SOX2 and p53 Expression Control Converges in PI3K/AKT Signaling with Versatile Implications for Stemness and Cancer


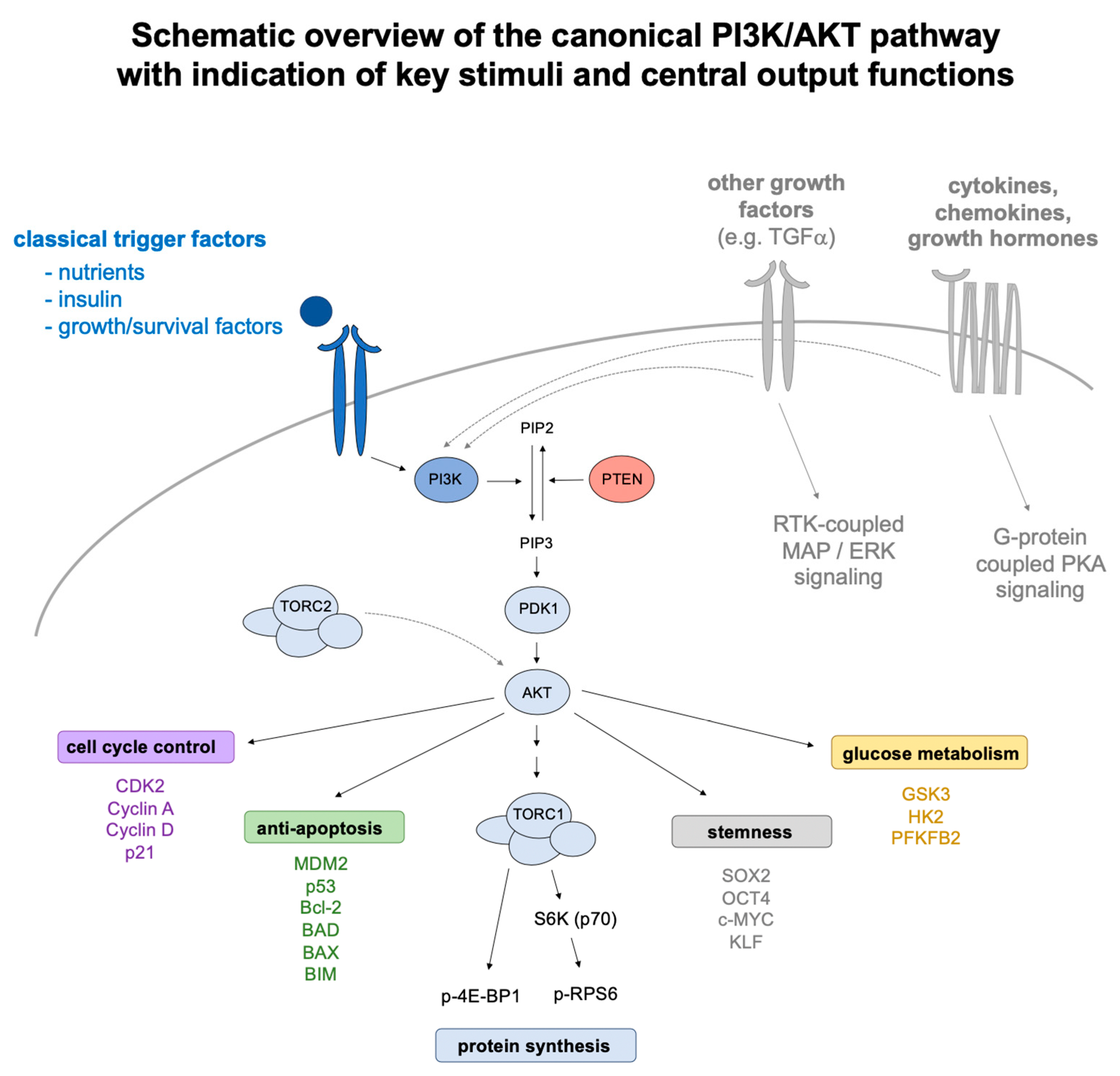{kind=link}
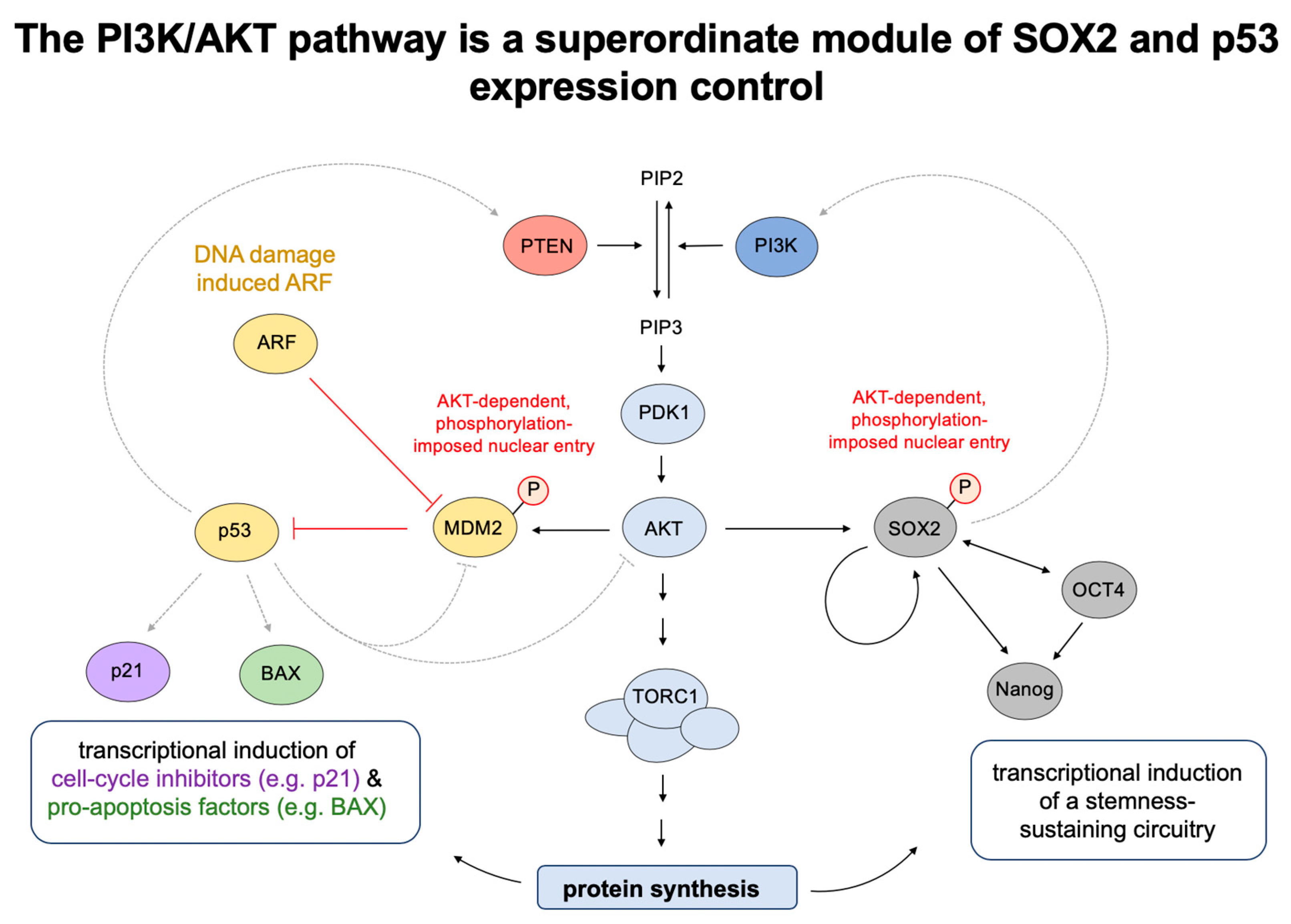{kind=link}
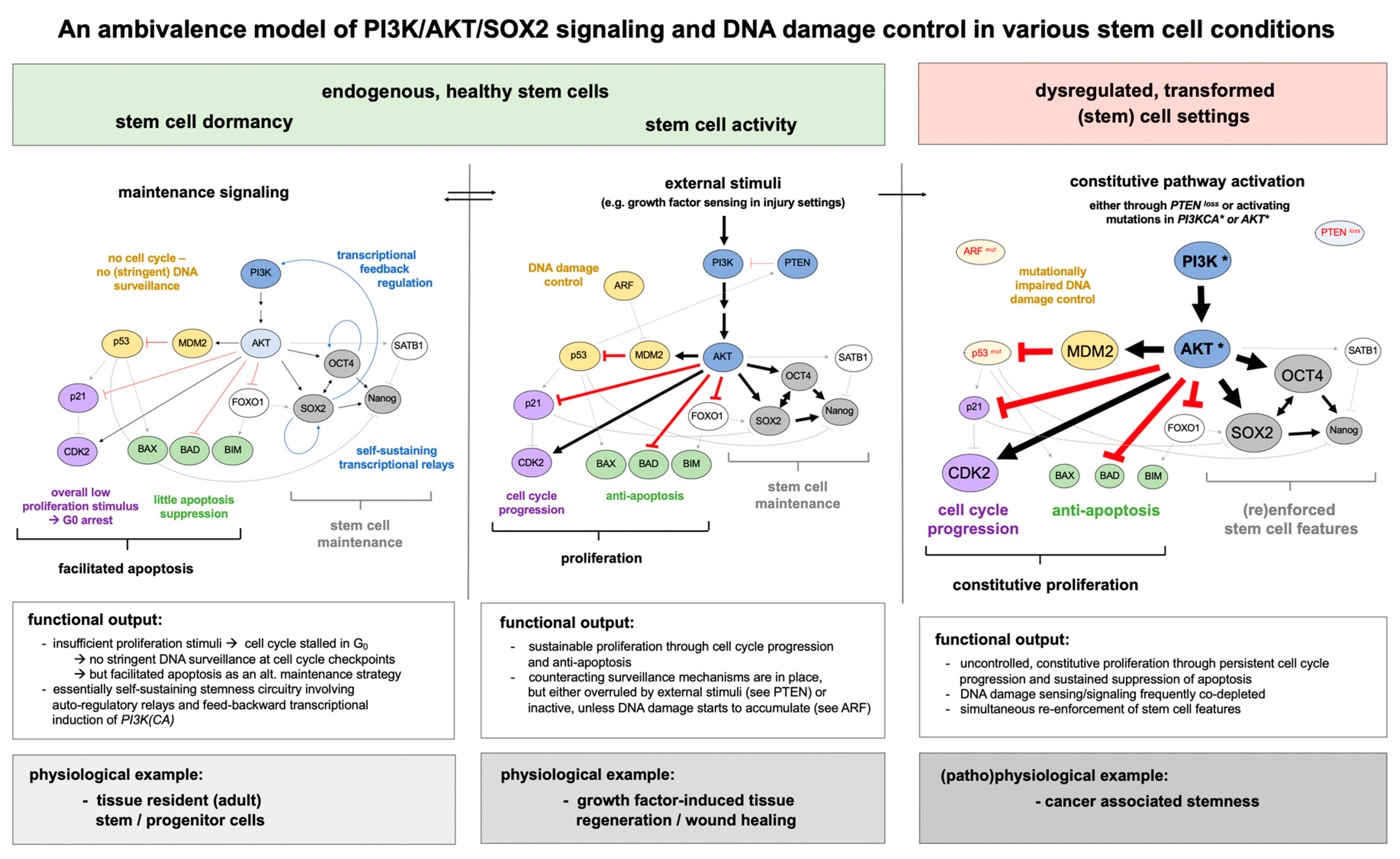{kind=link}
Abstract
:1. An Introduction to SOX2 Biology
2. Molecular–Functional Aspects of SOX2-Imposed Stemness
3. The PI3K/AKT/SOX2 Axis in Stemness, Reprogramming, and Cancer
4. DNA Damage Control in Stemness and Reprogramming
5. AKT/p53 Antagonisms Balance Cell Cycle Progression and DNA Damage Control
6. P53 Mutant-Dependent, Impaired Apoptosis and PI3K/AKT Enforced Growth Signaling Synergize in Cancer
7. PI3K/AKT/SOX2 Signaling and p53-Dependent Apoptosis Regulation in Stem Cells
8. Concluding Remarks and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Feng, R.; Wen, J. Overview of the roles of Sox2 in stem cell and development. Boil. Chem. 2015, 396, 883–891. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Masui, S.; Nakatake, Y.; Toyooka, Y.; Shimosato, D.; Yagi, R.; Takahashi, K.; Okochi, H.; Okuda, A.; Matoba, R.; Sharov, A.A.; et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat. Cell Biol. 2007, 9, 625–635. [Google Scholar] [CrossRef]
- Kopp, J.L.; Ormsbee, B.D.; Desler, M.; Rizzino, A. Small Increases in the Level of Sox2 Trigger the Differentiation of Mouse Embryonic Stem Cells. Stem Cells 2008, 26, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Avilion, A.A.; Nicolis, S.K.; Pevny, L.H.; Perez, L.; Vivian, N.; Lovell-Badge, R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genome Res. Dev. 2003, 17, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Nichols, J.; Smith, A.; Li, M. SoxB transcription factors specify neuroectodermal lineage choice in ES cells. Mol. Cell. Neurosci. 2004, 27, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K.A.; Hever, A.M.; Rainger, J.; Rogers, R.C.; Magee, A.; Fiedler, Z.; Keng, W.T.; Sharkey, F.H.; McGill, N.; Hill, C.J.; et al. Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum. Mol. Genet. 2006, 15, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Bardakjian, T.; Reis, L.M.; Tyler, R.C.; Semina, E.V. Novel SOX2 mutations and genotype-phenotype correlation in anophthalmia and microphthalmia. Am. J. Med Genet. Part A 2009, 149A, 2706–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, D.; Heavner, W.; Pevny, L.H. Combinatorial regulation of optic cup progenitor cell fate by SOX2 and PAX6. Development 2011, 138, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Taranova, O.V.; Magness, S.T.; Fagan, B.M.; Wu, Y.; Surzenko, N.; Hutton, S.R.; Pevny, L.H. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genome Res. Dev. 2006, 20, 1187–1202. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, K.J.L.; McKenzie, I.A.; Mill, P.; Smith, K.; Akhavan, M.; Barnabé-Heider, F.; Biernaskie, J.; Junek, A.; Kobayashi, N.R.; Toma, J.G.; et al. A dermal niche for multipotent adult skin-derived precursor cells. Nat. Cell Biol. 2004, 6, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. Sox2+ Adult Stem and Progenitor Cells Are Important for Tissue Regeneration and Survival of Mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuebben, E.L.; Rizzino, A. The dark side of SOX2: Cancer—A comprehensive overview. Oncotarget 2017, 8, 44917–44943. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Bauer, J.; Wise, P.; Krüger, M.; Simonsen, U.; Wehland, M.; Infanger, M.; Corydon, T.J. The role of SOX family members in solid tumours and metastasis. Semin. Cancer Boil. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.J.M.; Stoop, H.; Biermann, K.; Van Gurp, R.J.H.L.M.; Swartzman, E.; Cribbes, S.; Ferlinz, A.; Shannon, M.; Oosterhuis, J.; Looijenga, L.H.J. Expression and interdependencies of pluripotency factors LIN28, OCT3/4, NANOG and SOX2 in human testicular germ cells and tumours of the testis. Int. J. Androl. 2011, 34, e160–e174. [Google Scholar] [CrossRef] [PubMed]
