Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches


1
Bill Walsh Translational Cancer Research Laboratory, Kolling Institute, Northern Sydney Local Health District, St Leonards, Sydney, NSW 2065, Australia
2
Northern Clinical School, Faculty of Medicine and Health, University of Sydney, Sydney, NSW 2006, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(13), 4806; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134806
Submission received: 9 June 2020
/
Revised: 3 July 2020
/
Accepted: 5 July 2020
/
Published: 7 July 2020
(This article belongs to the Special Issue Genetically Engineered Mice to Study Cancer)
Abstract
:High-grade serous epithelial ovarian cancer (HGSC) is the most aggressive subtype of epithelial ovarian cancer. The identification of germline and somatic mutations along with genomic information unveiled by The Cancer Genome Atlas (TCGA) and other studies has laid the foundation for establishing preclinical models with high fidelity to the molecular features of HGSC. Notwithstanding such progress, the field of HGSC research still lacks a model that is both robust and widely accessible. In this review, we discuss the recent advancements and utility of HGSC genetically engineered mouse models (GEMMs) to date. Further analysis and critique on alternative approaches to modelling HGSC considers technological advancements in somatic gene editing and modelling prototypic organs, capable of tumorigenesis, on a chip.
1. Introduction
Epithelial ovarian cancer (OC) is the third most common gynaecologic cancer [1], with patients experiencing the worst prognosis and highest mortality rate of all gynaecological malignancies [2]. OC is three times more lethal than breast cancer [3] and mortality is projected to increase significantly [4]. OC lethality is attributed to a problematic triad of asymptomatic tumour growth, delayed onset of symptoms, and current dearth of effective screening methods that together make early tumour detection difficult [5,6]. After typical late-stage diagnosis, treatment options include tumour debulking surgery, followed by chemotherapy with taxane and platinum, which OC patients initially respond well to but the incidence of recurrence with concurrent decrease in sensitivity to chemotherapeutic options is high [7].
Preclinical mouse models are essential for investigating carcinogenesis and testing new therapies, however, have proven somewhat difficult to achieve in OC. This is partly due to OC encompassing several distinct disease subtypes, each with a unique histological and molecular pathology. The four OC subtypes which have been classified histologically are mucinous, clear cell, endometrioid, and serous. In 2004, a broad classification separating ovarian cancers to type I and type II tumours was introduced [8], prompting the division between high- and low-grade serous OC. Further genomic analyses allowed for the identification of clusters of germline and somatic mutations, revealing molecular phenotypes underlying the existing histological subtypes [8,9,10], the findings of which are outlined below in Table 1. These molecular phenotypes have been used to guide the development of genetically engineered mouse models (GEMMs) that attempt to recapitulate the initiation and development of OC. Table 1 provides a summary of GEMMs that have been developed to resemble mucinous, endometrioid and low-grade serous OC. To our knowledge, no GEMMs have been developed that resemble clear cell OC. The most effort has undoubtedly gone into the development of GEMMs for the most common and aggressive subtype of OC, high-grade serous cancer (HGSC), which will be the main focus of this review. HGSC GEMMs have been reviewed in greater detail previously [11] however a summary of the most recent models is detailed in Table 1.
2. High-Grade Serous Ovarian Carcinoma
HGSC accounts for the largest proportion of deaths from epithelial ovarian cancer, with over 70% of deaths being attributed to the disease [36,37]. HGSC is the most aggressive type of epithelial OC, with a median progression-free survival (PFS) rate of 18 months [7]. Much of the molecular pathophysiology of HGSC has been uncovered by The Cancer Genome Atlas (TCGA) [30], through which HGSC can be further divided into the following four subtypes based on TCGA gene expression profiles: differentiated, immunoreactive, mesenchymal, and proliferative. Interestingly, the initial TCGA cohort did not show statistically significant differences in survival time between subtypes but combining the subtypes with prognostic genetic information allowed for the prognostic Classification of Ovarian Cancer (CLOVAR) model to be developed [38].
Through prognostic genomics it was shown that the immunoreactive subtype had the longest survival, the proliferative subtype had significantly worse overall survival than the immunoreactive subtype but obtained the greatest benefit from bevacizumab treatment, and that mesenchymal subtype patients had significantly worse overall survival than patients of the immunoreactive subtype [39]. A study introducing a novel histopathological classification system that was closely correlated with the TCGA molecular subtypes was able to also demonstrate that patients with mesenchymal tumours may be more sensitive to paclitaxel [40]. However, it should be noted that further studies have carefully microdissected tumour and stromal tissue to demonstrate that the TCGA mesenchymal and immunoreactive subtypes are likely to be a reflection of increased contribution tumour infiltrating and surrounding stromal cells [41,42] and highlighting the potential impact of the tumour microenvironment on patient survival and response to therapy. Given that HGSC can be further divided into distinct subtypes that have distinct survival profiles and response to therapies, this may have downstream effects on future selection and use of HGSC GEMMs, particularly in the context of emerging immunotherapy and tumour microenvironment-targeting therapies.
Interactions between tumour cells and non-malignant stromal cells – such as endothelial cells, fibroblasts, adipocytes, mature and circulating progenitor immune cells – have been shown to promote tumour initiation, growth, and metastasis using both in vitro and in vivo models [43]. The established interplay further underpins the necessity for a GEMM that accurately recapitulates HGSC; allowing for dynamic investigations on prevention, early detection, and development of new treatment strategies that consider the interplay between the tumour and its surrounding microenvironment. Such in vivo models offer dynamic insights that would not be exposed using cell lines and are limited in patient-derived xenograft models. However, there are relatively few robust GEMMs of HGSC despite our knowledge of the key driver mutations – this is predominantly due to historical contention of the cell of origin and the lack of reliable and specific promoters for the main two cell-of-origin contenders [44]. Also, due to long latency, complex breeding strategies and the associated expense, the promising GEMMs that have been developed have not extended into widespread use by ovarian cancer researchers. This review will highlight recent HGSC GEMM developments, discuss whether or not they are serving their purpose as preclinical research tools, and discuss new approaches and future directions for GEMMs in preclinical HGSC research.
3. GEMMs of HGSC—Summary and Updates
The path to developing a successful HGSC GEMM has been confounded by an initial lack of understanding of the cell-of-origin for HGSC. The primary consensus was that epithelial OCs, true to their name, originated in the ovarian surface epithelia (OSE) [45], however more recently it has been proposed that HGSC originates in the fimbriae of the fallopian tube (FT = oviduct in mice) [33,46,47]. The matter is further complicated by limited OSE or oviduct-specific promoters that can be used to target genetic changes to these cell types using the CRE-loxP system. Many OC GEMMs were developed in the early 2000s and these are reviewed in greater detail by Howell [11] and summarised below, with a greater focus given to the more recently developed models.
Early promoter-driven GEMMs relied on promoters that target the developing reproductive system such as anti-Müllerian hormone receptor 2 (Amhr2/MISIIR). As such, these models did not specifically target the OSE or oviduct, leading to the development of uterine leiomyosarcomas, granulosa cell tumours, low-grade serous ovarian tumours or HGSC depending on the genes targeted [48,49,50].
The lack of highly specific promoters for the OSE led to the development of a surgical technique by Flesken-Nikitin et al. [51] to drive genetic changes to the OSE. Briefly, the technique involves intra-bursal delivery of adenoviral CRE (ADCRE) into the space between the ovary and the ovarian bursal membrane. The method demands a high level of technical competence and consideration towards the construct used, estrous stage of the animal, solutions used, and the multiplicity of infection of the adenovirus to obtain reproducible results [11]. In addition, cells in structures surrounding the ovarian bursa, including the oviduct can also potentially be exposed to ADCRE due to proximity or through CRE leakages that may occur along the needle path [52]. Therefore, many of the models developed using this technique tended to have lower penetrance than promoter-driven models.
