Theoretical Investigations on Interactions of Arylsulphonyl Indazole Derivatives as Potential Ligands of VEGFR2 Kinase

 , , ,
, , , 
Abstract
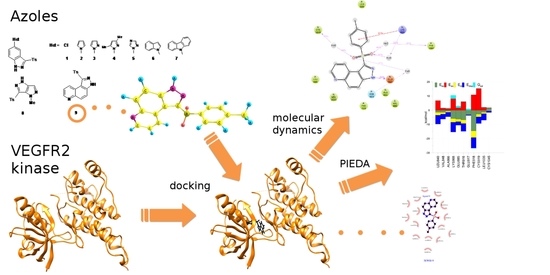
:
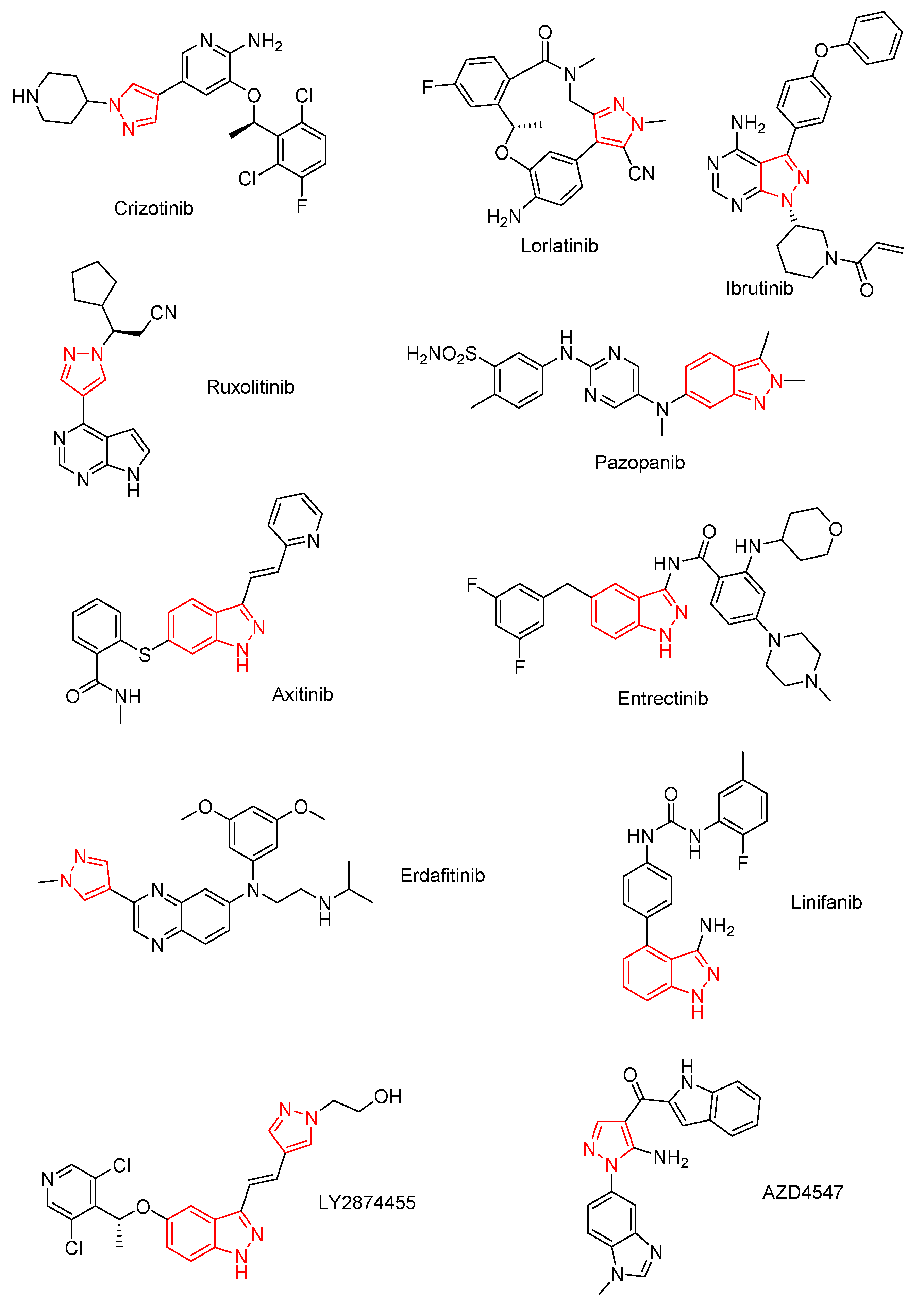
1. Introduction
2. Results and Discussion
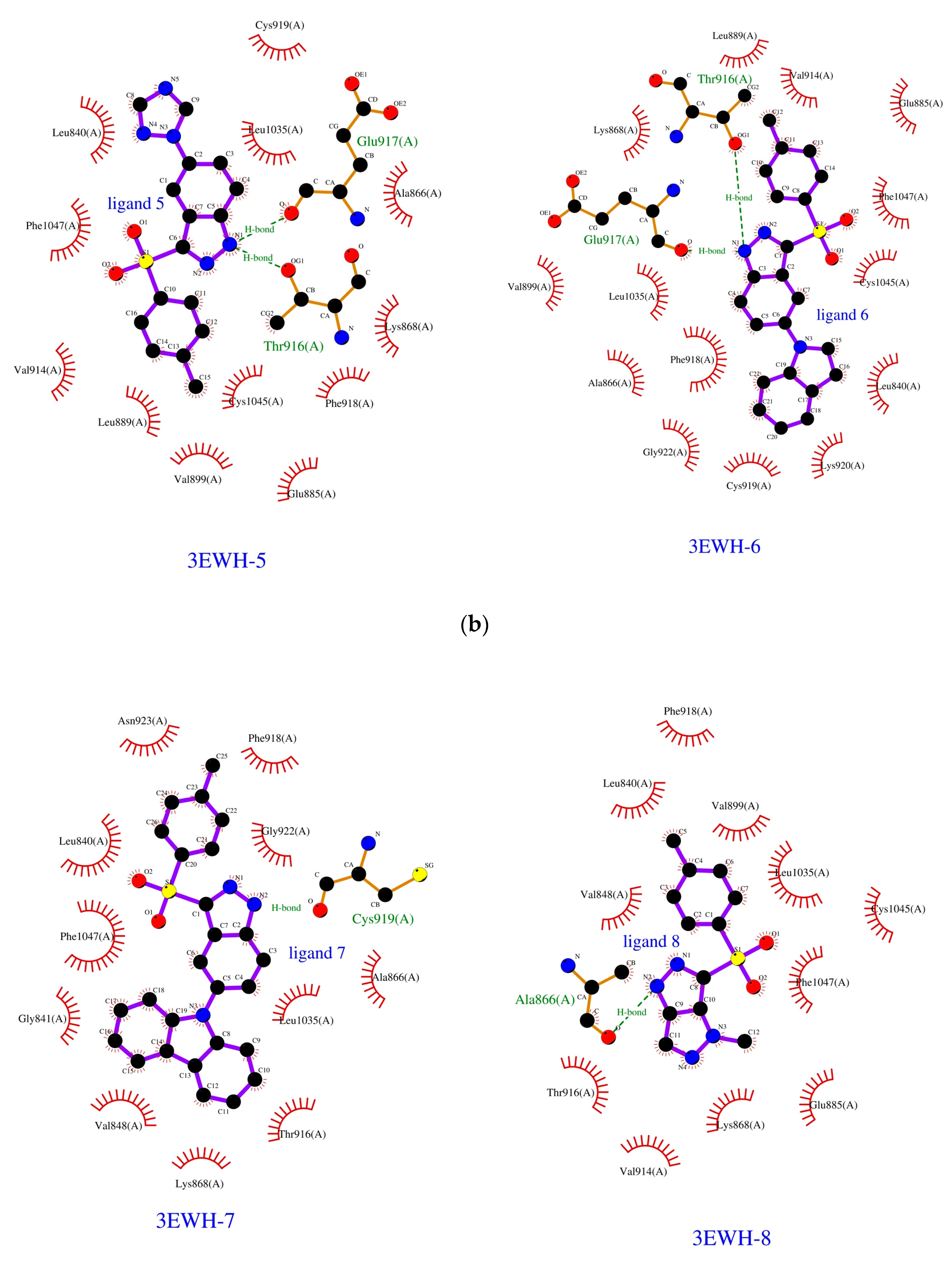
2.1. Molecular Docking
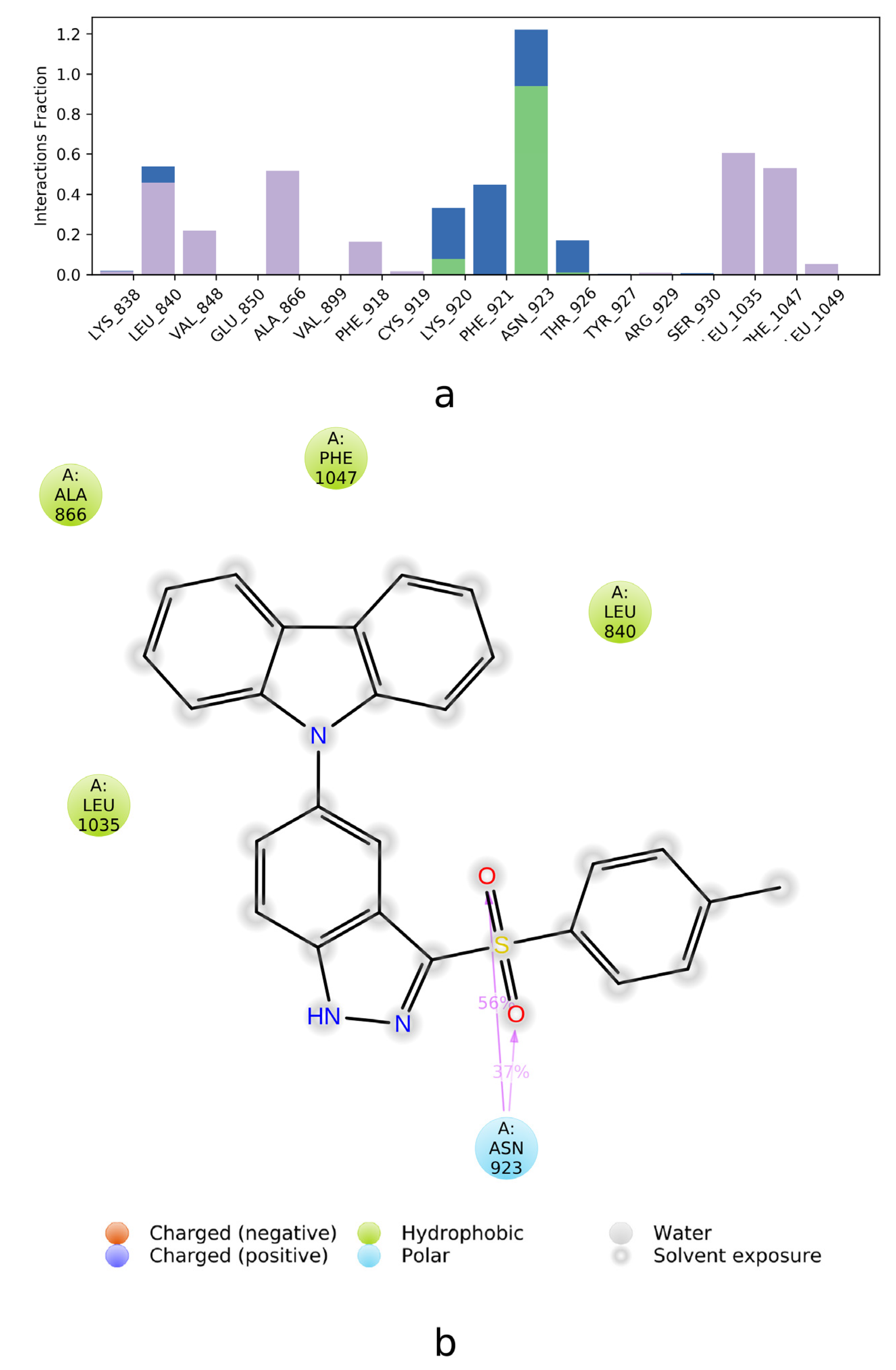
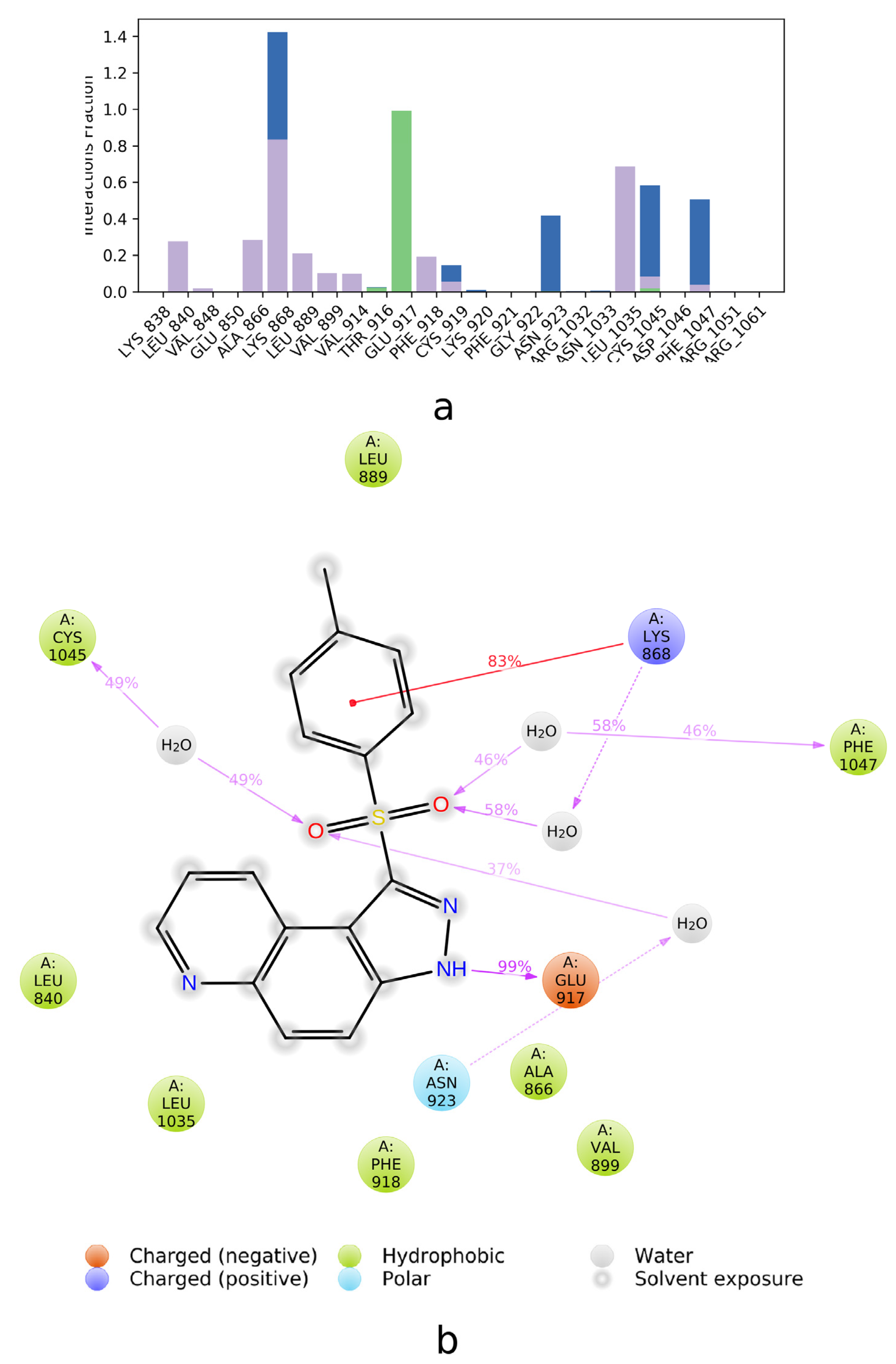
2.2. SAPT Analysis of Ligand-Amino Acid Complexes
2.3. PIEDA Analysis of Ligand-Protein Complexes
2.4. Estimation of the Interaction Energy
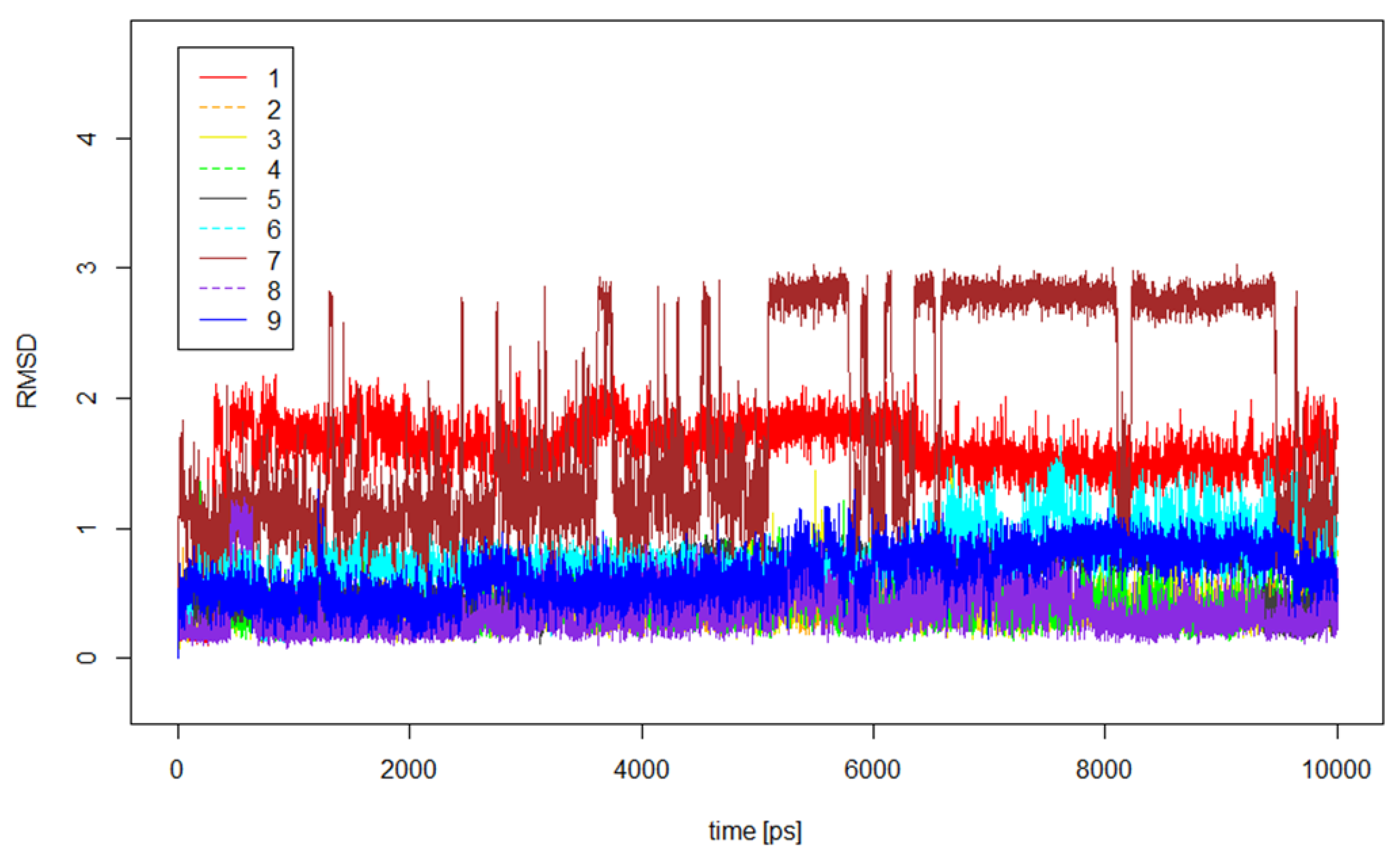
2.5. Molecular Dynamics Calculations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matthews, D.J.; Gerritsen, M.E. Targeting Protein Kinases for Cancer Therapy; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Shokat, K.M. (Ed.) Protein Kinase Inhibitors in Research and Medicine (Methods in Enzymology, Volume 548); Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors in the treatment of renal cell carcinomas. Pharmacol. Res. 2017, 120, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Rodriguez-Ariza, A.; Lopez-Pedrera, C.; Aranda, E.; Barbarroja, N. VEGF targeted therapy in acute myeloid leukemia. Crit. Rev. Oncol. Hemat. 2011, 80, 241–256. [Google Scholar] [CrossRef]
- Medinger, M.; Passweg, J. Role of tumour angiogenesis in haematological malignancies. CH Med. Wkly. 2014, 144, w14050. [Google Scholar] [CrossRef]
- Giraud, F.; Anizon, F.; Moreau, P. Advances in the synthesis and kinase inhibitory potencies of non-fused indazole derivatives. In Targets in Heterocyclic Systems: Chemistry and Properties; Attanasi, O.A., Noto, R., Spinelli, D., Eds.; Italian Society of Chemistry: Camerino, Italy, 2014; pp. 1–28. [Google Scholar]
- Avendaño, C.; Menéndez, J.C. Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. Recent Advances in the Development of Indazole-based Anticancer Agents. ChemMedChem 2018, 13, 1490–1507. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954. [Google Scholar] [CrossRef]
- Harris, P.A.; Stafford, J.A. Discovery of Pazopanib: A PAN Vascular Endothelial Growth Factor Kinase Inhibitor. In Kinase Inhibitor Drugs; Li, R., Stafford, J.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 57–77. [Google Scholar]
- Kania, R.S. Structure-based Design and Characterization of Axitinib. In Kinase Inhibitor Drugs; Li, R., Stafford, J.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 167–201. [Google Scholar]
- Smith, K.M.; Fagan, P.C.; Pomari, E.; Germano, G.; Frasson, C.; Walsh, C.; Silverman, I.; Bonvini, P.; Li, G. Antitumor Activity of Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor, in ETV6-NTRK3–Positive Acute Myeloid Leukemia. Mol. Cancer Ther. 2018, 17, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.K.; Davare, M.A.; Druker, B.J.; Tognon, C.E. Revisiting NTRKs as an emerging oncogene in hematological malignancies. Leukemia 2019, 33, 2563–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.I.A.; Cervantes, A.; Tabernero, J.; Infante, J.; Lara, P.N.; Spira, A.; Calvo, E.; Moreno, V.; Blay, J.; Lauer, R.; et al. Safety and activity of the pan–fibroblast growth factor receptor (FGFR) inhibitor erdafitinib in phase 1 study patients with advanced urothelial carcinoma. Ann. Oncol. 2016, 27, 781. [Google Scholar] [CrossRef]
- Roskoski, R. The role of fibroblast growth factor receptor (FGFR) protein-tyrosine kinase inhibitors in the treatment of cancers including those of the urinary bladder. Pharmacol. Res. 2020, 151, 104567. [Google Scholar] [CrossRef]
- Aversa, C.; Leone, F.; Zucchini, G.; Serini, G.; Geuna, E.R.; Milani, A.; Valdembri, D.; Martinello, R.; Montemurro, F. Linifanib: Current status and future potential in cancer therapy. Expert Rev. Anticancer Ther. 2015, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Guo, M.; Philips, M.A.; Qu, L.; Jiang, L.; Li, J.; Chen, X.; Chen, Z.; Chen, L.; Chen, Y. Crystal Structure of the FGFR4/LY2874455 Complex Reveals Insights into the Pan-FGFR Selectivity of LY2874455. PLoS ONE 2016, 11, e0162491. [Google Scholar] [CrossRef]
- Toton, E.; Ignatowicz, E.; Bernard, M.K.; Kujawski, J.; Rybczynska, M. Evaluation of apoptotic activity of new condensed pyrazole derivatives. J. Physiol. Pharmacol. 2013, 64, 115–123. [Google Scholar]
- Lehmann, T.P.; Kujawski, J.; Kruk, J.; Czaja, K.; Bernard, M.K.; Jagodziński, P.P. Cell-specific cytotoxic effect of pyrazole derivatives on breast cancer cell lines MCF7 and MDA-MB-231. J. Physiol. Pharmacol. 2017, 68, 201–207. [Google Scholar] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.rcsb.org/structure/3EWH (accessed on 5 March 2020).