- Bareiss, P.M.; Paczulla, A.; Wang, H.; Schairer, R.; Wiehr, S.; Kohlhofer, U.; Rothfuss, O.C.; Fischer, A.; Perner, S.; Staebler, A.; et al. SOX2 Expression Associates with Stem Cell State in Human Ovarian Carcinoma. Cancer Res. 2013, 73, 5544–5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Mizushima, T.; Yokoyama, Y.; Hirose, H.; Wu, X.; Qian, Y.; Ikehata, K.; Miyoshi, N.; Takahashi, H.; Haraguchi, N.; et al. Sox2 is associated with cancer stem-like properties in colorectal cancer. Sci. Rep. 2018, 8, 17639. [Google Scholar] [CrossRef]
- Al Mamun, M.; Mannoor, K.; Cao, J.; Qadri, F.; Song, X. SOX2 in cancer stemness: Tumor malignancy and therapeutic potentials. J. Mol. Cell Boil. 2018, 12, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmut, I.; Schnieke, A.; McWhir, J.; Kind, A.J.; Campbell, K.H.S. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Hochedlinger, K.; Jaenisch, R. Nuclear Transplantation, Embryonic Stem Cells, and the Potential for Cell Therapy. N. Engl. J. Med. 2003, 349, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Schepers, G.E.; Teasdale, R.D.; Koopman, P. Twenty Pairs of Sox. Dev. Cell 2002, 3, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Srivastava, Y.; Jauch, R. Molecular basis for the genome engagement by Sox proteins. Semin. Cell Dev. Boil. 2017, 63, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Bowles, J.; Schepers, G.; Koopman, P. Phylogeny of the SOX Family of Developmental Transcription Factors Based on Sequence and Structural Indicators. Dev. Boil. 2000, 227, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Develpoment 2013, 140, 4129–4144. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2007, 26, 101–106. [Google Scholar] [CrossRef]
- Sarkar, A.; Hochedlinger, K. The sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013, 12, 15–30. [Google Scholar] [CrossRef] [Green Version]
- Guth, S.I.E.; Wegner, M. Having it both ways: Sox protein function between conservation and innovation. Cell. Mol. Life Sci. 2008, 65, 3000–3018. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, H.; Lan, F.; Lee, A.; Chen, L.; Lin, C.; Yao, Y.; Li, L. Generation of Human-Induced Pluripotent Stem Cells from Gut Mesentery-Derived Cells by Ectopic Expression of OCT4/SOX2/NANOG. Cell. Reprogram. 2010, 12, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Zhang, Q.; Ye, X.; Liu, K.; Liu, L. Efficient Induction of Pluripotent Stem Cells from Granulosa Cells byOct4andSox2. Stem Cells Dev. 2014, 23, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Maherali, N.; Hochedlinger, K. Tgfβ Signal Inhibition Cooperates in the Induction of iPSCs and Replaces Sox2 and cMyc. Curr. Boil. 2009, 19, 1718–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichida, J.K.; Blanchard, J.; Lam, K.; Son, E.Y.; Chung, J.E.; Egli, D.; Loh, K.M.; Carter, A.C.; Di Giorgio, F.P.; Koszka, K.; et al. A Small-Molecule Inhibitor of Tgf-β Signaling Replaces Sox2 in Reprogramming by Inducing Nanog. Cell Stem Cell 2009, 5, 491–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Lin, T.; Wu, S. Reprogramming with Small Molecules instead of Exogenous Transcription Factors. Stem Cells Int. 2015, 2015, 794632. [Google Scholar] [CrossRef] [Green Version]
- Takayama, Y.; Clore, G.M. Interplay between Minor and Major Groove-binding Transcription Factors Sox2 and Oct1 in Translocation on DNA Studied by Paramagnetic and Diamagnetic NMR*. J. Boil. Chem. 2012, 287, 14349–14363. [Google Scholar] [CrossRef] [Green Version]
- Yesudhas, D.; Anwar, M.A.; Panneerselvam, S.; Kim, H.-K.; Choi, S. Evaluation of Sox2 binding affinities for distinct DNA patterns using steered molecular dynamics simulation. FEBS Open Bio 2017, 7, 1750–1767. [Google Scholar] [CrossRef]