More recently, promoters have been developed to target mutations to the oviductal epithelial cells. These include paired box 8 (Pax8) and oviductal glycoprotein 1 (Ovgp1). An inducible Pax8 promoter (Pax8-rtTA;TetO-Cre) was used by Perets et al. to target knockout of Brca1/2, Trp53 and Pten in the secretory cells of the mouse oviduct [34], thereby effectively modelling HGSC originating in the fallopian tube (FT) at a 100% transformation rate when using homozygous Brca1 or heterozygous Brca2 mutations. Mutations to Brca1/2 resulted in significantly lower latency (13 weeks) than Tp53-/-/Pten-/- only mutants, with the models developing features consistent with serous tubular intra-epithelial carcinoma (STIC) lesions (secretory cell proliferation, loss of polarity, cellular atypia, and serous histology) and metastases patterns characteristic of HGSC. The success of this model is underpinned by the fact that PAX8 is a marker of secretory cells in the FT, but not the ovaries [53,54] and expressed in HGSC tumours [55,56] in humans and mice. However, the use of Pax8 as a promotor for the development of oviduct derived HGSC GEMMs is limited by its lack of specificity as Pax8 is also highly expressed in the thyroid and kidney. Models using Pax8 driven perturbations in Tp53 and T121 have resulted in enlarged thymi resulting in respiratory distress and subsequent early lethality [35].
Ovgp1 encodes for a large epithelial glycoprotein that is secreted from non-ciliated epithelial cells and has 100-fold higher expression in the oviduct compared to other tissues [57] and represents a more specific promoter for the development of oviduct-derived models of HGSC. Zhai et al. [34] crossed an existing Ovgp1-iCreERT2 transgenic line [25] with variations in alleles of Rb1, Trp53, Brca1, Nf1 and Pten. Mice with aberrant Brca1, Trp53, Rb1 and Nf1 (BPRN) and mice with aberrant Brca1, Trp53 and Nf1 (BPN) mice had the highest penetrance characteristic of HGSC, with the former demonstrating shortest latency. Further characterisation of BPRN showed these mice develop tumours highly correlating in transcriptional signatures with human HGSCs of the immunoreactive and mesenchymal TCGA subtypes [58]. However, it should be noted that off-target tumours occurred in a sizeable proportion of BPRN mice and included lymphoma, thyroid, lung, skin, and breast malignancies; this is not surprising given that some mice contained germline alterations in Brca1 and Trp53 and the long latency to tumour development. Finally, tumour development in mice harbouring Brca1, Trp53 and Pten aberrations (BPP mice) was the most rapid and tumours displayed phenotypes not seen in the other genotypes such as mucinous metaplasia. Interestingly, these are the same genetic aberrations Perets et al. used in their Pax8-driven HGSC model. However, they did not observe these features, raising the possibility that Pax8 and Ovgp1 are initiating carcinogenic changes in different cell populations.
Although later GEMMs have focused on FT/oviduct-derived HGSC, emerging molecular profiling suggests that the OSE may still represent the cell of origin for a significant proportion of HGSC [59,60]. These findings have been corroborated by proteomic analysis [61], and it is predicted that the two subtypes of HGSC demarcated by cell-of-origin may respond differently to therapy [62], with OSE-derived tumours demonstrating higher resistance to chemotherapy [61]. To explicate the matter, Zhang et al. [35] targeted identical genetic aberrations (Trp53 mutation and Rb inactivation) to either the oviduct or the OSE to directly compare tumours arising from different cell lineages. This study was instrumental in not only showing that HGSC can arise in both the oviduct and the OSE, but that the cell of origin impacts tumour growth, metastasis and response to therapy. Importantly, as part of this study, they managed to develop a new promoter-driven model of OSE-derived HGSC using Lgr5; a gene involved with OSE homeostasis and repair, with concentrated expression in the ovarian hilum in adult mice [63]. By successfully inducing HGSC in Lgr5CreERT2 mice, Zhang et al. provide further evidence for the dual origin of HGSC tumours.
As demonstrated by the above models, GEMMs attempting to recapitulate serous epithelial OC have focused on known molecular pathognomonic markers, such as mutations or loss of TP53, BRCA1/2, and RB, as well as alterations in genes not commonly associated with HGSC such as PTEN. It becomes clear from these GEMMs that to achieve the required genetic changes to induce HGSC in mice extensive breeding programs are required, which may act as an impediment for many researchers to use as they prove to be challenging, time-consuming, and expensive. To overcome this, SV40Tag has been widely used in the development of many GEMMs of cancer, including OC [64,65,66]. It is particularly relevant in HGSC as SV40Tag leads to the inactivation of p53 and Rb. Previous studies have demonstrated that SV40Tag activation with ADCRE or via the Amhr2 promoter can induce ovarian tumours in a relatively efficient manner [64,67,68]. However, the dearth of studies comparing this model to the molecular phenotype of human HGSC and the lack of OSE or FT specificity of the Amhr2 promoter may provide limitations to its use as a model to recapitulate human HGSC.
Despite recent improvements to GEMMs of HGSC their widespread use has not eventuated, we surmise that this is due to several factors – extensive breeding requirements to introduce up to 4 or more genetic alterations to the OSE or FTE, long latency to tumorigenesis (in excess of 12 months in some instances), and instances of low penetrance and metastatic spread.
4. Alternative Approaches and Future Directions
GEMMs offer the ideal system to recapitulate spontaneous tumour development through a process of transformation of normal cells to development of precursor lesions before formation of invasive and metastatic tumours, which is why GEMMs remain the platform of choice for investigations into HGSC prevention strategies and early carcinogenesis. However, GEMMs may not be essential to researchers wanting to test new therapies and further investigate the biology of established tumours, metastases, and tumour microenvironment. To this end, there are other approaches in use and emerging that may appeal to researchers as an alternative to GEMMs due to the models’ higher efficiency and reduced cost (summarised in Figure 1).
The tumour microenvironment is increasingly recognised to play a significant role in all cancers [69,70,71], including HGSC [72,73,74]. Therefore, a preclinical model that not only recapitulates the genomics of HGSC but also the intricacies of the tumour microenvironment is highly desirable. Considering the importance of the immune system in tumorigenesis and the increased interest in the use of immunotherapies to treat cancer, the use of human cell lines and patient-derived xenografts (PDXs) is limited and syngeneic mouse cell lines may be a better option to epitomise the interactions between the tumour and stroma.
4.1. Syngeneic Cell Lines
Several syngeneic ovarian cancer cell lines have been developed as models of HGSC. Tumour cell engraftment commonly occurs via intra-peritoneal (i.p.) injection in order to mimic the widespread metastatic disease seen in HGSC patients. The other major methods of engraftment include sub-cutaneous (s.c.) and sub-bursal injection. S.c. injection allows for rapid tumour development and ease in measuring tumour volume with a caliper, whilst i.p. and intrabursal injection allow for a physiologically relevant microenvironment [75]. However, models using i.p. and intrabursal injection commonly result in widespread peritoneal disease with widespread metastases [75], which while more clinically relevant can be more complex when analysing changes in tumour growth. When considering an engraftment location, it is imperative to consider the features of the ovarian cancer cell line being used as investigations have uncovered variations in cell line tumorigenicity when comparing s.c., i.p., and intrabursal xenografts [76]. Interestingly, when comparing gene expression of s.c. to i.p. tumours it was shown that there was no overlap between the s.c. and i.p. datasets and those cell lines showing preference for i.p. growth expressed genes more commonly represented in primary cancers [76]. Syngeneic ovarian cancer cell lines are most commonly injected i.p. and a summary of survival times for recipient mice is presented in Table 2.