- Cee, V.J.; Cheng, A.C.; Romero, K.; Bellon, S.; Mohr, C.; Whittington, D.A.; Bak, A.; Bready, J.; Caenepeel, S.; Coxon, A.; et al. Pyridyl-pyrimidine benzimidazole derivatives as potent, selective, and orally bioavailable inhibitors of Tie-2 kinase. Bioorg. Med. Chem. Lett. 2009, 19, 424–427. [Google Scholar] [CrossRef]
- Pathak, P.; Shukla, P.K.; Kumar, V.; Kumar, A.; Verma, A. Quinazoline clubbed 1,3,5-triazine derivatives as VEGFR2 kinase inhibitors: Design, synthesis, docking, in vitro cytotoxicity and in ovo antiangiogenic activity. Inflammopharmacology 2018, 26, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Muñoz, C.; Alzate-Morales, J.H.; Cunha, S.; Gano, L.; Bergmann, R.; Steinbach, J.; Kniess, T. Synthesis, in silico, in vitro, and in vivo investigation of 5-[11C]methoxy-substituted sunitinib, a tyrosine kinase inhibitor of VEGFR-2. Eur. J. Med. Chem. 2012, 58, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Temirak, A.; Shaker, Y.M.; Ragab, F.A.F.; Ali, M.M.; Ali, H.I.; El Diwani, H.I. Part I. Synthesis, biological evaluation and docking studies of new 2-furylbenzimidazoles as antiangiogenic agents. Eur. J. Med. Chem. 2014, 87, 868–880. [Google Scholar] [CrossRef]
- Abdelhafez, O.M.; Ali, H.I.; Amin, K.M.; Abdallae, M.M.; Ahmed, E.Y. Design, synthesis and anticancer activity of furochromone and benzofuran derivatives targeting VEGFR-2 tyrosine kinase. RSC Adv. 2015, 5, 25312–25324. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, T.K.; Batran, R.Z.; Elseginy, S.A.; Ali, M.M.; Mahmoud, A.E. Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis. Bioorg. Chem. 2019, 85, 253–273. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Wang, Y.; Fan, Y.; Chen, X.; Yang, Y.; Hua, Y.; Xie, W.; Lu, T.; Tang, W.; et al. Protein–ligand interaction-guided discovery of novel VEGFR-2 inhibitors. J. Biomol. Struct. Dyn. 2019, 2, 1–16. [Google Scholar] [CrossRef]
- Zhang, S.J.; Ding, Z.-S.; Jiang, F.S.; Ge, Q.-F.; Guo, D.-W.; Lid, H.-B.; Hu, W.-X. Synthesis, anticancer evaluation and docking study of vadimezan derivatives with carboxyl substitution. Med. Chem. Commun. 2014, 5, 512–520. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Zhang, D.; Wang, L.; Lu, T.; Jiao, Y. Discovery of Novel Potent VEGFR-2 Inhibitors Exerting Significant Antiproliferative Activity against Cancer Cell Lines. J. Med. Chem. 2018, 61, 140–157. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E., III; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting and Cholesky decomposition approximations in symmetry-adapted perturbation theory: Implementation and application to probe the nature of π-π interactions in linear acenes. J. Chem. Phys. 2010, 132, 184111. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, E.G.; Parrish, R.M.; Sherrill, C.D.; Turney, J.M.; Schaefer, H.F. Large-scale symmetry-adapted perturbation theory computations via density fitting and Laplace transformation techniques: Investigating the fundamental forces of DNA-intercalator interactions. J. Chem. Phys. 2011, 135, 174107. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef] [PubMed]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. The importance of three-body terms in the fragment molecular orbital method. J. Chem. Phys. 2004, 120, 6832–6840. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, D.G.; Kitaura, K. Pair interaction energy decomposition analysis. J. Comput. Chem. 2007, 28, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Śliwa, P.; Kurczab, R.; Kafel, R.; Drabczyk, A.; Jaśkowska, J. Recognition of repulsive and attractive regions of selected serotonin receptor binding site using FMO-EDA approach. J. Mol. Model. 2019, 25, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Available online: http://openmopac.net/MOPAC2016.html (accessed on 20 March 2020).