- Fang, X.; Yoon, J.-G.; Li, L.; Yu, W.; Shao, J.; Hua, D.; Zheng, S.; Hood, L.; Goodlett, D.R.; Foltz, G.; et al. The SOX2 response program in glioblastoma multiforme: An integrated ChIP-seq, expression microarray, and microRNA analysis. BMC Genom. 2011, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Zhong, G.; Wu, J.; Chen, H.; Jia, Y. SOX2 recruits KLF4 to regulate nasopharyngeal carcinoma proliferation via PI3K/AKT signaling. Oncogenesis 2018, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-Y.; Bogu, G.; Soh, B.S.; Stanton, L.W. The Long Noncoding RNA RMST Interacts with SOX2 to Regulate Neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, Z.E.; Hamilton, D.J.; Hwang, T.; Parsonnet, N.V.; Rinn, J.L.; Wuttke, D.S.; Batey, R.T. The Sox2 transcription factor binds RNA. Nat. Commun. 2020, 11, 1805–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, L.; Wei, Y.; Lin, Y.; Wang, X.; Lai, Y.; Yin, M.; Chen, Y.; Guo, X.; Wu, S.; Zhu, Y.; et al. Concurrent binding to DNA and RNA facilitates the pluripotency reprogramming activity of Sox2. Nucleic Acids Res. 2020, 48, 3869–3887. [Google Scholar] [CrossRef]
- Li, J.; Pan, G.; Cui, K.; Liu, Y.; Xu, S.; Pei, D. A Dominant-negative Form of Mouse SOX2 Induces Trophectoderm Differentiation and Progressive Polyploidy in Mouse Embryonic Stem Cells. J. Boil. Chem. 2007, 282, 19481–19492. [Google Scholar] [CrossRef] [Green Version]
- Baltus, G.A.; Kowalski, M.P.; Zhai, H.; Tutter, A.V.; Quinn, D.; Wall, D.; Kadam, S. Acetylation of Sox2 Induces its Nuclear Export in Embryonic Stem Cells. Stem Cells 2009, 27, 2175–2184. [Google Scholar] [CrossRef]
- Schaefer, T.; Wang, H.; Mir, P.; Konantz, M.; Pereboom, T.C.; Paczulla, A.M.; Merz, B.; Fehm, T.; Perner, S.; Rothfuss, O.C.; et al. Molecular and functional interactions between AKT and SOX2 in breast carcinoma. Oncotarget 2015, 6, 43540–43556. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Ji, J.; Deng, R.; Tang, J.; Yang, F.; Feng, G.-K.; Chen, W.-D.; Wu, X.-Q.; Qian, X.-J.; Ding, K.; et al. DC120, a novel AKT inhibitor, preferentially suppresses nasopharyngeal carcinoma cancer stem-like cells by downregulating Sox2. Oncotarget 2015, 6, 6944–6958. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, L.; Zhang, H.; Huang, Y.; Fang, L.; Li, M.; Brown, P.J.; Arrowsmith, C.H.; Li, J.; Wong, J. AKT drives SOX2 overexpression and cancer cell stemness in esophageal cancer by protecting SOX2 from UBR5-mediated degradation. Oncogene 2019, 38, 5250–5264. [Google Scholar] [CrossRef]
- Schaefer, T.; Ramadoss, A.; Leu, S.; Tintignac, L.; Tostado, C.; Bink, A.; Schürch, C.; Müller, J.; Schärer, J.; Moffa, G.; et al. Regulation of glioma cell invasion by 3q26 gene products PIK3CA, SOX2 and OPA1. Brain Pathol. 2019, 29, 336–350. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.-H.; Majithia, A.; Huang, X.; Kimmel, A.R. Growth control via TOR kinase signaling, an intracellular sensor of amino acid and energy availability, with crosstalk potential to proline metabolism. Amino Acids 2008, 35, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyuhas, O. Ribosomal Protein S6 Phosphorylation. Int. Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar] [CrossRef]
- Josse, L.; Xie, J.; Proud, C.G.; Smales, C.M. mTORC1 signalling and eIF4E/4E-BP1 translation initiation factor stoichiometry influence recombinant protein productivity from GS-CHOK1 cells. Biochem. J. 2016, 473, 4651–4664. [Google Scholar] [CrossRef] [Green Version]
- Heuvel, S.v.d.; Harlow, E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science 1993, 262, 2050–2054. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Wang, H.; Liu, S.; Hu, N.; Li, X. Akt Regulated Phosphorylation of GSK-3β/Cyclin D1, p21 and p27 Contributes to Cell Proliferation Through Cell Cycle Progression from G1 to S/G2M Phase in Low-Dose Arsenite Exposed HaCat Cells. Front. Pharmacol. 2019, 10, 1176. [Google Scholar] [CrossRef] [Green Version]
