Regulation of Human Platelet Activation and Prevention of Arterial Thrombosis in Mice by Auraptene through Inhibition of NF-κB Pathway


 , and
, and 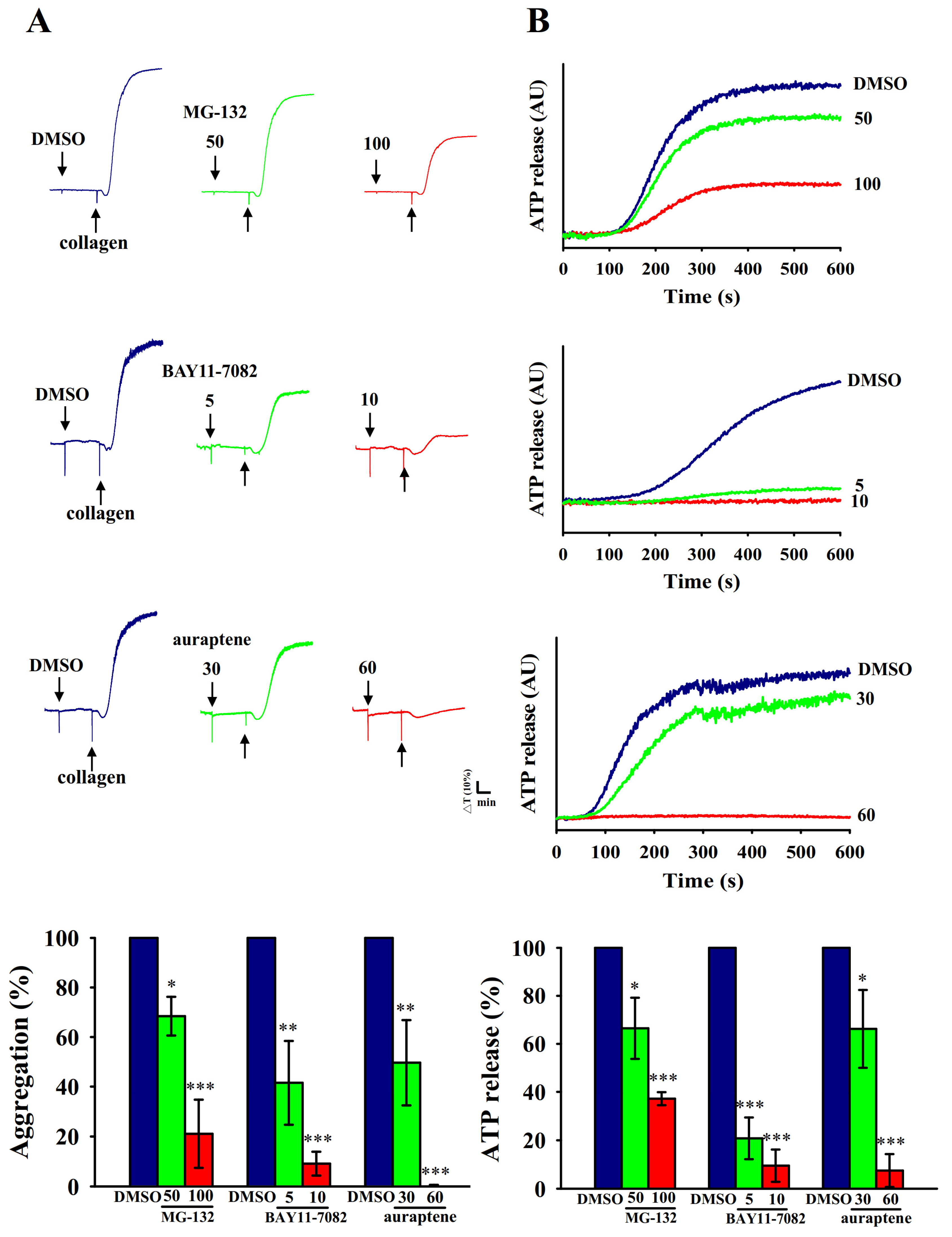{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Inhibitory Profiles of Auraptene and NF-κB Inhibitors in Human Platelet Aggregation and ATP-Release Reaction Stimulated by Collagen
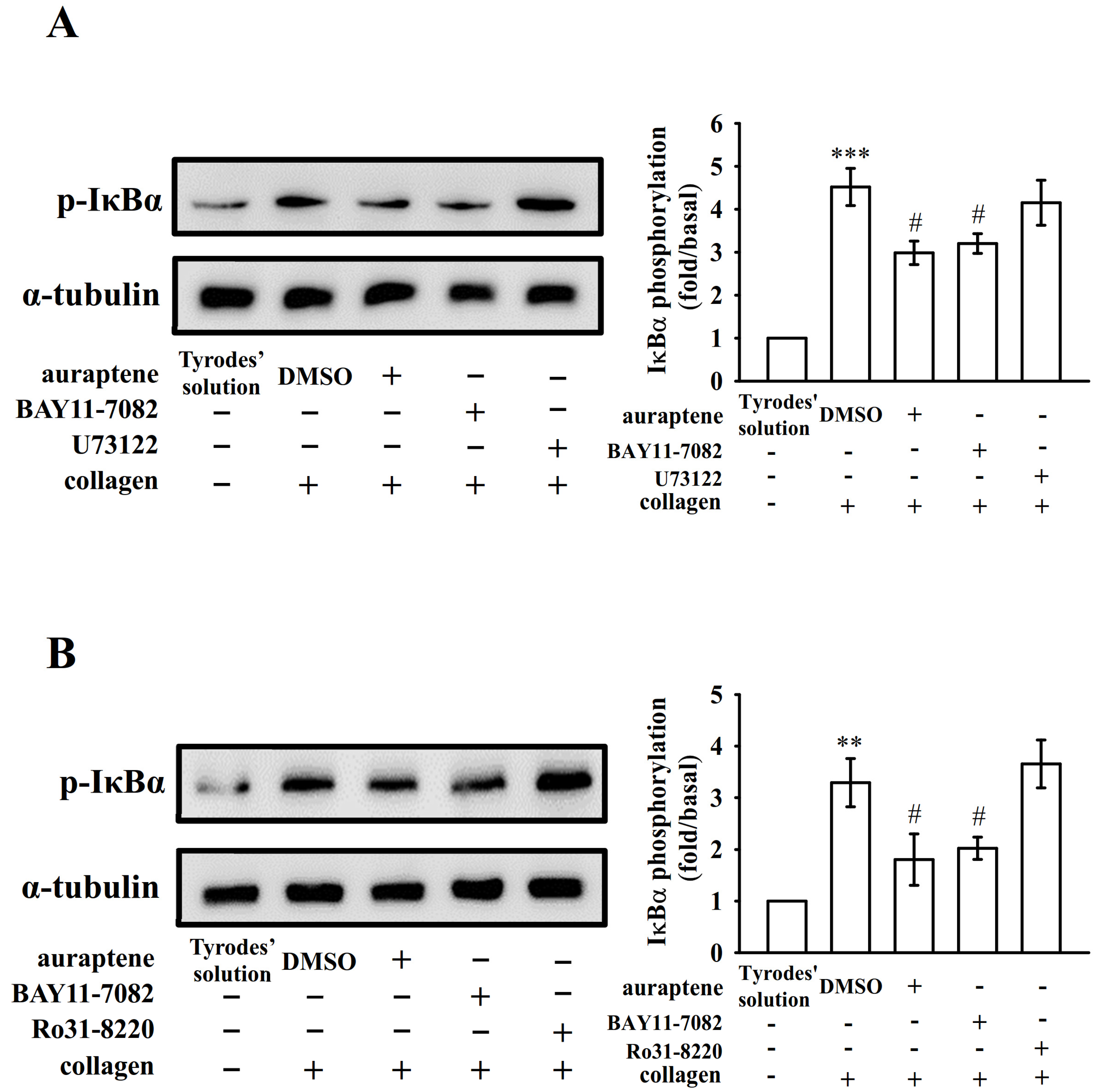
2.2. Platelet Activation Triggers NF-κB Signals
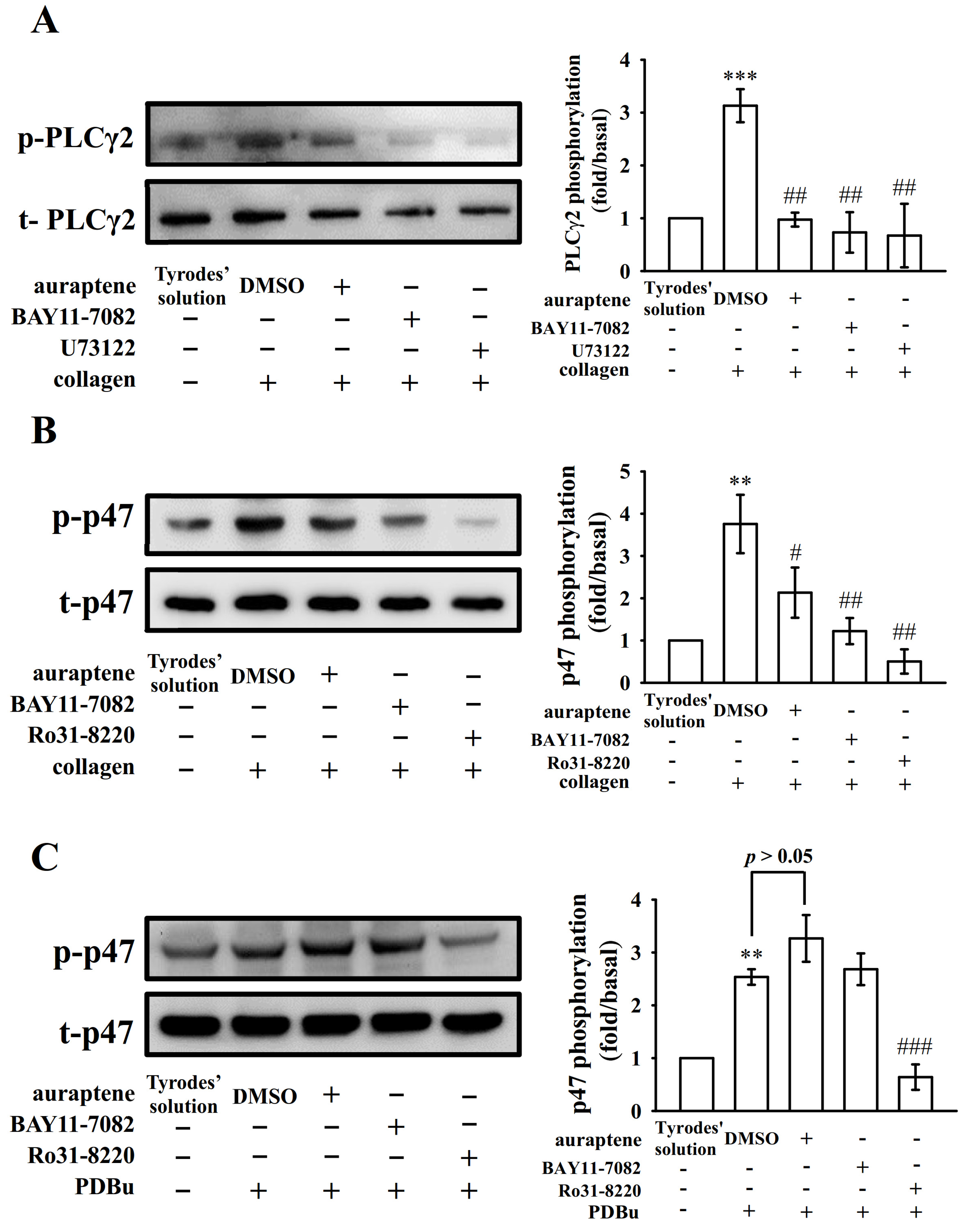
2.3. The Relationship between NF-κB Signaling and PLCγ2-PKC Activation in Human Platelets
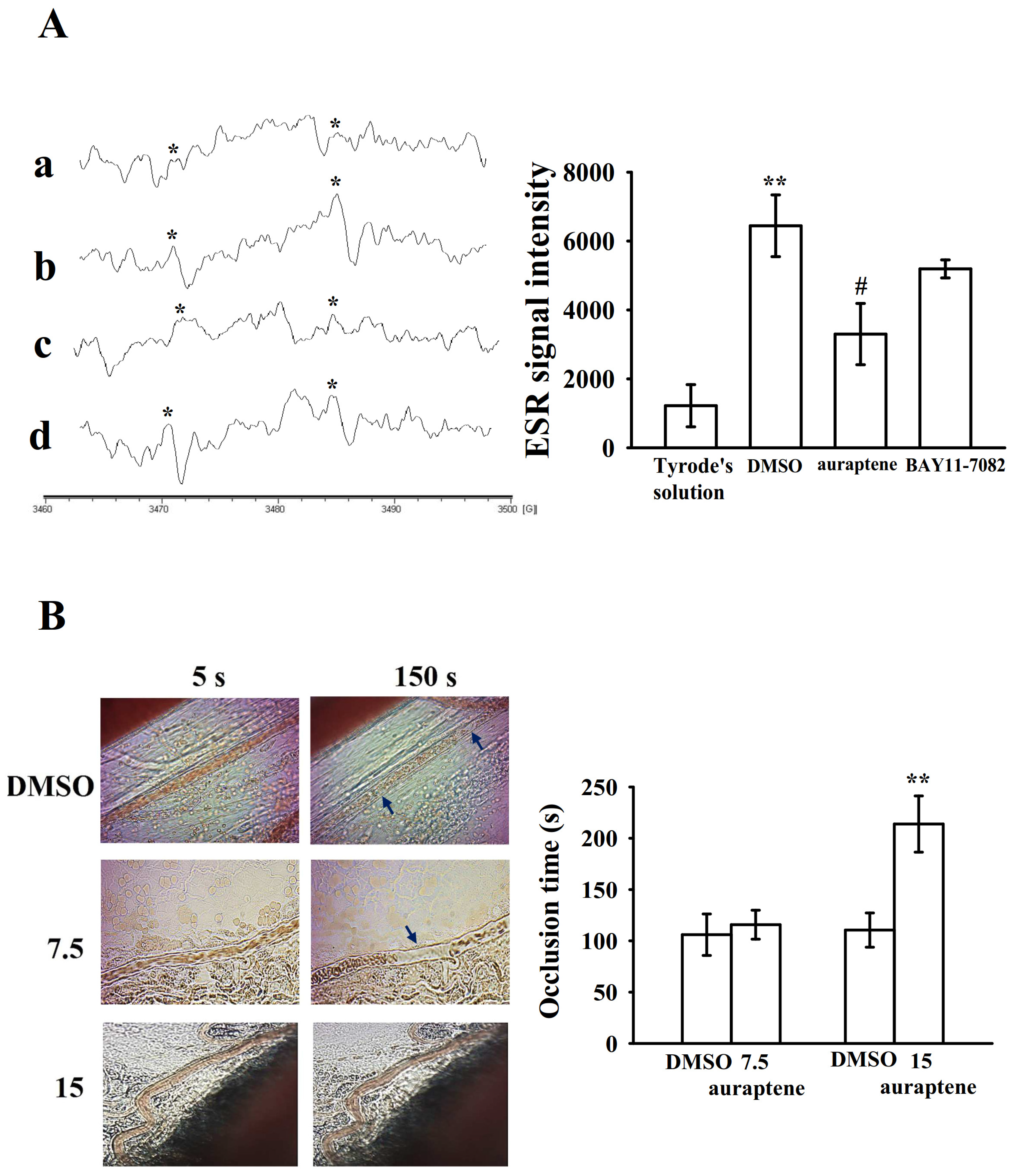
2.4. Effectiveness of BAY11-7082 and Auraptene in Collagen-Stimulated Hydroxyl Radicals (HO•) Formation
2.5. Regulatory In Vivo Activity of Auraptene in Vascular Thrombus Formation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Platelet Aggregation
4.3. Confocal Laser Fluorescence Microscopy
4.4. Immunoblotting Study
4.5. Measurement of Hydroxyl Radicals (HO•) by Electron Spin Resonance (ESR) Spectrometry
4.6. Vascular Thrombus in Mouse Mesenteric Microvessels Irradiated by Sodium Fluorescein
4.7. Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| BSA | Bovine serum albumin |
| CVDs | Cardiovascular diseases |
| DAG | Diacylglycerol |