The ID8 cell line is derived from explanted ovaries of C57Bl/6 mice, from which OSE was grown in vitro in the presence of EGF [80] and is the most widely used and recognised syngeneic mouse OC cell line. However, genomic analysis has shown that ID8 tumours are not highly representative of human HGSC, as they lack pathognomonic HGSC mutations in Trp53, Brca1, Brca2, Nf1, and Rb1 [77]. The study further showed that p53 remains transcriptionally active in ID8 tumours and that ID8 cells were able to form Rad51 foci in response to DNA double strand breaks, thereby demonstrating homologous recombination (HR) competence which are both features uncharacteristic of human HGSC. Walton et al. [77] were able to utilise the CRISPR/Cas9 system to engineer two mutated ID8 cell lines, one with a single Trp53 knockout (Trp53-/-) and another double knockout model with the addition of Brca2-/-(Trp53-/-;Brca2-/-). It was shown that the Trp53-/- and Trp53-/-;Brca2-/- models had significantly shorter median survival than the native ID8 orthotopic model when grown i.p. in mice, with the single knockout having the shortest median survival of the two (Table 2), demonstrating the importance of Trp53 knockout in survival latency. Morphological features of the Trp53-/- tumours were similar to the wild-type ID8 tumours, with the exception of increased peritoneal and diaphragmatic metastatic deposits in the former, whilst Trp53-/-;Brca2-/- cells formed significantly less ascites than native ID8 or the single knockout mice. As with HGSC tumours, Trp53-/-;Brca2-/- tumours did not display HR competence. Interestingly, when comparing the tumour microenvironment of the Trp53-/- and Trp53-/-;Brca2-/- models with the native ID8 model, each variation displayed phenotypically different immune infiltrates. Tumours derived within the single knockout model had significantly higher levels of macrophage infiltration than ID8 and Trp53-/-;Brca2-/- tumours; whilst the double knockout model developed tumours with the presence of CD3+ predominant lymphoid aggregates, with CD8+ populations on the periphery. There was no evidence of lymphoid aggregates in the Trp53-/- or ID8 tumours. The phenotypic variation in immune cell infiltrates highlights the importance of using an accurate syngeneic cell line to develop a reliable immunocompetent model, capable of accurately reflecting the interactions between the tumour and the microenvironment.
A follow up study [78], again using ID8 cells, included edits to Brca1 (Trp53-/-;Brca1-/-), Nf1 (Trp53-/-;Nf1-/-), Pten (Trp53-/-;Pten-/-) and a triple knockout with Pten (Trp53-/-;Brca2-/-;Pten-/-). When comparing survival in mice with Trp53-/- tumours, it was shown that there was no significant difference in survival of mice with Trp53-/-;Brca1-/- tumours (46 days), but Trp53-/-;Nf1-/- and Trp53-/-;Pten-/- tumours displayed significantly shorter survival (Table 2). Tumours generated from the triple deletion (Trp53-/-;Brca2-/-;Pten-/-) had significantly shorter survival (40 days) than Trp53-/- and Trp53-/-;Brca2-/- tumours (57 days), but longer survival than Trp53-/-;Pten-/-. Mutants containing edits to Brca1/2 were significantly more sensitive to PARP inhibition with rucaparib treatment in vitro than those with single mutations in Trp53 only. There was no significant difference in rucaparib sensitivity between Trp53-/-;Brca2-/- and Trp53-/-;Brca2-/-;Pten-/- cells in vitro and tumours in vivo. Whilst results for cisplatin therapy mirrored those of rucaparib in vitro, in vivo cisplatin administration lead to significant differences in survival times with mice harbouring the Trp53-/-;Brca2-/- tumours exhibiting the longest survival and Trp53-/-;Nf1-/- and Trp53-/-;Pten-/- tumours having the worst survival. Thereby demonstrating that tumours derived from differing mutations, will respond differently to treatment and highlighting the importance of choosing multiple syngeneic cell lines for preclinical testing of therapies. Further characterisation of these cell lines is required to determine which human HGSC subtype they most closely resemble.
Four polyclonal cell lines derived from the Pax8-rtTA; TetO-Cre; Brca1loxP/loxP; Trp53mut; PtenloxP/loxP GEMM [33] backcrossed onto a C57Bl/6 background were used to develop a syngeneic mouse model of FT-originating HGSC [79]. The study verified genomic concordance between syngeneic models derived from the polyclonal cells and human HGSC in terms of key orthologous gene copy number alterations (CNAs) and genes encoding for transcription factors. It was further shown through transcriptional network analysis that the polyclonal cell transplants lead to the development of tumours that reflect the transcriptional patterns of the human HGSC tumour microenvironment. Immune cell profiles varied between the metastases of each polyclonal cell line, with one (HGS1) showing the closest resemblance to human HGSC metastases, with immune cell analysis of tumours showing that there were significantly fewer CD3+ T cells in all four polyclonal cell-derived mouse tumours compared to human HGSC. Upon assessing the stroma, it was shown that the models demonstrated enhanced expression of collagens, matrisome glycoproteins, proteoglycans, ECM regulators, and ECM-secreted factors compared to omental controls. As with human HGSC tumours, those developed in the syngeneic mouse models had an increased expression of matrisome genes, comparable ECM stiffness, and a correlating increase in the Matrix Index [81]. The upregulation of six matrisome molecules (fibronectin 1, versican, collagens 1A1 and 11A1, cathepsin V, and cartilage oligometric matrix protein) is associated with disease score [81] in human HGSC; all six were detected in the aforementioned polyclonal cell-derived syngeneic models but to varying extents.
The examples listed above demonstrate how a syngeneic mouse model can be used to circumvent the extensive and time-consuming breeding programs required to establish a GEMM, by using a more cost-effective transplantable model of HGSC derived from either the OSE or FT.
4.2. Organoids
Organoids are a way of modelling 3D multicellular interactions in vitro and have recently been gaining more attention for cancer modelling. Briefly, organoids are grown in culture using pluripotent stem cells or organ progenitors that differentiate and self-organise to form an organ-like structure that behaves much like the archetypical organ in vivo. Such cellular organisation is feasible due to the use of a 3D cell culture matrix comprised of Matrigel, small molecules and growth factors that mimics the basement membrane. Organoid lines derived from surgically resected primary human HGSC tumours and metastatic lesions have been developed from both FT and OSE origin [82]. It should be noted that there was a high level of morphological variation in the HGSC organoids, with varying degrees of cellular organisation, and an absence of vessel and connective tissue elements. Majority of the organoids demonstrated aneuploidy and CNAs characteristic of OC. However, there were a minority subgroup that did not. Interestingly, CNAs were maintained after prolonged passage, making them attractive for in vitro use. The organoids showed conserved aberrations characteristic of HGSC, such as mutations in TP53, and loss of RB1 and PTEN and a maintained methylation profile after passaging. It was further demonstrated that HGSC-modelling organoids can be genetically modified in vitro in a stable manner. Organoid lines can be retransplanted into an immunocompromised mouse as an orthotopic xenograft and have been shown to develop tumours that maintain high fidelity with the original tumour and organoid in bladder [38] and liver [83] cancer. Therefore, future studies should aim to develop models of HGSC by transplanting in vitro modified organoids into a xenograft model.
Another efficient manner of using organoids for HGSC modelling in vivo is similar to the syngeneic cell lines discussed above, wherein the organoid is developed directly from a GEMM, transformed in vitro and then transplanted back into recipient mice. This method has been carried out effectively by Zhang et al. [35] to develop FT- and OSE-derived organoids, by targeting Pax8 and Lgr5+ respectively, and shown to develop tumours comparable to human HGSC tumours. The utility of this model is compounded by being able to transform organoids in vitro to direct mutations to the right cell of origin and circumvent off-target issues associated with the use of the Pax8 and Lgr5+ promoters. Another promising feature of organoids includes the ability to form human microtissues from primary cancer cells [84].
