–769. [Google Scholar] [CrossRef]
- Izadpanah, S.; Shabani, P.; Aghebati-Maleki, A.; Baghbani, E.; Baghbanzadeh, A.; Fotouhi, A.; Bakhshinejad, B.; Aghebati-Maleki, L.; Baradaran, B. Insights into the roles of miRNAs; miR-193 as one of small molecular silencer in osteosarcoma therapy. Biomed. Pharmacother. 2019, 111, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jing, J.; Cheng, L. Emerging roles of non-coding RNAs in the pathogenesis, diagnosis and prognosis of osteosarcoma. Investig. New Drugs 2018, 36, 1116–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Wang, Y.; Jing, J. The Roles of Circular RNAs in Osteosarcoma. Med. Sci. Monit. 2019, 25, 6378–6382. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.W.; Wang, J.L.; Zhao, N.; Guan, Y.; He, W. Single nucleotide polymorphism of hsa-miR-124a affects risk and prognosis of osteosarcoma. Cancer Biomark. 2016, 17, 249–257. [Google Scholar] [CrossRef]
- Bofill-De Ros, X.; Yang, A.; Gu, S. IsomiRs: Expanding the miRNA repression toolbox beyond the seed. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194373. [Google Scholar] [CrossRef] [PubMed]
- Rainusso, N.; Cleveland, H.; Hernandez, J.A.; Quintanilla, N.M.; Hicks, J.; Vasudevan, S.; Marco, R.A.W.; Allen-Rhoades, W.; Wang, L.L.; Yustein, J.T. Generation of patient-derived tumor xenografts from percutaneous tumor biopsies in children with bone sarcomas. Pediatr. Blood Cancer 2019, 66, e27579. [Google Scholar] [CrossRef]
- Stewart, E.; Federico, S.M.; Chen, X.; Shelat, A.A.; Bradley, C.; Gordon, B.; Karlstrom, A.; Twarog, N.R.; Clay, M.R.; Bahrami, A.; et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017, 549, 96–100. [Google Scholar] [CrossRef]
- Nanni, P.; Landuzzi, L.; Manara, M.C.; Righi, A.; Nicoletti, G.; Cristalli, C.; Pasello, M.; Parra, A.; Carrabotta, M.; Ferracin, M.; et al. Bone sarcoma patient-derived xenografts are faithful and stable preclinical models for molecular and therapeutic investigations. Sci. Rep. 2019, 9, 12174. [Google Scholar] [CrossRef]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Ambati, S.R.; Shieh, J.H.; Pera, B.; Lopes, E.C.; Chaudhry, A.; Wong, E.W.; Saxena, A.; Su, T.L.; Moore, M.A. BO-1055, a novel DNA cross-linking agent with remarkable low myelotoxicity shows potent activity in sarcoma models. Oncotarget 2016, 7, 43062–43075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manara, M.C.; Valente, S.; Cristalli, C.; Nicoletti, G.; Landuzzi, L.; Zwergel, C.; Mazzone, R.; Stazi, G.; Arimondo, P.B.; Pasello, M.; et al. A Quinoline-Based DNA Methyltransferase Inhibitor as a Possible Adjuvant in Osteosarcoma Therapy. Mol. Cancer Ther. 2018, 17, 1881–1892. [Google Scholar] [CrossRef] [Green Version]
- Xian, M.; Cao, H.; Cao, J.; Shao, X.; Zhu, D.; Zhang, N.; Huang, P.; Li, W.; Yang, B.; Ying, M.; et al. Bortezomib sensitizes human osteosarcoma cells to adriamycin-induced apoptosis through ROS-dependent activation of p-eIF2alpha/ATF4/CHOP axis. Int. J. Cancer 2017, 141, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Raimondi, L.; Salamanna, F.; Carina, V.; Costa, V.; Bellavia, D.; Alessandro, R.; Fini, M.; Giavaresi, G. Relevance of 3d culture systems to study osteosarcoma environment. J. Exp. Clin. Cancer Res. 2018, 37, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Nail, L.R.; Brennan, M.; Rosset, P.; Deschaseaux, F.; Piloquet, P.; Pichon, O.; Le Caignec, C.; Crenn, V.; Layrolle, P.; Herault, O.; et al. Comparison of Tumor- and Bone Marrow-Derived Mesenchymal Stromal/Stem Cells from Patients with High-Grade Osteosarcoma. Int. J. Mol. Sci. 2018, 19, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuvaneshwar, K.; Harris, M.; Gusev, Y.; Madhavan, S.; Iyer, R.; Vilboux, T.; Deeken, J.; Yang, E.; Shankar, S. Genome sequencing analysis of blood cells identifies germline haplotypes strongly associated with drug resistance in osteosarcoma patients. BMC Cancer 2019, 19, 357. [Google Scholar] [CrossRef]
- Johnson, J.A.; Bootman, J.L. Drug-related morbidity and mortality. A cost-of-illness model. Arch. Intern. Med. 1995, 155, 1949–1956. [Google Scholar] [CrossRef]
- Lazarou, J.; Pomeranz, B.H.; Corey, P.N. Incidence of adverse drug reactions in hospitalized patients: A meta-analysis of prospective studies. JAMA 1998, 279, 1200–1205. [Google Scholar] [CrossRef]