| DMSO | Dimethyl sulfoxide |
| ESR | Electron spin resonance |
| NF-κB | Nuclear factor-κB |
| PDBu | Phorbol-12,13-dibutyrate |
| PGE1 | Prostaglandin E1 |
| PLCγ2 | Phospholipase Cγ2 |
| PKC | Protein kinase C |
References
- Kojok, K.; El-Kadiry, A.E.; Merhi, Y. Role of NF-κB in Platelet Function. Int. J. Mol. Sci. 2019, 20, 4185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, X.; Shi, X.; Zhu, M.; Wang, J.; Huang, S.; Huang, X.; Wang, H.; Li, L.; Deng, H.; et al. Platelet integrin alphaIIbbeta3: Signal transduction, regulation, and its therapeutic targeting. J. Hematol. Oncol. 2019, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Kim, S.D.; Lee, W.M.; Endale, M.; Kamruzzaman, S.M.; Oh, W.J.; Cho, J.Y.; Kim, S.K.; Cho, H.J.; Park, H.J.; et al. A noble function of BAY 11-7082: Inhibition of platelet aggregation mediated by an elevated cAMP-induced VASP, and decreased ERK2/JNK1 phosphorylations. Eur. J. Pharmacol. 2010, 627, 85–91. [Google Scholar] [CrossRef]
- Fuentes, E.; Rojas, A.; Palomo, I. NF-κB signaling pathway as target for antiplatelet activity. Blood Rev. 2016, 30, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Morris, S.; Epps, J.; Carroll, R. Demonstration of an activation regulated NF-kappaB/I-kappaBalpha complex in human platelets. Thromb. Res. 2002, 106, 199–203. [Google Scholar] [CrossRef]