- Raha, K.; Merz, K.M., Jr. Large-scale validation of a quantum mechanics based scoring function: Predicting the binding affinity and the binding mode of a diverse set of protein-ligand complexes. J. Med. Chem. 2005, 48, 4558–4575. [Google Scholar] [CrossRef]
- Desmond Molecular Dynamics System, D.E. Shaw Research, New York, NY, 2020. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2020.
- Czaja, K.; Kujawski, J.; Kamel, K.; Bernard, M.K. Selected arylsulphonyl pyrazole derivatives as potential Chk1 kinase ligands—computational investigations. J. Mol. Model. 2020, 26, 144. [Google Scholar] [CrossRef]
- Stewart, J.P.P. Optimization of parameters for semi-empirical methodsVI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1994. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1996, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like SmallMolecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose–Hoover thermostat. J. Chem. Phys. 1985, 83, 4069. [Google Scholar] [CrossRef]
- Rogge, S.M.J.; Vanduyfhuys, L.; Ghysels, A.; Waroquier, M.; Verstraelen, T.; Maurin, G.; Van Speybroeck, V. A Comparison of Barostats for the Mechanical Characterization of Metal—Organic Frameworks. J. Chem. Theory Comput. 2015, 11, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pose | Estimated Binding Affinity of the Docked Azoles 1–9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand 1 | Ligand 2 | Ligand 3 | Ligand 4 | Ligand 5 | Ligand 6 | Ligand 7 | Ligand 8 | Ligand 9 | |
| 1 | −8.900 | −9.700 | −9.600 | −10.300 | −9.500 | −10.900 | −11.600 | −8.000 | −9.900 |
| 2 | −8.600 | −8.900 | −9.100 | −10.000 | −8.400 | −10.300 | −11.100 | −7.700 | −9.400 |
| HB | Contacts Calculated for Docked Azoles 1–9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand 1 | Ligand 2 | Ligand 3 | Ligand 4 | Ligand 5 | Ligand 6 | Ligand 7 | Ligand 8 | Ligand 9 | |
| N-H…O=CAla866 | × | × | × | × | × | × | × | 2.377 | × |
| CH…+H3NLys868 | × | × | × | × | × | × | 2.110 | × | × |
| CH3…O=CGlu885 | 2.932 | 2.587 | 2.968 | 2.877 | 2.943 | × | × | × | 2.533 |
| N-H…OThr916 | 2.607 | 2.577 | 2.604 | 2.619 | 2.608 | 2.656 | × | × | 2.684 |
| Npyridinic…H-OThr916 | 2.166 | 2.103 | 2.158 | 2.147 | 2.159 | 2.165 | × | × | 2.170 |
| N-H…O=CGlu917 | 2.274 | 2.266 | 2.249 | 2.263 | 2.255 | 2.263 | × | × | 2.298 |
| N-H…OCys919 | × | × | × | × | × | × | 2.067 | × | × |
| Amino Acid | Calculated Value of Total SAPT0 Energy for Docked Azoles 1–9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand 1 | Ligand 2 | Ligand 3 | Ligand 4 | Ligand 5 | Ligand 6 | Ligand 7 | Ligand 8 | Ligand 9 | |
| Ala866 | −3.350 | −3.700 | −3.350 | −3.550 | −3.400 | −3.400 | −2.270 | −6.270 | −3.830 |
| Glu885 | −1.450 | −0.780 | −1.660 | −1.400 | −1.500 | −1.190 | 0.200 | 10.230 | −0.820 |
| Thr916 | −7.760 | −6.980 | −7.740 | −7.330 | −7.870 | −7.690 | −0.640 | −5.240 | −7.200 |
| Glu917 | −9.250 | −9.160 | −9.090 | −8.820 | −9.380 | −8.980 | −2.630 | −0.840 | −9.030 |
| Phe918 | −1.110 | −2.210 | −1.890 | −2.080 | −1.810 | −3.680 | −2.470 | −0.840 | −1.310 |
| Cys919 | 2.740 | 2.020 | 3.940 | 3.750 | 1.640 | 1.040 | −5.700 | −0.690 | 5.470 |
| Phe1047 | −0.830 | −1.560 | −1.160 | −0.300 | −1.210 | −1.280 | −9.950 | −1.330 | 1.060 |
| Ligand | Calculated Value of Total Tie Energy for Docked Azoles 1–9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand 1 | Ligand 2 | Ligand 3 | Ligand 4 | Ligand 5 | Ligand 6 | Ligand 7 | Ligand 8 | Ligand 9 | |