- Signer, R.A.J.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef]
- Blanco, S.; Bandiera, R.; Popis, M.; Hussain, S.; Lombard, P.; Aleksic, J.; Sajini, A.; Tanna, H.; Cortés-Garrido, R.; Gkatza, N.; et al. Stem cell function and stress response are controlled by protein synthesis. Nature 2016, 534, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Dehkordi, A.N.; Babaheydari, F.M.; Chehelgerdi, M.; Dehkordi, S.R. Skin tissue engineering: Wound healing based on stem-cell-based therapeutic strategies. Stem Cell Res. Ther. 2019, 10, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CAH1047R induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef]
- Schaefer, T.; Lengerke, C. SOX2 protein biochemistry in stemness, reprogramming, and cancer: The PI3K/AKT/SOX2 axis and beyond. Oncogene 2019, 39, 278–292. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Paulson, A.; Charafe-Jauffret, E.; Ginestier, C.; Brown, M.; Dutcher, J.; Clouthier, S.G.; Wicha, M.S. Regulation of Mammary Stem/Progenitor Cells by PTEN/Akt/β-Catenin Signaling. PLoS Boil. 2009, 7, e1000121. [Google Scholar] [CrossRef]
- Gargini, R.; Cerliani, J.P.; Escoll, M.; Antón, I.M.; Wandosell, F. Cancer Stem Cell-Like Phenotype and Survival Are Coordinately Regulated by Akt/FoxO/Bim Pathway. Stem Cells 2015, 33, 646–660. [Google Scholar] [CrossRef]
- Fang, L.; Zhang, L.; Wei, W.; Jin, X.; Wang, P.; Tong, Y.; Li, J.; Du, J.X.; Wong, J. A Methylation-Phosphorylation Switch Determines Sox2 Stability and Function in ESC Maintenance or Differentiation. Mol. Cell 2014, 55, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Jeong, C.-H.; Cho, Y.-Y.; Kim, M.-O.; Kim, S.-H.; Cho, E.-J.; Lee, S.-Y.; Jeon, Y.-J.; Lee, K.Y.; Yao, K.; Keum, Y.-S.; et al. Phosphorylation of Sox2 Cooperates in Reprogramming to Pluripotent Stem Cells. Stem Cells 2010, 28, 2141–2150. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA Damage Response. Cold Spring Harb. Perspect. Boil. 2010, 3, a000745. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Kastan, M.B. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.H.; Swift, S.L.; White, H.; Misso, K.; Kleijnen, J.; Quek, R.G. A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int. J. Oncol. 2019, 55, 597–616. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.R.; He, Y.; Hong, T.S.; Corcoran, R.B.; Clark, J.W.; Ryan, D.P.; Zou, L.; Ting, D.T.; Catenacci, D.V.; Chao, J.; et al. Analysis of DNA Damage Response Gene Alterations and Tumor Mutational Burden Across 17,486 Tubular Gastrointestinal Carcinomas: Implications for Therapy. Oncologist 2019, 24, 1340–1347. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Gerson, S.L. DNA Repair Defects in Stem Cell Function and Aging. Annu. Rev. Med. 2005, 56, 495–508. [Google Scholar] [CrossRef]
- Tiwari, V.; Wilson, D.M. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef] [Green Version]
- Hoeijmakers, J. Genome maintenance mechanisms are critical for preventing cancer as well as other aging-associated diseases. Mech. Ageing Dev. 2007, 128, 460–462. [Google Scholar] [CrossRef]