4.3. Somatic Gene Editing
A common criticism of GEMMs in cancer is that many of them are developed by using promoters that are expressed embryonically, resulting in loss of tumour suppressor genes and/or expression of oncogenes early on in development rather than in the adult organ. This can be circumvented by using inducible CRE systems such as the TetOCre or tamoxifen-inducible CRE. However, as seen in the recent paper by Zhang et al. [35], at times there is inherent “leakiness” of CRE that may interfere and lead to activation of genes early and in the wrong locations. With the increase in use of gene editing technologies, such as CRISPR/Cas9, new GEMMs are being developed that involve somatic gene edits in living adult animals, which is more akin to how cancers arise in the adult.
The CRISPR/Cas9 system is a powerful tool capable of recognising specific gene sequences and inducing a double-stranded break through non-homologous end joining (NHEJ) to induce somatic gene edits without having to undertake the onerous, time-consuming and expensive crossbreeding involved with developing a GEMM. Further, by inducing such edits somatically in adult mice, CRISPR/Cas9 allows for a more accurate representation of the sporadic mutations typical of tumorigenesis. Modifications to the Cas9 nuclease has allowed for a wide scope of applications that have been comprehensively reviewed by Anzalone et al. [85]. A significant drawback of this system, however, is the complexity involved in somatic CRISPR delivery. Direct DNA transfection has been shown in multiple tissue types, including the liver [86], pancreas [87], and brain [88] but is limited to a few organ types. To overcome these limitations, most CRISPR models to date rely on viral vectors such as adenovirus and adeno-associated viruses. Viral mediated delivery has been used to induce combinatorial alterations in a lung cancer model in vivo [89], demonstrating that the tool could be harnessed to induce the multiple pathognomonic HGSC genomic aberrations. An important consideration for using CRISPR/Cas9 systems for somatic gene editing is low in vivo editing efficiency of 1-5% or lower. Although the complete mechanisms underlying low editing efficiency remain to be elucidated, studies have shown that primary cells prefer to undergo apoptosis [90] or use NHEJ [91]. It has been postulated that this is due to wild-type p53 being activated by Cas9 mediated double strand breaks to initiate growth arrest and apoptosis [92,93]. There has yet to be a CRISPR/Cas9 HGSC model developed to date. However, given that viral delivery has previously been used to deliver CRE recombinase to the OSE/oviduct, somatically edited GEMMs of HGSC would be feasible.
4.4. Tumour-on-a-Chip
The tumour-on-a-chip systems are microfluidic devices that aim to recapitulate features of tumour physiology. The system substrate is commonly glass or optically clear polymers with perfused, hollow microchannels. Typically, two or more cell types are set up within these channels and the system can be set up to have the cell-to-cell interactions occurring directly or indirectly. The intricacy of this system and the addition of microfluidics adds a dimension of physiological mimicry and gives the researcher precise control over variables [94]. The simplest version of a tumor-on-a-chip system is comprised of a tumour spheroid placed in a microfluidic system, whilst more complex versions can include 3D cancer tissues in contact with non-cancerous tissues and spheroids surrounded by healthy tissues [95]. These models are bolstered by the possibility of using human derived spheroids or organoids for a more accurate recapitulation of patient tumours with lung cancer models having been used to investigate the interactions between tumour cells and CAFs [96] and drug sensitivity [97]. The tumour-on-a-chip system has been used to model HGSC using CRISPR-Cas9 edited TP53 knockouts in dog oviductal epithelia, wherein cells lost normal morphology, exhibited increased proliferation and DNA double strand breaks [98]. Upon genetic analysis the TP53 knockouts had a decreased expression of PTEN and RB1 but no changes on BRCA1/2. This study is a promising first step in modelling HGSC on a chip, however further studies investigating interactions between the tumour and stroma are necessary to further validate this method.
5. Conclusions
Compared to other tumour types, GEMMs of HGSC have been notoriously difficult to develop. Contention surrounding the cell of origin, the lack of robust and specific promoters, extensive breeding requirements, and long latency to tumour development have all contributed to these difficulties. Despite the development of several models that closely resemble HGSC, the aforementioned challenges have meant that these GEMMs have not been widely adopted for preclinical research. New technologies are rapidly emerging that will lead to the development of better GEMMs or provide strong alternatives for their use in preclinical research.
Author Contributions
Conceptualization, R.Z., V.M.H. and E.K.C.; writing—original draft preparation, R.Z.; writing—review and editing, V.M.H. and E.K.C. All authors have read and agreed to the published version of the manuscript.
Funding
R.Z. is funded by the Jane Sherrard Post-Doctoral Fellowship; V.M.H. is funded by the McNish Fellowship; E.K.C. is funded by the Proud Family Fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| HGSC | High-grade serous epithelial ovarian cancer |
| TCGA | The Cancer Genome Atlas |
| GEMM | Genetically engineered mouse model |
| OC | Ovarian cancer |
| OSE | Ovarian surface epithelia |
| Amhr2 | Anti-Mullerian hormone receptor 2 gene |
| ADCRE | Adenoviral CRE |
| Pax8 | Paired box 8 gene |
| Ovgp1 | Oviductal glycoprotein 1 gene |
| PDX | Patient-derived xenograft |
| i.p. | Intra-peritoneal |
| s.c. | Sub-cutaneous |
| CAN | Copy number alterations |
| ECM | Extracellular matrix |
| CLOVAR | Classification of Ovarian Cancer |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, B.W.; Kleihues, P. World Cancer Report; IARC Press: Lyon, France, 2003. [Google Scholar]