- Malaver, E.; Romaniuk, M.A.; D’Atri, L.P.; Pozner, R.G.; Negrotto, S.; Benzadon, R.; Schattner, M. NF-kappaB inhibitors impair platelet activation responses. J. Thromb. Haemost. 2009, 7, 1333–1343. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, S.L.; Casey, A.E.; Pollock, S.J.; Gertz, J.M.; McMillan, D.H.; Narasipura, S.D.; Mody, N.A.; King, M.R.; Maggirwar, S.B.; Francis, C.W.; et al. Platelets and megakaryocytes contain functional nuclear factor-kappaB. Arter. Thromb. Vasc. Biol. 2010, 30, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Gambaryan, S.; Kobsar, A.; Rukoyatkina, N.; Herterich, S.; Geiger, J.; Smolenski, A.; Lohmann, S.M.; Walter, U. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J. Biol. Chem. 2010, 285, 18352–18363. [Google Scholar] [CrossRef] [Green Version]
- Sibbing, D.; von Beckerath, N.; Morath, T.; Stegherr, J.; Mehilli, J.; Sarafoff, N.; Braun, S.; Schulz, S.; Schömig, A.; Kastrati, A. Oral anticoagulation with coumarin derivatives and antiplatelet effects of clopidogrel. Eur. Heart J. 2010, 31, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Rashmi, V.; Odhav, B. Review on natural coumarin lead compounds for their pharmacological