| TIE | −42.900 | −61.600 | −49.300 | −56.200 | −56.800 | −66.500 | −45.700 | −36.500 | −48.400 |
| Amino Acid | Calculated Value of Etot Energy for Docked Azoles 1–9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand 1 | Ligand 2 | Ligand 3 | Ligand 4 | Ligand 5 | Ligand 6 | Ligand 7 | Ligand 8 | Ligand 9 | |
| Lys838 | nd | −3.050 | −3.090 | −4.640 | nd | −4.790 | nd | nd | nd |
| Ala866 | −2.270 | −2.780 | −2.670 | −3.000 | −2.120 | −2.120 | −1.680 | −0.220 | −3.030 |
| Lys868 | −3.910 | −6.380 | −5.460 | −6.050 | −3.370 | −4.870 | −4.770 | −2.150 | −4.850 |
| Glu885 | −6.930 | −6.410 | −4.960 | −5.460 | −7.170 | −7.170 | nd | −3.490 | −7.520 |
| Thr916 | −8.940 | −8.400 | −9.200 | −8.890 | −9.000 | −9.000 | −1.700 | −3.600 | −8.640 |
| Glu917 | −3.970 | −2.200 | −1.690 | −1.390 | −4.870 | −4.870 | −1.210 | −0.660 | −2.720 |
| Phe918 | −17.160 | −17.790 | −16.700 | −16.540 | −18.820 | −19.200 | −6.700 | −1.720 | −15.910 |
| Cys919 | 4.930 | 3.730 | 4.760 | 4.770 | 4.400 | 4.400 | −7.300 | −0.650 | 6.110 |
| Phe1047 | −3.210 | −4.390 | −3.410 | −2.670 | −3.620 | 3.620 | −10.470 | −3.900 | −1.450 |
| Contacts | Contacts Length Calculated for Optimized Azoles 1–9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand 1 | Ligand 2 | Ligand 3 | Ligand 4 | Ligand 5 | Ligand 6 | Ligand 7 | Ligand 8 | Ligand 9 | |
| N-H…O=CAla866 | × | × | × | × | × | × | × | 3.311 | × |
| CH…+H3NLys868 | × | × | × | × | × | × | 2.645 | × | × |
| CH3…O=CGlu885 | 2.377 | 2.107 | 2.130 | 2.443 | 2.313 | × | × | × | 2.213 |
| N-H…OThr916 | 3.710 | 2.498 | 2.909 | 2.512 | 4.258 | 2326 | × | × | 2.284 |
| Npyridinic…H-OThr916 | 4.213 | 3.841 | 3.879 | 3.097 | 4.457 | 2.609 | × | × | 3.235 |
| N-H…O=CGlu917 | 1.803 | 2.058 | 2.258 | 2.235 | 1.917 | 2.072 | × | × | 2.487 |
| N-H…OCys919 | × | × | × | × | × | × | 1.411 | × | × |
| Compound | HOF of Ligand (ΔHfcomplex(L)) | HOF of Protein (ΔHfcomplex(P)) | HOF of Complex (ΔHf(PL)) | ΔHint |
|---|---|---|---|---|
| 1 | −0.240 | −2438.770 | −2532.830 | −93.810 |
| 2 | 41.550 | −2117.010 | −2167.680 | −92.220 |
| 3 | 129.410 | −1993.880 | −1930.300 | −65.830 |
| 4 | 42.660 | −2 353.080 | −2 403.600 | −93.190 |
| 5 | 69.860 | −2154.820 | −2199.390 | −114.430 |
| 6 | 57.370 | −2264.260 | −2308.590 | −101.710 |
| 7 | 75.330 | −1875.310 | −1923.270 | −123.290 |
| 8 | 43.520 | −1562.010 | −1607.010 | −88.510 |
| 9 | 30.330 | −3622.170 | −3915.320 | −323.470 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czaja, K.; Kujawski, J.; Śliwa, P.; Kurczab, R.; Kujawski, R.; Stodolna, A.; Myślińska, A.; Bernard, M.K. Theoretical Investigations on Interactions of Arylsulphonyl Indazole Derivatives as Potential Ligands of VEGFR2 Kinase. Int. J. Mol. Sci. 2020, 21, 4793. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134793
Czaja K, Kujawski J, Śliwa P, Kurczab R, Kujawski R, Stodolna A, Myślińska A, Bernard MK. Theoretical Investigations on Interactions of Arylsulphonyl Indazole Derivatives as Potential Ligands of VEGFR2 Kinase. International Journal of Molecular Sciences. 2020; 21(13):4793. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134793
Chicago/Turabian StyleCzaja, Kornelia, Jacek Kujawski, Paweł Śliwa, Rafał Kurczab, Radosław Kujawski, Anna Stodolna, Agnieszka Myślińska, and Marek K. Bernard. 2020. "Theoretical Investigations on Interactions of Arylsulphonyl Indazole Derivatives as Potential Ligands of VEGFR2 Kinase" International Journal of Molecular Sciences 21, no. 13: 4793. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134793