- Yoneda, A.; Lendorf, M.E.; Couchman, J.R.; Multhaupt, H.A. Breast and Ovarian Cancers: A Survey and Possible Roles for the Cell Surface Heparan Sulfate Proteoglycans. J. Histochem. Cytochem. 2012, 60, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Coburn, S.B.; Bray, F.; Sherman, M.E.; Trabert, B. International Patterns and Trends in Ovarian Cancer Incidence, Overall and by Histologic Subtype. Int. J. Cancer 2017, 140, 2451–2460. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, I.J.; Menon, U. Progress and Challenges in Screening for Early Detection of Ovarian Cancer. Mol. Cell Proteom. 2004, 3, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Badgwell, D.; Bast, R.C., Jr. Early Detection of Ovarian Cancer. Dis. Markers 2007, 23, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond Chemotherapy: Targeted Therapies in Ovarian Cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef]
- Shih, I.M.; Kurman, R.J. Ovarian Tumorigenesis: A Proposed Model Based on Morphological and Molecular Genetic Analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Köbel, M.; Kalloger, S.E.; Boyd, N.; McKinney, S.; Mehl, E.; Palmer, C.; Leung, S.; Bowen, N.J.; Lonescu, D.N.; Prentice, L.M.; et al. Ovarian Carcinoma Subtypes Are Different Diseases: Implications for Biomarker Studies. PLoS Med. 2008, 5, e232. [Google Scholar] [CrossRef]
- Ayhan, A.; Kurman, R.J.; Vang, R.; Logani, S.; Seidman, J.D.; Shih, I.M. Defining the Cut Point between Low-Grade and High-Grade Ovarian Serous Carcinomas: A Clinicopathologic and Molecular Genetic Analysis. Am. J. Surg. Pathol. 2009, 33, 1220–1224. [Google Scholar] [CrossRef] [Green Version]
- Howell, V.M. Genetically Engineered Mouse Models for Epithelial Ovarian Cancer: Are We There Yet? Semin. Cell Dev. Biol. 2014, 27, 106–117. [Google Scholar] [CrossRef]
- Cheasley, D.; Wakefield, M.J.; Ryland, G.L.; Allan, P.E.; Alsop, K.; Amarasinghe, K.C.; Ananda, S.; Anglesio, M.S.; Au-Yeung, G.; Bowtell, D.D.; et al. The Molecular Origin and Taxonomy of Mucinous Ovarian Carcinoma. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gemignani, M.L.; Schlaerth, A.C.; Bogomolniy, F.; Barakat, R.R.; Lin, O.; Soslow, R.; Venkatraman, E.; Boyd, J. Role of Kras and Braf Gene Mutations in Mucinous Ovarian Carcinoma. Gynecol. Oncol. 2003, 90, 378–381. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Kommoss, S.; Tolcher, M.C.; Clarke, B.; Galletta, L.; Porter, H.; Damaraju, S.; Fereday, S.; Winterhoff, B.J.; Senz, J.; et al. Molecular Characterization of Mucinous Ovarian Tumours Supports a Stratified Treatment Approach with Her2 Targeting in 19% of Carcinomas. J. Pathol. 2013, 229, 111–120. [Google Scholar] [CrossRef]
- Kelemen, L.E.; Lawrenson, K.; Tyrer, J.; Li, Q.; Lee, J.M.; Seo, J.H.; Phelan, C.M.; Beesley, J.; Chen, X.; Aben, K.K.; et al. Genome-Wide Significant Risk Associations for Mucinous Ovarian Carcinoma. Nat. Genet. 2015, 47, 888–897. [Google Scholar]
- Ren, Y.A.; Mullany, L.K.; Liu, Z.; Herron, A.J.; Wong, K.K.; Richards, J.S. Mutant P53 Promotes Epithelial Ovarian Cancer by Regulating Tumor Differentiation, Metastasis, and Responsiveness to Steroid Hormones. Cancer Res. 2016, 76, 2206–2218. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Yang, W.; et al. Arid1a Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Anglesio, M.S.; Carey, M.S.; Kobel, M.; Mackay, H.; Huntsman, D.G. Clear Cell Carcinoma of the Ovary: A Report from the First Ovarian Clear Cell Symposium, June 24th, 2010. Gynecol. Oncol. 2011, 121, 407–415. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Mandai, M.; Oura, T.; Matsumura, N.; Hamanishi, J.; Baba, T.; Matsui, S.; Murphy, S.K.; Konishi, I. Identification of an Ovarian Clear Cell Carcinoma Gene Signature That Reflects Inherent Disease Biology and the Carcinogenic Processes. Oncogene 2010, 29, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, Y.; Tsuda, H.; Inoue, T.; Berkowitz, R.S.; Mok, S.C. Pten Expression in Clear Cell Adenocarcinoma of the Ovary. Gynecol. Oncol. 2006, 101, 71–75. [Google Scholar] [CrossRef]
- McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; Yang, W.; Senz, J.; Chow, C.; Heravi-Moussavi, A.; Morin, G.B.; Mes-Masson, A.M.; Carey, M.S.; et al. Subtype-Specific Mutation of Ppp2r1a in Endometrial and Ovarian Carcinomas. J. Pathol. 2011, 223, 567–573. [Google Scholar] [CrossRef]
- Catasús, L.; Bussaglia, E.; Rodríguez, I.; Gallardo, A.; Pons, C.; Irving, J.A.; Prat, J. Molecular Genetic Alterations in Endometrioid Carcinomas of the Ovary: Similar Frequency of Beta-Catenin Abnormalities but Lower Rate of Microsatellite Instability and Pten Alterations Than in Uterine Endometrioid Carcinomas. Hum. Pathol. 2004, 35, 1360–1368. [Google Scholar] [CrossRef]
- Dinulescu, D.M.; Ince, T.A.; Quade, B.J.; Shafer, S.A.; Crowley, D.; Jacks, T. Role of K-Ras and Pten in the Development of Mouse Models of Endometriosis and Endometrioid Ovarian Cancer. Nat. Med. 2005, 11, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Hendrix-Lucas, N.; Kuick, R.; Zhai, Y.; Schwartz, D.R.; Akyol, A.; Hanash, S.; Misek, D.E.; Katabuchi, H.; Williams, B.O.; et al. Mouse Model of Human Ovarian Endometrioid Adenocarcinoma Based on Somatic Defects in the Wnt/Β-Catenin and Pi3k/Pten Signaling Pathways. Cancer Cell 2007, 11, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Zhai, Y.; Kuick, R.; Karnezis, A.N.; Garcia, P.; Naseem, A.; Hu, T.C.; Fearon, E.R.; Cho, K.R. Impact of Oviductal Versus Ovarian Epithelial Cell of Origin on Ovarian Endometrioid Carcinoma Phenotype in the Mouse. J. Pathol. 2016, 240, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nakayama, K.; Ishibashi, T.; Ishikawa, N.; Ishikawa, M.; Katagiri, H.; Minamoto, T.; Sato, E.; Sanuki, K.; Yamashita, H.; et al. Kras/Braf Analysis in Ovarian Low-Grade Serous Carcinoma Having Synchronous All Pathological Precursor Regions. Int. J. Mol. Sci. 2016, 17, 625. [Google Scholar] [CrossRef] [Green Version]
- Singer, G.; Oldt, R., 3rd; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih, I.M. Mutations in Braf and Kras Characterize the Development of Low-Grade Ovarian Serous Carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [Green Version]