activity. Biomed. Res. Int. 2013, 2013, 963248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curini, M.; Cravotto, G.; Epifano, F.; Giannone, G. Chemistry and biological activity of natural and synthetic prenyloxycoumarins. Curr. Med. Chem. 2006, 13, 199–222. [Google Scholar] [CrossRef]
- Hsia, C.W.; Tsai, C.L.; Sheu, J.R.; Lu, W.J.; Hsia, C.H.; Velusamy, M.; Jayakumar, T.; Li, J.Y. Suppression of human platelet activation via integrin αIIbβ3 outside-in independent signal and reduction of the mortality in pulmonary thrombosis by auraptene. Int. J. Mol. Sci. 2019, 20, 5585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Hayden, M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef]
- Ali, F.Y.; Davidson, S.J.; Moraes, L.A.; Traves, S.L.; Paul-Clark, M.; Bishop-Bailey, D.; Warner, T.D.; Mitchell, J.A. Role of nuclear receptor signaling in platelets: Antithrombotic effects of PPARbeta. FASEB J. 2006, 20, 326–328. [Google Scholar] [CrossRef]
- Moraes, L.A.; Paul-Clark, M.J.; Rickman, A.; Flower, R.J.; Goulding, N.J.; Perretti, M. Ligand-specific glucocorticoid receptor activation in human platelets. Blood 2005, 106, 4167–4175. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Adams, T.; Zhi, H.; Yu, M.; Wen, R.; Newman, P.J.; Wang, D.; Newman, D.K. Restoration of responsiveness of phospholipase Cγ2-deficient platelets by enforced expression of phospholipase Cγ1. PLoS ONE 2015, 10, e0119739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, D.S.; Hsiao, G.; Shen, M.Y.; Tsai, Y.J.; Chen, T.F.; Sheu, J.R. ESR spin trapping of a carbon-centered free radical from agonist-stimulated human platelets. Free Radic. Biol. Med. 2005, 39, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wang, H.; Zhang, G.; Lu, Y.; An, X.; Ren, S.; Wang, Y.; Chen, Y.; White, J.G.; Zhang, C.; et al. Platelet IkappaB kinase-beta deficiency increases mouse arterial neointima formation via delayed glycoprotein Ibalpha shedding. Arter. Thromb. Vasc. Biol. 2013, 33, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.F.; Lee, J.J.; Chang, C.C.; Lin, K.H.; Wang, S.H.; Sheu, J.R. Platelet protease-activated receptor (PAR)4, but not PAR1, associated with neutral sphingomyelinase responsible for thrombin-stimulated ceramide-NF-κB signaling in human platelets. Haematologica 2013, 98, 793–801. [Google Scholar] [CrossRef]
- Crosby, D.; Poole, A.W. Physical and functional interaction between protein kinase C Delta and Fyn tyrosine kinase in human platelets. J. Biol. Chem. 2003, 278, 24533–24541. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, G.; Lin, K.H.; Chang, Y.; Chen, T.L.; Tzu, N.H.; Chou, D.S.; Sheu, J.R. Protective mechanisms of inosine in platelet activation and cerebral ischemic damage. Arter. Thromb. Vasc. Biol. 2005, 25, 1998–2004. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsia, C.-W.; Wu, M.-P.; Shen, M.-Y.; Hsia, C.-H.; Chung, C.-L.; Sheu, J.-R. Regulation of Human Platelet Activation and Prevention of Arterial Thrombosis in Mice by Auraptene through Inhibition of NF-κB Pathway. Int. J. Mol. Sci. 2020, 21, 4810. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134810
Hsia C-W, Wu M-P, Shen M-Y, Hsia C-H, Chung C-L, Sheu J-R. Regulation of Human Platelet Activation and Prevention of Arterial Thrombosis in Mice by Auraptene through Inhibition of NF-κB Pathway. International Journal of Molecular Sciences. 2020; 21(13):4810. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134810
Chicago/Turabian StyleHsia, Chih-Wei, Ming-Ping Wu, Ming-Yi Shen, Chih-Hsuan Hsia, Chi-Li Chung, and Joen-Rong Sheu. 2020. "Regulation of Human Platelet Activation and Prevention of Arterial Thrombosis in Mice by Auraptene through Inhibition of NF-κB Pathway" International Journal of Molecular Sciences 21, no. 13: 4810. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134810