A Possible Role for Arylsulfatase G in Dermatan Sulfate Metabolism

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
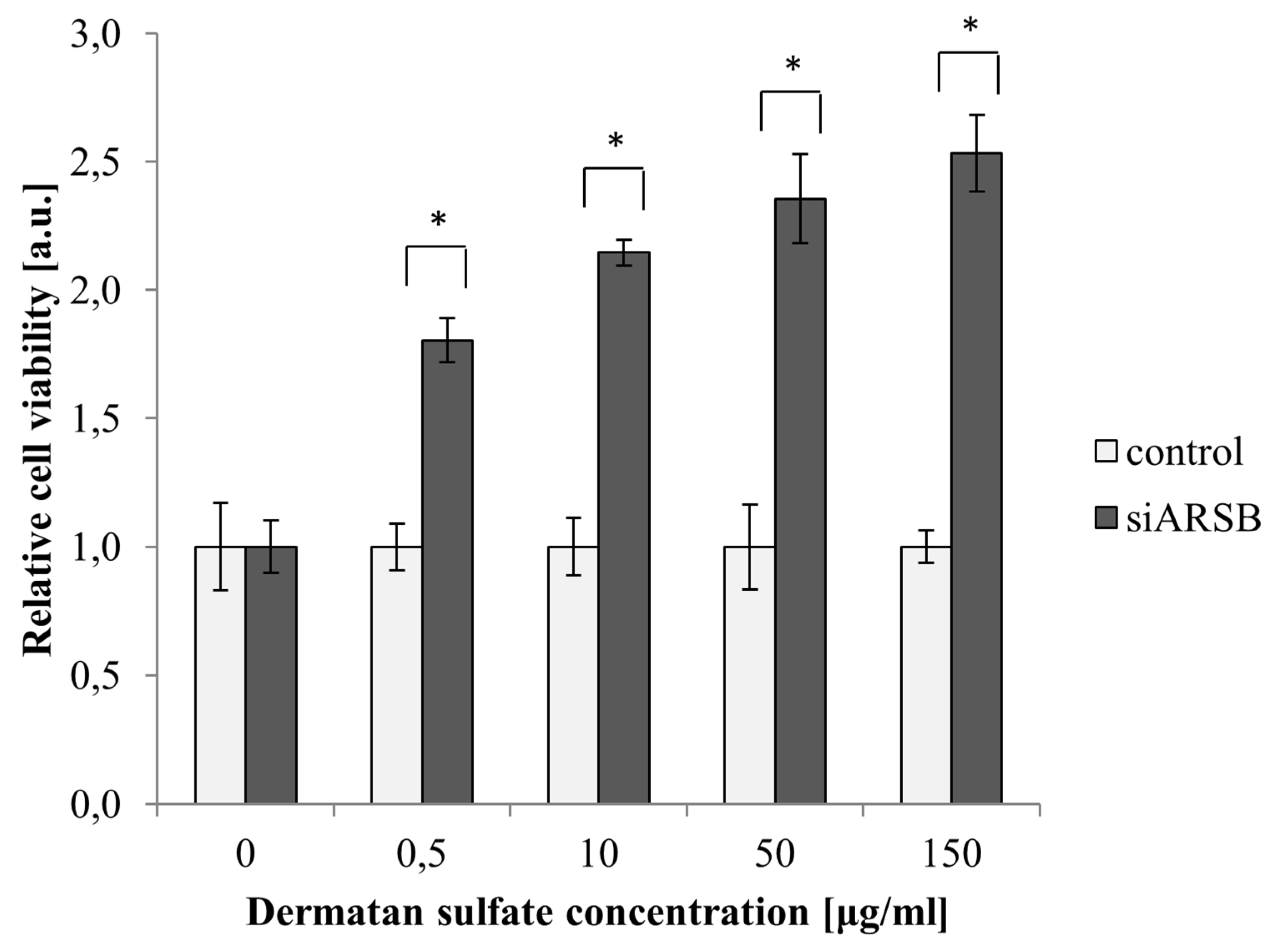