- Mullany, L.K.; Fan, H.Y.; Liu, Z.; White, L.D.; Marshall, A.; Gunaratne, P.; Anderson, M.L.; Creighton, C.J.; Xin, L.; Deavers, M.; et al. Molecular and Functional Characteristics of Ovarian Surface Epithelial Cells Transformed by Krasg12d and Loss of Pten in a Mouse Model in Vivo. Oncogene 2011, 30, 3522–3536. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; DeFazio, A.; et al. Driver Mutations in Tp53 Are Ubiquitous in High Grade Serous Carcinoma of the Ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of Brca1 and Brca2 Mutations with Survival, Chemotherapy Sensitivity, and Gene Mutator Phenotype in Patients with Ovarian Cancer. Jama 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Milea, A.; George, S.H.; Matevski, D.; Jiang, H.; Madunic, M.; Berman, H.K.; Gauthier, M.L.; Gallie, B.; Shaw, P.A. Retinoblastoma Pathway Deregulatory Mechanisms Determine Clinical Outcome in High-Grade Serous Ovarian Carcinoma. Mod. Pathol. 2014, 27, 991–1001. [Google Scholar] [CrossRef] [Green Version]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the Fallopian Tube Secretory Epithelium Leads to High-Grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-Grade Serous Carcinomas Arise in the Mouse Oviduct Via Defects Linked to the Human Disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Dolgalev, I.; Zhang, T.; Ran, H.; Levine, D.A.; Neel, B.G. Both Fallopian Tube and Ovarian Surface Epithelium Are Cells-of-Origin for High-Grade Serous Ovarian Carcinoma. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilks, C.B.; Ionescu, D.N.; Kalloger, S.E.; Köbel, M.; Irving, J.; Clarke, B.; Santos, J.; Le, N.; Moravan, V.; Swenerton, K. Tumor Cell Type Can Be Reproducibly Diagnosed and Is of Independent Prognostic Significance in Patients with Maximally Debulked Ovarian Carcinoma. Hum. Pathol. 2008, 39, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D. The Genesis and Evolution of High-Grade Serous Ovarian Cancer. Nat. Rev. Cancer 2010, 10, 803–808. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically Relevant Gene Signatures of High-Grade Serous Ovarian Carcinoma. J. Clin. Invest. 2013, 123, 517–525. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and Therapeutic Relevance of Molecular Subtypes in High-Grade Serous Ovarian Cancer. J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Jing, Y.; Zhang, M.; Zhang, Z.; Ma, P.; Peng, H.; Shi, K.; Gao, W.Q.; Zhuang, G. Stroma-Associated Master Regulators of Molecular Subtypes Predict Patient Prognosis in Ovarian Cancer. Sci. Rep. 2015, 5, 16066. [Google Scholar] [CrossRef] [Green Version]
- Schwede, M.; Waldron, L.; Mok, S.C.; Wei, W.; Basunia, A.; Merritt, M.A.; Mitsiades, C.S.; Parmigiani, G.; Harrington, D.P.; Quackenbush, J.; et al. The Impact of Stroma Admixture on Molecular Subtypes and Prognostic Gene Signatures in Serous Ovarian Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.H.; Bast, R.C. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef] [Green Version]
- Auersperg, N.; Wong, A.S.; Choi, K.C.; Kang, S.K.; Leung, P.C. Ovarian Surface Epithelium: Biology, Endocrinology, and Pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Jarboe, E.; Folkins, A.; Nucci, M.R.; Kindelberger, D.; Drapkin, R.; Miron, A.; Lee, Y.; Crum, C.P. Serous Carcinogenesis in the Fallopian Tube: A Descriptive Classification. Int. J. Gynecol. Pathol. 2008, 27, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih, I.M. Tp53 Mutations in Serous Tubal Intraepithelial Carcinoma and Concurrent Pelvic High-Grade Serous Carcinoma—Evidence Supporting the Clonal Relationship of the Two Lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.Y.; Liu, Z.; Paquet, M.; Wang, J.; Lydon, J.P.; DeMayo, F.J.; Richards, J.S. Cell Type–Specific Targeted Mutations of Kras and Pten Document Proliferation Arrest in Granulosa Cells Versus Oncogenic Insult to Ovarian Surface Epithelial Cells. Cancer Res. 2009, 69, 6463–6472. [Google Scholar] [CrossRef] [Green Version]
- Jamin, S.P.; Arango, N.A.; Mishina, Y.; Hanks, M.C.; Behringer, R.R. Requirement of Bmpr1a for Müllerian Duct Regression During Male Sexual Development. Nat. Genet. 2002, 32, 408–410. [Google Scholar] [CrossRef]
- Xing, D.; Scangas, G.; Nitta, M.; He, L.; Xu, X.; Ioffe, Y.J.; Aspuria, P.J.; Hedvat, C.Y.; Anderson, M.L.; Oliva, E.; et al. A Role for Brca1 in Uterine Leiomyosarcoma. Cancer Res. 2009, 69, 8231–8235. [Google Scholar] [CrossRef] [Green Version]
- Flesken-Nikitin, A.; Choi, K.C.; Eng, J.P.; Shmidt, E.N.; Nikitin, A.Y. Induction of Carcinogenesis by Concurrent Inactivation of P53 and Rb1 in the Mouse Ovarian Surface Epithelium. Cancer Res. 2003, 63, 3459–3463. [Google Scholar]
- Šale, S.; Orsulic, S. Models of Ovarian Cancer Metastasis: Murine Models. Drug Discov. Today Dis. Models 2006, 3, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Mittag, J.; Winterhager, E.; Bauer, K.; Grummer, R. Congenital Hypothyroid Female Pax8-Deficient Mice Are Infertile Despite Thyroid Hormone Replacement Therapy. Endocrinology 2007, 148, 719–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, N.J.; Logani, S.; Dickerson, E.B.; Kapa, L.B.; Akhtar, M.; Benigno, B.B.; McDonald, J.F. Emerging Roles for Pax8 in Ovarian Cancer and Endosalpingeal Development. Gynecol. Oncol. 2007, 104, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Tacha, D.; Zhou, D.; Cheng, L. Expression of Pax8 in Normal and Neoplastic Tissues: A Comprehensive Immunohistochemical Study. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Laury, A.R.; Hornick, J.L.; Perets, R.; Krane, J.F.; Corson, J.; Drapkin, R.; Hirsch, M.S. Pax8 Reliably Distinguishes Ovarian Serous Tumors from Malignant Mesothelioma. Am. J. Surg. Pathol. 2010, 34, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, E.B.; Verhage, H.G.; Jaffe, R.C. Complementary Deoxyribonucleic Acid Cloning and Molecular Characterization of an Estrogen-Dependent Human Oviductal Glycoprotein1. Biol. Reprod. 1994, 51, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCool, K.W.; Freeman, Z.T.; Zhai, Y.; Wu, R.; Hu, K.; Liu, C.J.; Tomlins, S.A.; Fearon, E.R.; Magnuson, B.; Kuick, R.; et al. Murine Oviductal High-Grade Serous Carcinomas Mirror the Genomic Alterations, Gene Expression Profiles, and Immune Microenvironment of Their Human Counterparts. Cancer Res. 2020, 80, 877–889. [Google Scholar] [CrossRef]