- McNeely, T.; Leone, M.; Yanai, H.; Beerman, I. DNA damage in aging, the stem cell perspective. Hum. Genet. 2019, 139, 309–331. [Google Scholar] [CrossRef]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Boil. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, H.; Regoes, R.R.; Boddupalli, C.S.; Bonhoeffer, S.; Manz, M.G. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J. Exp. Med. 2011, 208, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent Hematopoietic Stem Cells Accumulate DNA Damage during Aging that Is Repaired upon Entry into Cell Cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Van Zant, G.; Szilvassy, S.J. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood 2005, 106, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-Dependent Alterations of the DNA Methylation Landscape Underlie Hematopoietic Stem Cell Aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Prasher, J.M.; Lalai, A.S.; Heijmans-Antonissen, C.; Ploemacher, R.E.; Hoeijmakers, J.H.J.; Touw, I.P.; Niedernhofer, L.J. Reduced hematopoietic reserves in DNA interstrand crosslink repair-deficient Ercc1−/− mice. EMBO J. 2005, 24, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Kim, J.; Sykes, S.M.; Shimamura, A.; Stuckert, P.; Zhu, K.; Hamilton, A.; Deloach, M.K.; Kutok, J.L.; Akashi, K.; et al. Hematopoietic Stem Cell Defects in Mice with Deficiency of Fancd2 or Usp1. Stem Cells 2010, 28, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.; Kook, S.H.; Robinson, A.R.; Niedernhofer, L.J.; Lee, B.C. Cell autonomous and nonautonomous mechanisms drive hematopoietic stem/progenitor cell loss in the absence of DNA repair. Stem Cells 2013, 31, 511–525. [Google Scholar] [CrossRef] [Green Version]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A Distinctive DNA Damage Response in Human Hematopoietic Stem Cells Reveals an Apoptosis-Independent Role for p53 in Self-Renewal. Cell Stem Cell 2010, 7, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Rasko, I.; Georgieva, M.; Farkas, G.; Santha, M.; Coates, J.; Burg, K.; Mitchell, D.L.; Johnson, R.T. New patterns of bulk DNA repair in ultraviolet irradiated mouse embryo carcinoma cells following differentiation. Somat. Cell Mol. Genet. 1993, 19, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G.; Leake, A.; Armstrong, L.; Lako, M.; Von Zglinicki, T. Stress Defense in Murine Embryonic Stem Cells Is Superior to That of Various Differentiated Murine Cells. Stem Cells 2004, 22, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Swistowska, A.M.; Lee, J.W.; Liu, Y.; Liu, S.-T.; Da Cruz, A.B.; Rao, M.; Souza-Pinto, N.; Zeng, X.; Bohr, V.A.; et al. Human Embryonic Stem Cells Have Enhanced Repair of Multiple Forms of DNA Damage. Stem Cells 2008, 26, 2266–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N.P.; Crockford, T.L.; Cabuy, E.; Vindigni, A.; et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Stambrook, P.J. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc. Natl. Acad. Sci. USA 2004, 101, 14443–14448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Christmann, M.; Fraser, S.T.; Kaina, B. Mouse embryonic stem cells are hypersensitive to apoptosis triggered by the DNA damage O6-methylguanine due to high E2F1 regulated mismatch repair. Cell Death Differ. 2007, 14, 1422–1432. [Google Scholar] [CrossRef]
- Ghimire, S.; Van Der Jeught, M.; Neupane, J.; Roost, M.S.; Anckaert, J.; Popovic, M.; Van Nieuwerburgh, F.; Mestdagh, P.; Vandesompele, J.; Deforce, D.; et al. Comparative analysis of naive, primed and ground state pluripotency in mouse embryonic stem cells originating from the same genetic background. Sci. Rep. 2018, 8, 5884. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, R.B.; Stringer, J.R.; Shao, C.; Tischfield, J.; Stambrook, P.J. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc. Natl. Acad. Sci. USA 2002, 99, 3586–3590. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Cervantes, R.; Tichy, E.; Tischfield, J.; Stambrook, P.J. Protecting genomic integrity in somatic cells and embryonic stem cells. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2007, 614, 48–55. [Google Scholar] [CrossRef]