- Landrigan, C.P.; Parry, G.J.; Bones, C.B.; Hackbarth, A.D.; Goldmann, D.A.; Sharek, P.J. Temporal trends in rates of patient harm resulting from medical care. N. Engl. J. Med. 2010, 363, 2124–2134. [Google Scholar] [CrossRef] [Green Version]
- Trusheim, M.R.; Berndt, E.R.; Douglas, F.L. Stratified medicine: Strategic and economic implications of combining drugs and clinical biomarkers. Nat. Rev. Drug Discov. 2007, 6, 287–293. [Google Scholar] [CrossRef]
- Garbayo, E.; Pascual-Gil, S.; Rodriguez-Nogales, C.; Saludas, L.; Estella-Hermoso de Mendoza, A.; Blanco-Prieto, M.J. Nanomedicine and drug delivery systems in cancer and regenerative medicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020. [Google Scholar] [CrossRef]
- Pereira-Silva, M.; Alvarez-Lorenzo, C.; Concheiro, A.; Santos, A.C.; Veiga, F.; Figueiras, A. Nanomedicine in osteosarcoma therapy: Micelleplexes for delivery of nucleic acids and drugs toward osteosarcoma-targeted therapies. Eur. J. Pharm. Biopharm. 2020, 148, 88–106. [Google Scholar] [CrossRef]
- Manolio, T.A.; Abramowicz, M.; Al-Mulla, F.; Anderson, W.; Balling, R.; Berger, A.C.; Bleyl, S.; Chakravarti, A.; Chantratita, W.; Chisholm, R.L.; et al. Global implementation of genomic medicine: We are not alone. Sci. Transl. Med. 2015, 7, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Individualized Therapy for Relapsed Malignancies in Childhood (INFORM). Available online: https://www.kitz-heidelberg.de/en/for-physicians/clinical-studies/molecular-diagnostics-studies/inform/ (accessed on 30 March 2020).
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UK 100,000 Genomes Cancer Project. Available online: https://www.genomicsengland.co.uk/about-genomics-england/the-100000-genomes-project/ (accessed on 30 March 2020).
- Turnbull, C.; Scott, R.H.; Thomas, E.; Jones, L.; Murugaesu, N.; Pretty, F.B.; Halai, D.; Baple, E.; Craig, C.; Hamblin, A.; et al. The 100 000 Genomes Project: Bringing whole genome sequencing to the NHS. BMJ 2018, 361, k1687. [Google Scholar] [CrossRef] [Green Version]
- Geisinger MyCode Community Health Initiative. Available online: https://www.geisinger.org/mycode (accessed on 30 March 2020).
- Ginsburg, G.S.; Phillips, K.A. Precision Medicine: From Science to Value. Health Aff. (Millwood) 2018, 37, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Chisholm, R.L.; Ozenberger, B.; Roden, D.M.; Williams, M.S.; Wilson, R.; Bick, D.; Bottinger, E.P.; Brilliant, M.H.; Eng, C.; et al. Implementing genomic medicine in the clinic: The future is here. Genet. Med. 2013, 15, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Owusu Obeng, A.; Fei, K.; Levy, K.D.; Elsey, A.R.; Pollin, T.I.; Ramirez, A.H.; Weitzel, K.W.; Horowitz, C.R. Physician-Reported Benefits and Barriers to Clinical Implementation of Genomic Medicine: A Multi-Site IGNITE-Network Survey. J. Pers. Med. 2018, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Hay, M.A.; Severson, E.A.; Miller, V.A.; Liebner, D.A.; Vergilio, J.; Millis, S.Z.; Chen, J.L. Identifying Opportunities and Challenges for Patients With Sarcoma as a Result of Comprehensive Genomic Profiling of Sarcoma Specimens. JCO Precis. Oncol. 2020, 4, 176–182. [Google Scholar] [CrossRef]
- Li, Z.; Dou, P.; Liu, T.; He, S. Application of Long Noncoding RNAs in Osteosarcoma: Biomarkers and Therapeutic Targets. Cell Physiol. Biochem. 2017, 42, 1407–1419. [Google Scholar] [CrossRef]
- Khan, F.A.; Pandupuspitasari, N.S.; Huang, C.-J.; Ao, Z.; Jamal, M.; Zohaib, A.; Hakim, M.R.; Zhao, S. CRISPR/Cas9 therapeutics: A cure for cancer and other genetic diseases. Oncotarget 2016, 7, 52541–52552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zewde, M.; Kiyotani, K.; Park, J.H.; Fang, H.; Yap, K.L.; Yew, P.Y.; Alachkar, H.; Kato, T.; Mai, T.H.; Ikeda, Y.; et al. The era of immunogenomics/immunopharmacogenomics. J. Hum. Genet. 2018, 63, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Chang, T.C.; Carter, R.A.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; Peng, J.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Autosomal Dominant (Gene Involved) | Incidence of HGOS | Estimated Fold-Risk | Autosomal Recessive (Gene Involved) | Incidence of HGOS |