- Lawrenson, K.; Fonseca, M.A.; Segato, F.; Lee, J.M.; Corona, R.I.; Seo, J.H.; Coetzee, S.; Lin, Y.G.; Pejovic, T.; Mhawech-Fauceglia, P.; et al. Integrated Molecular Profiling Studies to Characterize the Cellular Origins of High-Grade Serous Ovarian Cancer. bioRxiv 2018, 330597. [Google Scholar] [CrossRef] [Green Version]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular Analysis of High-Grade Serous Ovarian Carcinoma with and without Associated Serous Tubal Intra-Epithelial Carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative Proteomic Profiling of Ovarian Cancer Cell Lines Reveals Precursor Cell Associated Proteins and Functional Status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Hao, D.; Li, J.; Jia, S.; Meng, Y.; Zhang, C.; Wang, L.; Di, L.J. Integrated Analysis Reveals Tubal- and Ovarian-Originated Serous Ovarian Cancer and Predicts Differential Therapeutic Responses. Clin. Cancer Res. 2017, 23, 7400–7411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, A.; Tan, S.; Singh, G.; Rizk, P.; Swathi, Y.; Tan, T.Z.; Huang, R.Y.J.; Leushacke, M.; Barker, N. Lgr5 Marks Stem/Progenitor Cells in Ovary and Tubal Epithelia. Nat. Cell Biol. 2014, 16, 745–757. [Google Scholar] [CrossRef]
- Connolly, D.C.; Bao, R.; Nikitin, A.Y.; Stephens, K.C.; Poole, T.W.; Hua, X.; Harris, S.S.; Vanderhyden, B.C.; Hamilton, T.C. Female Mice Chimeric for Expression of the Simian Virus 40 Tag under Control of the Misiir Promoter Develop Epithelial Ovarian Cancer. Cancer Res. 2003, 63, 1389–1397. [Google Scholar]
- Hensley, H.; Quinn, B.A.; Wolf, R.L.; Litwin, S.L.; Mabuchi, S.; Williams, S.J.; Williams, C.; Hamilton, T.C.; Connolly, D.C. Magnetic Resonance Imaging for Detection and Determination of Tumor Volume in a Genetically Engineered Mouse Model of Ovarian Cancer. Cancer Biol. Ther. 2007, 6, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, S.; Altomare, D.A.; Connolly, D.C.; Klein-Szanto, A.; Litwin, S.; Hoelzle, M.K.; Hensley, H.H.; Hamilton, T.C.; Testa, J.R. Rad001 (Everolimus) Delays Tumor Onset and Progression in a Transgenic Mouse Model of Ovarian Cancer. Cancer Res. 2007, 67, 2408–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabova, L.; Yin, C.; Bupp, S.; Guerin, T.M.; Schlomer, J.J.; Householder, D.B.; Baran, M.L.; Yi, M.; Song, Y.; Sun, W.; et al. Perturbation of Rb, P53, and Brca1 or Brca2 Cooperate in Inducing Metastatic Serous Epithelial Ovarian Cancer. Cancer Res. 2012, 72, 4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laviolette, L.A.; Garson, K.; Macdonald, E.A.; Senterman, M.K.; Courville, K.; Crane, C.A.; Vanderhyden, B.C. 17β-Estradiol Accelerates Tumor Onset and Decreases Survival in a Transgenic Mouse Model of Ovarian Cancer. Endocrinology 2010, 151, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Bissell, M.J.; LaBarge, M.A. Context, Tissue Plasticity, and Cancer: Are Tumor Stem Cells Also Regulated by the Microenvironment? Cancer Cell 2005, 7, 17–23. [Google Scholar]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal Fibroblasts in Cancer Initiation and Progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Bissell, M.J.; Kenny, P.A.; Radisky, D.C. Microenvironmental Regulators of Tissue Structure and Function Also Regulate Tumor Induction and Progression: The Role of Extracellular Matrix and Its Degrading Enzymes. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2005. [Google Scholar]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. Tgf-Β Modulates Ovarian Cancer Invasion by Upregulating Caf-Derived Versican in the Tumor Microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A Collagen-Remodeling Gene Signature Regulated by Tgf-Β Signaling Is Associated with Metastasis and Poor Survival in Serous Ovarian Cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, D.C.; Hensley, H.H. Xenograft and Transgenic Mouse Models of Epithelial Ovarian Cancer and Non-Invasive Imaging Modalities to Monitor Ovarian Tumor Growth in Situ: Applications in Evaluating Novel Therapeutic Agents. Curr. Protoc. Pharmacol. 2009, 45, Unit14.12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, L.; Kim, M.K.; Lyle, L.T.; Bunch, K.P.; House, C.D.; Ning, F.; Noonan, A.M.; Annunziata, C.M. Characterization of Ovarian Cancer Cell Lines as in Vivo Models for Preclinical Studies. Gynecol. Oncol. 2016, 142, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. Crispr/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef] [Green Version]
- Walton, J.B.; Farquharson, M.; Mason, S.; Port, J.; Kruspig, B.; Dowson, S.; Stevenson, D.; Murphy, D.; Matzuk, M.; Kim, J.; et al. Crispr/Cas9-Derived Models of Ovarian High Grade Serous Carcinoma Targeting Brca1, Pten and Nf1, and Correlation with Platinum Sensitivity. Sci. Rep. 2017, 7, 16827. [Google Scholar] [CrossRef]
- Maniati, E.; Berlato, C.; Gopinathan, G.; Heath, O.; Kotantaki, P.; Lakhani, A.; McDermott, J.; Pegrum, C.; Delaine-Smith, R.M.; Pearce, O.M.; et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep. 2020, 30, 525–540. [Google Scholar] [CrossRef] [Green Version]
- Roby, K.F.; Taylor, C.C.; Sweetwood, J.P.; Cheng, Y.; Pace, J.L.; Tawfik, O.; Persons, D.L.; Smith, P.G.; Terranova, P.F. Development of a Syngeneic Mouse Model for Events Related to Ovarian Cancer. Carcinogenesis 2000, 21, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Böhm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An Organoid Platform for Ovarian Cancer Captures Intra- and Interpatient Heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with Crispr–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Öllinger, R.; Friedrich, M.; Ehmer, U.; Barenboim, M.; Steiger, K.; Heid, I.; Mueller, S.; Maresch, R.; Engleitner, T.; et al. Crispr/Cas9 Somatic Multiplex-Mutagenesis for High-Throughput Functional Cancer Genomics in Mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13982–13987. [Google Scholar] [CrossRef] [Green Version]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Öllinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed Pancreatic Genome Engineering and Cancer Induction by Transfection-Based Crispr/Cas9 Delivery in Mice. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic Crispr/Cas9-Mediated Tumour Suppressor Disruption Enables Versatile Brain Tumour Modelling. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Maddalo, D.; Manchado, E.; Concepcion, C.P.; Bonetti, C.; Vidigal, J.A.; Han, Y.C.; Ogrodowski, P.; Crippa, A.; Rekhtman, N.; de Stanchina, E.; et al. In Vivo Engineering of Oncogenic Chromosomal Rearrangements with the Crispr/Cas9 System. Nature 2014, 516, 423–427. [Google Scholar] [CrossRef] [Green Version]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.J.; Mitros, T.; Muñoz, D.P.; Boffelli, D.; et al. Selection-Free Genome Editing of the Sickle Mutation in Human Adult Hematopoietic Stem/Progenitor Cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome Editing with Cas9 in Adult Mice Corrects a Disease Mutation and Phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [Green Version]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. Crispr–Cas9 Genome Editing Induces a P53-Mediated DNA Damage Response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. P53 Inhibits Crispr–Cas9 Engineering in Human Pluripotent Stem Cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, D.; MHattersley, S.; DStafford, N.; JHaswell, S.; Greenman, J. Development of Microfluidic-Based Analytical Methodology for Studying the Effects of Chemotherapy Agents on Cancer Tissue. Curr. Anal. Chem. 2013, 9, 2–8. [Google Scholar] [CrossRef]