- Armstrong, L.; Tilgner, K.; Saretzki, G.; Atkinson, S.P.; Stojkovic, M.; Moreno, R.; Przyborski, S.; Lako, M. Human Induced Pluripotent Stem Cell Lines Show Stress Defense Mechanisms and Mitochondrial Regulation Similar to Those of Human Embryonic Stem Cells. Stem Cells 2010, 28, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.H.; Mason, M.J.; Xie, W.; Volinia, S.; Singer, M.; Peterson, C.; Ambartsumyan, G.; Aimiuwu, O.; Richter, L.; Zhang, J.; et al. Induced Pluripotent Stem Cells and Embryonic Stem Cells Are Distinguished by Gene Expression Signatures. Cell Stem Cell 2009, 5, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simara, P.; Tesarova, L.; Rehakova, D.; Matula, P.; Stejskal, S.; Hampl, A.; Koutna, I. DNA double-strand breaks in human induced pluripotent stem cell reprogramming and long-term in vitro culturing. Stem Cell Res. Ther. 2017, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- González, F.; Georgieva, D.; Vanoli, F.; Shi, Z.-D.; Stadtfeld, M.; Ludwig, T.; Jasin, M.; Huangfu, D. Homologous recombination DNA repair genes play a critical role in reprogramming to a pluripotent state. Cell Rep. 2013, 3, 651–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53–p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef]
- Marión, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [Green Version]
- Utikal, J.S.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef]
- Digweed, M.; DeMuth, I.; Rothe, S.; Scholz, R.; Jordan, A.; Grötzinger, C.; Schindler, D.; Grompe, M.; Sperling, K. SV40 large T-antigen disturbs the formation of nuclear DNA-repair foci containing MRE11. Oncogene 2002, 21, 4873–4878. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Ye, Z.; Hommond, H.H.; Yu, X.; Lin, J.; Chen, G.; Zou, J.; Cheng, L. Improved Efficiency and Pace of Generating Induced Pluripotent Stem Cells from Human Adult and Fetal Fibroblasts. Stem Cells 2008, 26, 1998–2005. [Google Scholar] [CrossRef] [Green Version]
- Laurent, L.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic Changes in the Copy Number of Pluripotency and Cell Proliferation Genes in Human ESCs and iPSCs during Reprogramming and Time in Culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Arora, D.; Singh, A. Systems biology approach deciphering the biochemical signaling pathway and pharmacokinetic study of PI3K/mTOR/p53-Mdm2 module involved in neoplastic transformation. Netw. Model. Anal. Health Inform. Bioinform. 2017, 7, 2. [Google Scholar] [CrossRef]
- Lane, D.P.; Levine, A. P53 Research: The Past Thirty Years and the Next Thirty Years. Cold Spring Harb. Perspect. Boil. 2010, 2, a000893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, T.; Sontag, E.; Chen, P.A.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Boil. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Hofmann, K.; Boulton, S.; Gartner, A. The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Boil. 2001, 11, 1722–1727. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ko, L.J.; Jayaraman, L.; Prives, C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genome Res. Dev. 1996, 10, 2438–2451. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.L.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Comparative Binding of p53 to its Promoter and DNA Recognition Elements. J. Mol. Boil. 2005, 348, 589–596. [Google Scholar] [CrossRef]
- Wee, K.B.; Surana, U.; Aguda, B.D. Oscillations of the p53-Akt Network: Implications on Cell Survival and Death. PLoS ONE 2009, 4, e4407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levav-Cohen, Y.; Goldberg, Z.; Tan, K.H.; Alsheich-Bartok, O.; Zuckerman, V.; Haupt, S.; Haupt, Y. The p53-Mdm2 Loop: A Critical Juncture of Stress Response. Membr. Biog. 2014, 85, 161–186. [Google Scholar] [CrossRef]