|---|---|---|---|---|
| Li-Fraumeni Syndrome (TP53) | 3% [17] 5–11% [18] | 107 [19] | Rothmund-Thomson Syndrome (RECQL4) | 32% [20] 48% [21] |
| Hereditary Retinoblastoma (RB1) | 12% [17] | 200–400* [22] 69–400* [19] | Rapadilino Syndrome (RECQL4) | 13% [22] 40% [21] |
| Diamond-Blackfan anemia (genes encoding ribosomal proteins, of which the most common is RPS19, and GATA1) | <1% [23] 14% [24] | 42 [24] | Bloom Syndrome (RECQL3) | <12% [25] |
| Werner Syndrome (RECQL2) | 7% [22] |
| ClinicalTrials.Gov NCT Identifier | Drug | Mechanism of Drug Action and Trial Description | Stage of Development (Time Period) |
|---|---|---|---|
| NCT03210714 | Erdafitinib (JNJ-42756493) | Inhibition of FGFR with negative effects on tumor cell growth and angiogenesis. This trial studies the activity of erdafitinib in patients with FGFR gene mutations, who are affected by disseminated tumors. | Phase II (11/2017–12/2024) |
| NCT03213665 | Tazemetostat (EPZ-6438) | Inhibition of the activity of human polycomb repressive complex 2 containing wild-type histone-lysine N-methyltransferases EZH1 and EZH2, with a consequent inhibition of tumor cells growth. This trial is intended to assess the activity of tazemetostat in patients with EZH2, SMARCB1, or SMARCA4 gene mutations, who are affected by disseminated tumors. | Phase II (07/2017–09/2024) |
| NCT03213678 | Samotolisib (LY3023414) | Inhibition of PI3K/AKT/mTOR pathway, producing negative effects on the growth of cancer cells. This trial evaluates the activity of samotolisib in patients with TSC or PI3K/mTOR mutations, who developed metastasis or local recurrences, or who are refractory to conventional treatments. | Phase II (07/2017–09/2024) |
| NCT03220035 | Vemurafenib (PLX40321; trade name Zelboraf®) | Inhibition of the mutated B-Raf protein, interrupting its stimulation of cell growth. This trial evaluates how vemurafenib works in treating patients with BRAF V600 mutations who are affected by tumors that have disseminated in the body or do not respond to conventional treatments. | Phase II (07/2017–12/2023) |
| NCT03233204 | Olaparib (AZD-2281, MK-7339, trade name Lynparza®) | Inhibition of PARP1 and consequent impairment of DNA repair activity. This trial assesses the efficacy of olaparib in treating patients with hereditary BRCA1 or BRCA2 mutations, leading to defects in DNA damage repair activity. The trial recruits patients who have developed metastasis or local recurrences, or who are refractory to conventional treatments. | Phase II (07/2017–09/2024) |
| NCT03526250 | Palbociclib (PD-0332991, trade name Ibrance®) | Selective inhibition of the cyclin-dependent kinases CDK4 and CDK6. By inhibiting CDK4 and CDK6, palbociclib may stop the growth of cancer cells. This trial evaluates how palbociclib works in treating patients with RB1-positive tumors which have disseminated in the body or do not respond to conventional treatments. | Phase II (06/2018–06/2025) |
| NCT03698994 | Ulixertinib (BVD-523; VRT752271) | Inhibition of ERK1/2 kinases, belonging to the MAPK pathway. By inhibiting cancer cells harboring mutations in the MAPK pathway, ulixertinib may stop tumor growth. This trial is intended to estimate the activity of ulixertinib in treating patients with disseminated tumors, who present genetic alterations in the MAPK signaling pathway. | Phase II (10/2018–12/2025) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hattinger, C.M.; Patrizio, M.P.; Luppi, S.; Serra, M. Pharmacogenomics and Pharmacogenetics in Osteosarcoma: Translational Studies and Clinical Impact. Int. J. Mol. Sci. 2020, 21, 4659. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134659
Hattinger CM, Patrizio MP, Luppi S, Serra M. Pharmacogenomics and Pharmacogenetics in Osteosarcoma: Translational Studies and Clinical Impact. International Journal of Molecular Sciences. 2020; 21(13):4659. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134659
Chicago/Turabian StyleHattinger, Claudia Maria, Maria Pia Patrizio, Silvia Luppi, and Massimo Serra. 2020. "Pharmacogenomics and Pharmacogenetics in Osteosarcoma: Translational Studies and Clinical Impact" International Journal of Molecular Sciences 21, no. 13: 4659. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134659