- Trujillo-de Santiago, G.; Flores-Garza, B.G.; Tavares-Negrete, J.A.; Lara-Mayorga, I.M.; González-Gamboa, I.; Zhang, Y.S.; Rojas-Martínez, A.; Ortiz-López, R.; Álvarez, M.M. The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors. Materials 2019, 12, 2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, L.; Zhu, Z.; Xu, Z.; He, T.; Li, E.; Guo, Z.; Liu, F.; Jiang, C.; Wang, Q. Cancer Associated Fibroblast-Derived Hepatocyte Growth Factor Inhibits the Paclitaxel-Induced Apoptosis of Lung Cancer A549 Cells by up-Regulating the Pi3k/Akt and Grp78 Signaling on a Microfluidic Platform. PLoS ONE 2015, 10, e0129593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Tu, T.Y.; Kim, C.; Thiery, J.P.; Kamm, R.D. Identification of Drugs as Single Agents or in Combination to Prevent Carcinoma Dissemination in a Microfluidic 3d Environment. Oncotarget 2015, 6, 36603–36614. [Google Scholar] [CrossRef] [Green Version]
- de Almeida Monteiro Melo Ferraz, M.; Nagashima, J.B.; Venzac, B.; Le Gac, S.; Songsasen, N. A Dog Oviduct-on-a-Chip Model of Serous Tubal Intraepithelial Carcinoma. Sci. Rep. 2020, 10, 1575. [Google Scholar] [CrossRef] [Green Version]
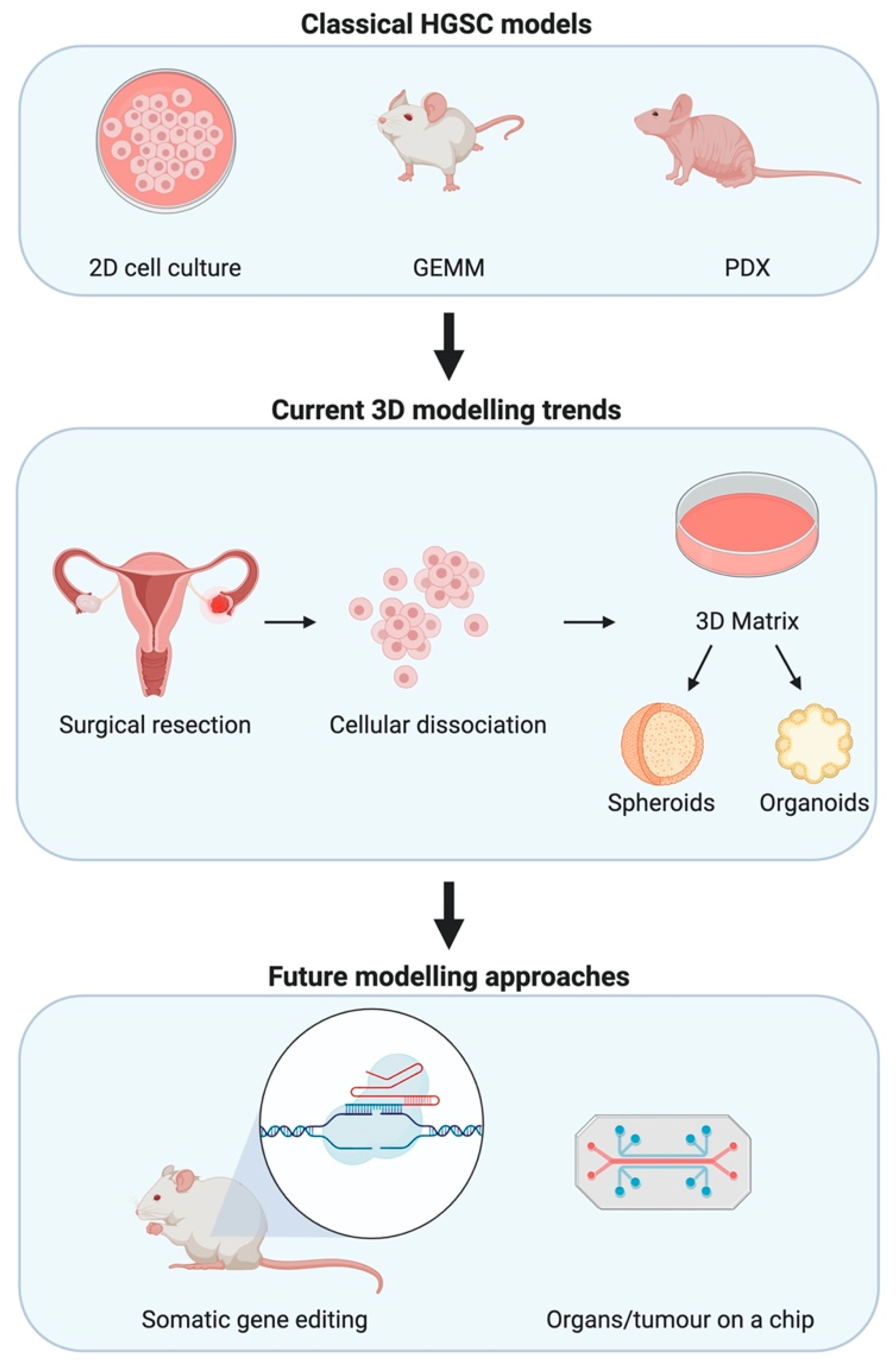
Figure 1.
Modelling high-grade serous epithelial ovarian cancer (HGSC) for research. Classical methods to investigate HGSC have depended on 2D mono- and co-cultures, GEMMs, and patient-derived xenografts (PDXs). Advances in in-vitro modelling lead to trends in 3D modelling wherein primary cells are used to form spheroids and organoids that aim to recapitulate interactions between cell types. Whilst technological advances seek to join such 3D modelling advances with microfluidics allowing for organs and tumours on a chip. Somatic gene editing facilitated by advances in CRISPR/Cas9 biotechnology allows for faster oncogenic mutations that are more representative of the real-world scenario.
Figure 1.
Modelling high-grade serous epithelial ovarian cancer (HGSC) for research. Classical methods to investigate HGSC have depended on 2D mono- and co-cultures, GEMMs, and patient-derived xenografts (PDXs). Advances in in-vitro modelling lead to trends in 3D modelling wherein primary cells are used to form spheroids and organoids that aim to recapitulate interactions between cell types. Whilst technological advances seek to join such 3D modelling advances with microfluidics allowing for organs and tumours on a chip. Somatic gene editing facilitated by advances in CRISPR/Cas9 biotechnology allows for faster oncogenic mutations that are more representative of the real-world scenario.


{kind=link}
Table 1.
The subtypes of Epithelial Ovarian Cancer in women, their frequencies and molecular features and the associated genetically engineered mouse models (GEMMs).
Table 1.
The subtypes of Epithelial Ovarian Cancer in women, their frequencies and molecular features and the associated genetically engineered mouse models (GEMMs).
| Subtype | Frequency | Molecular Features | GEMMs |
|---|---|---|---|
| Mucinous | <5% | CDKN2A copy number alterations [12] KRAS, TP53, RNF43, BRAF, PIK3CA, ARID1A [12,13] mutations ERBB2 [12], HER2 [14], HOXD9 [15] amplification | Amhr2-Cre; LSL-KrasG12D/+PtenloxP/loxP; Trp53R172H/+ [16] |
| Clear cell | ~10% | ARID1A [17] and PIK3CA [18] mutations Ubiquitous HNF1β expression [19] Loss of PTEN expression [20] | No GEMMs |
| Endometrioid | ~10% | ARID1a [17] and PPP2R1A [21] mutations PTEN alterations [22] | (ADCRE) - LSL-K-rasG12D/+PtenloxP/loxP [23] (ADCRE) - ApcloxP/loxP;PtenloxP/loxP [24] Ovgp1-iCreERT2;ApcloxP/loxP; PtenloxP [25] |
| Low-grade serous | <5% | KRAS, BRAF, ERBB2 mutations [26,27] | Amhr2-Cre; LSL-KrasG12D/+; PtenloxP/loxP [28] |
| High-grade serous | ~70% | TP53 [29,30] and BRCA1/2 [30,31] mutations CCNE1 and RB1 [32] aberrations | Older GEMMs reviewed in detail (Howell) Most recent HGSC GEMMS: Pax8-rtTA; TetO-Cre; Brca1loxP/loxP; Trp53mut; PtenloxP/loxP [33] Ovgp1-iCreERT2; Brca1loxP/loxP; Trp53loxP/loxP; Rb1loxP/loxP; Nf1 loxP/loxP [34] Lgr5-Cre; Trp53R172H/+; T121 [35] |
Table 2.
Median survival of allograft models.
| Syngeneic Cell Line | Engraftment Location | Median Survival (days) |
|---|---|---|
| ID8 | i.p. | 101 * |
| ID8Trp53-/- | i.p. | 47 * |
| ID8Trp53-/-;Brca2-/- | i.p. | 57 * |
| ID8Trp53-/-;Brca1-/- | i.p. | 46 ^ |
| ID8Trp53-/-;Pten-/- | i.p. | 34 ^ |
| ID8Trp53-/-;Pten+/- | i.p. | 40.5 ^ |
| ID8Trp53-/-;Nf1-/- | i.p. | 36.5 ^ |
| ID8Trp53-/-;Brca2-/-;Pten-/- | i.p. | 40 ^ |
| 60577: p53-/-;Brca1-/- | i.p. | 36 # |
| 30200: p53-/-;Brca1-/- | i.p. | 77 # |
| HGS1: Pax8-Cre;p53-/-;Pten-/-;Brca2-/- | i.p. | 91 # |
| HGS2: Pax8-Cre;p53-/-;Pten-/-;Brca2-/- | i.p. | 80.5 # |
| HGS3: Pax8-Cre;p53-/-;Pten-/-;Brca2+/- | i.p. | 87.5 # |
| HGS4: Pax8-Cre;p53-/-;Pten-/-;Brca2+/- | i.p. | 80.5 # |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zakarya, R.; Howell, V.M.; Colvin, E.K. Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches. Int. J. Mol. Sci. 2020, 21, 4806. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134806
AMA Style
Zakarya R, Howell VM, Colvin EK. Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches. International Journal of Molecular Sciences. 2020; 21(13):4806. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134806
Chicago/Turabian StyleZakarya, Razia, Viive M. Howell, and Emily K. Colvin. 2020. "Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches" International Journal of Molecular Sciences 21, no. 13: 4806. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134806
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.