- Gottlieb, T.M.; Leal, J.F.M.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN Transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber-Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.-W.; et al. The Ink4a Tumor Suppressor Gene Product, p19Arf, Interacts with MDM2 and Neutralizes MDM2’s Inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: Implications for targeting mTOR during malignancy. Oncogene 2011, 31, 1949–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Abraham, A.G.; O’Neill, E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem. Soc. Trans. 2014, 42, 798–803. [Google Scholar] [CrossRef]
- Freed-Pastor, W.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ching, C.B.; Hansel, D.E. Expanding therapeutic targets in bladder cancer: The PI3K/Akt/mTOR pathway. Lab. Investig. 2010, 90, 1406–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordes, I.; Kluth, M.; Zygis, D.; Rink, M.; Chun, F.K.-H.; Eichelberg, C.; Dahlem, R.; Fisch, M.; Höppner, W.; Wagner, W.; et al. PTENdeletions are related to disease progression and unfavourable prognosis in early bladder cancer. Histopathology 2013, 63, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Catasus, L.; Gallardo, A.; Cuatrecasas, M.; Prat, J. Concomitant PI3K–AKT and p53 alterations in endometrial carcinomas are associated with poor prognosis. Mod. Pathol. 2009, 22, 522–529. [Google Scholar] [CrossRef]
- Molinolo, A.A.; Hewitt, S.; Amornphimoltham, P.; Keelawat, S.; Rangdaeng, S.; Garcia, A.M.; Raimondi, A.R.; Jufe, R.; Itoiz, M.; Gao, Y.; et al. Dissecting the Akt/Mammalian Target of Rapamycin Signaling Network: Emerging Results from the Head and Neck Cancer Tissue Array Initiative. Clin. Cancer Res. 2007, 13, 4964–4973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigaki, H.; Baba, Y.; Watanabe, M.; Murata, A.; Ishimoto, T.; Iwatsuki, M.; Iwagami, S.; Nosho, K.; Baba, H. PIK3CA Mutation Is Associated with a Favorable Prognosis among Patients with Curatively Resected Esophageal Squamous Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Mangone, F.R.; Bobrovnitchaia, I.G.; Salaorni, S.; Manuli, E.; Nagai, M.A. PIK3CA exon 20 mutations are associated with poor prognosis in breast cancer patients. Clinics 2012, 67, 1285–1290. [Google Scholar] [CrossRef]
- Boyault, S.; Drouet, Y.; Navarro, C.; Bachelot, T.; Lasset, C.; Treilleux, I.; Tabone, E.; Puisieux, A.; Wang, Q. Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res. Treat. 2011, 132, 29–39. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Terzian, T.; Suh, Y.-A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genome Res. Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef] [Green Version]
- Suh, Y.-A.; Post, S.M.; Elizondo-Fraire, A.C.; Maccio, D.R.; Jackson, J.G.; El-Naggar, A.K.; Van Pelt, C.; Terzian, T.; Lozano, G.; Maccio, D.R. Multiple stress signals activate mutant p53 in vivo. Cancer Res. 2011, 71, 7168–7175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandioler-Eckersberger, D.; Ludwig, C.; Rudas, M.; Kappel, S.; Janschek, E.; Wenzel, C.; Schlagbauer-Wadl, H.; Mittlböck, M.; Gnant, M.; Steger, G.; et al. TP53 mutation and p53 overexpression for prediction of response to neoadjuvant treatment in breast cancer patients. Clin. Cancer Res. 2000, 6, 50–56. [Google Scholar] [PubMed]
- Olivier, M.; Langer, A.; Klaar, S.; Eyfjord, J.; Lidereau, R.; Bieche, I.; Varley, J.; Bignon, Y.; Winqvist, R.; Jukkola-Vuorinen, A.; et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Duman, B.B.; Sahin, B.; Acikalin, A.; Ergin, M.; Zorludemir, S. PTEN, Akt, MAPK, p53 and p95 expression to predict trastuzumab resistance in HER2 positive breast cancer. J. Balk. Union Oncol. 2013, 18, 44–50. [Google Scholar]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Boil. 2015, 22, 875–880. [Google Scholar] [CrossRef]
- Ge, Y.; Wu, S.; Zhang, Z.; Li, X.; Li, F.; Yan, S.; Liu, H.; Huang, J.; Zhao, Y. Inhibition of p53 and/or AKT as a new therapeutic approach specifically targeting ALT cancers. Protein Cell 2019, 10, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e8. [Google Scholar] [CrossRef] [Green Version]
- Bamodu, O.A.; Chang, H.-L.; Ong, J.-R.; Lee, W.-H.; Yeh, C.-T.; Tsai, J.-T. Elevated PDK1 Expression Drives PI3K/AKT/MTOR Signaling Promotes Radiation-Resistant and Dedifferentiated Phenotype of Hepatocellular Carcinoma. Cells 2020, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Krizhanovsky, V.; Lowe, S.W. The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef] [Green Version]
- Duxin, J.P.; Walter, J.C. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Boil. 2015, 37, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.-S.; Pondarre, C.; et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, T.J.; Luna, R.M.D.O.; Cho, S.; Amelse, L.L.; Chavez-Reyes, A.; Lozano, G. Loss of one but not twomdm2 null alleles alters the tumour spectrum inp53 null mice. J. Pathol. 1999, 188, 322–328. [Google Scholar] [CrossRef]
- Abbas, H.A.; Maccio, D.R.; Coskun, S.; Jackson, J.G.; Hazen, A.L.; Sills, T.M.; You, M.J.; Hirschi, K.K.; Lozano, G. Mdm2 Is Required for Survival of Hematopoietic Stem Cells/Progenitors via Dampening of ROS-Induced p53 Activity. Cell Stem Cell 2010, 7, 606–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, H.; Simonsson, S. Core transcription factors, Oct4, Sox2 and Nanog, individually form complexes with nucleophosmin (Npm1) to control embryonic stem (ES) cell fate determination. Aging 2010, 2, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Liu, S.; Wang, P.; Zhao, S.; Wang, F.; Bing, L.; Zhang, Y.; Ling, E.-A.; Gao, J.; Hao, A. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology 2011, 59, 763–775. [Google Scholar] [CrossRef]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2004, 7, 165–171. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Kippin, T.E.; Martens, D.J.; Van Der Kooy, D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genome Res. Dev. 2005, 19, 756–767. [Google Scholar] [CrossRef] [Green Version]
- Orford, K.W.; Scadden, D.T. Deconstructing stem cell self-renewal: Genetic insights into cell-cycle regulation. Nat. Rev. Genet. 2008, 9, 115–128. [Google Scholar] [CrossRef]
- Marqués-Torrejón, M.Á.; Porlan, E.; Banito, A.; Gómez-Ibarlucea, E.; Lopez-Contreras, A.J.; Fernandez-Capetillo, O.; Vidal, A.; Gil, J.; Torres, J.; Fariñas, I. Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell Stem Cell 2012, 12, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagi, S.; Saito, T.; Mizutani, K.-I.; Masuyama, N.; Gotoh, Y.; Iwama, A.; Nakauchi, H.; Masui, S.; Niwa, H.; Nishimoto, M.; et al. The Sox-2 Regulatory Regions Display Their Activities in Two Distinct Types of Multipotent Stem Cells. Mol. Cell. Boil. 2004, 24, 4207–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Vassetzky, Y.; Dokudovskaya, S. mTORC1 pathway in DNA damage response. Biochim. Biophys. Acta (BBA) Mol. Cell. Res. 2018, 1865, 1293–1311. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaefer, T.; Steiner, R.; Lengerke, C. SOX2 and p53 Expression Control Converges in PI3K/AKT Signaling with Versatile Implications for Stemness and Cancer. Int. J. Mol. Sci. 2020, 21, 4902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144902
Schaefer T, Steiner R, Lengerke C. SOX2 and p53 Expression Control Converges in PI3K/AKT Signaling with Versatile Implications for Stemness and Cancer. International Journal of Molecular Sciences. 2020; 21(14):4902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144902
Chicago/Turabian StyleSchaefer, Thorsten, Rebekah Steiner, and Claudia Lengerke. 2020. "SOX2 and p53 Expression Control Converges in PI3K/AKT Signaling with Versatile Implications for Stemness and Cancer" International Journal of Molecular Sciences 21, no. 14: 4902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144902