2.1. Viability of PASM Cells Is Increased by Growth with DS
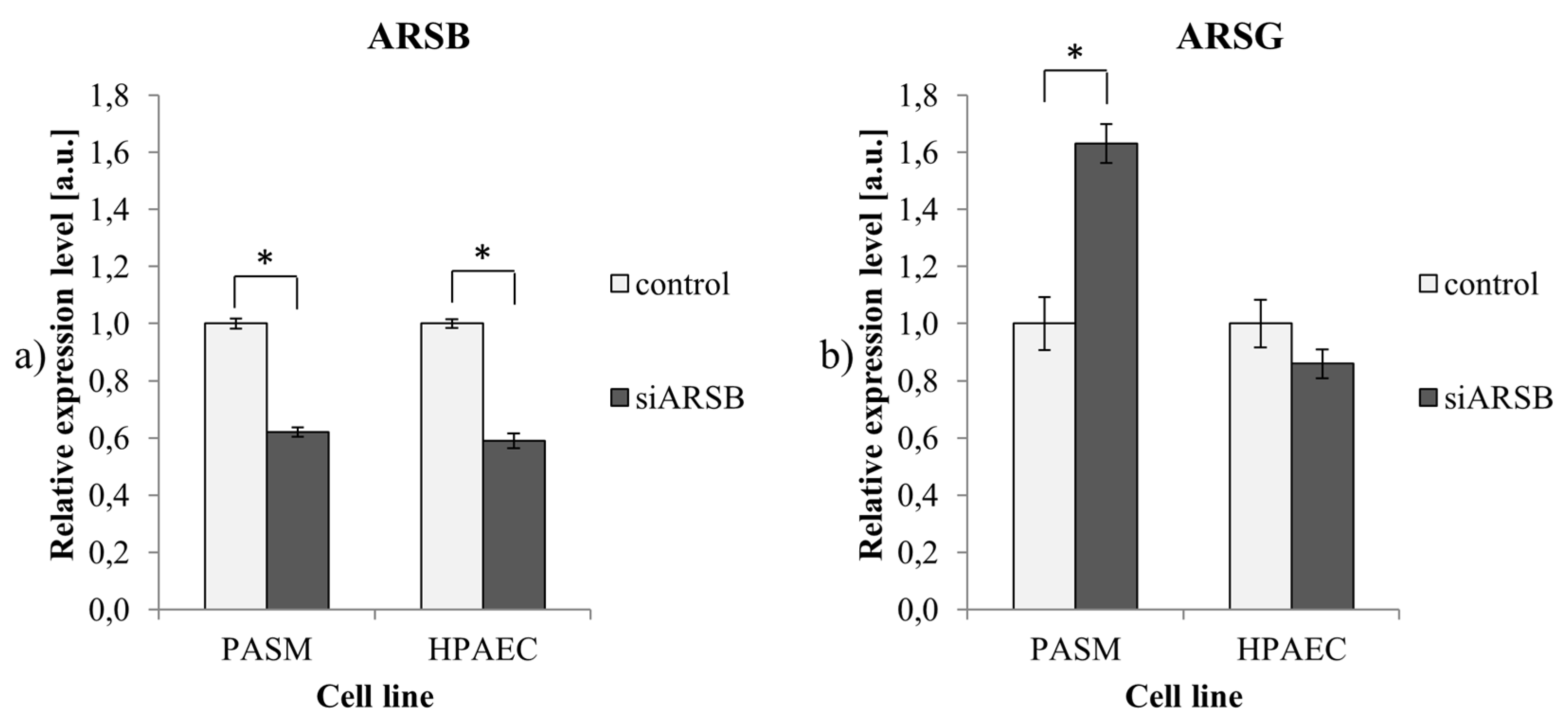
2.2. Reduced ARSB Expression is Accompanied by Upregulation of ARSG in PASM Cells
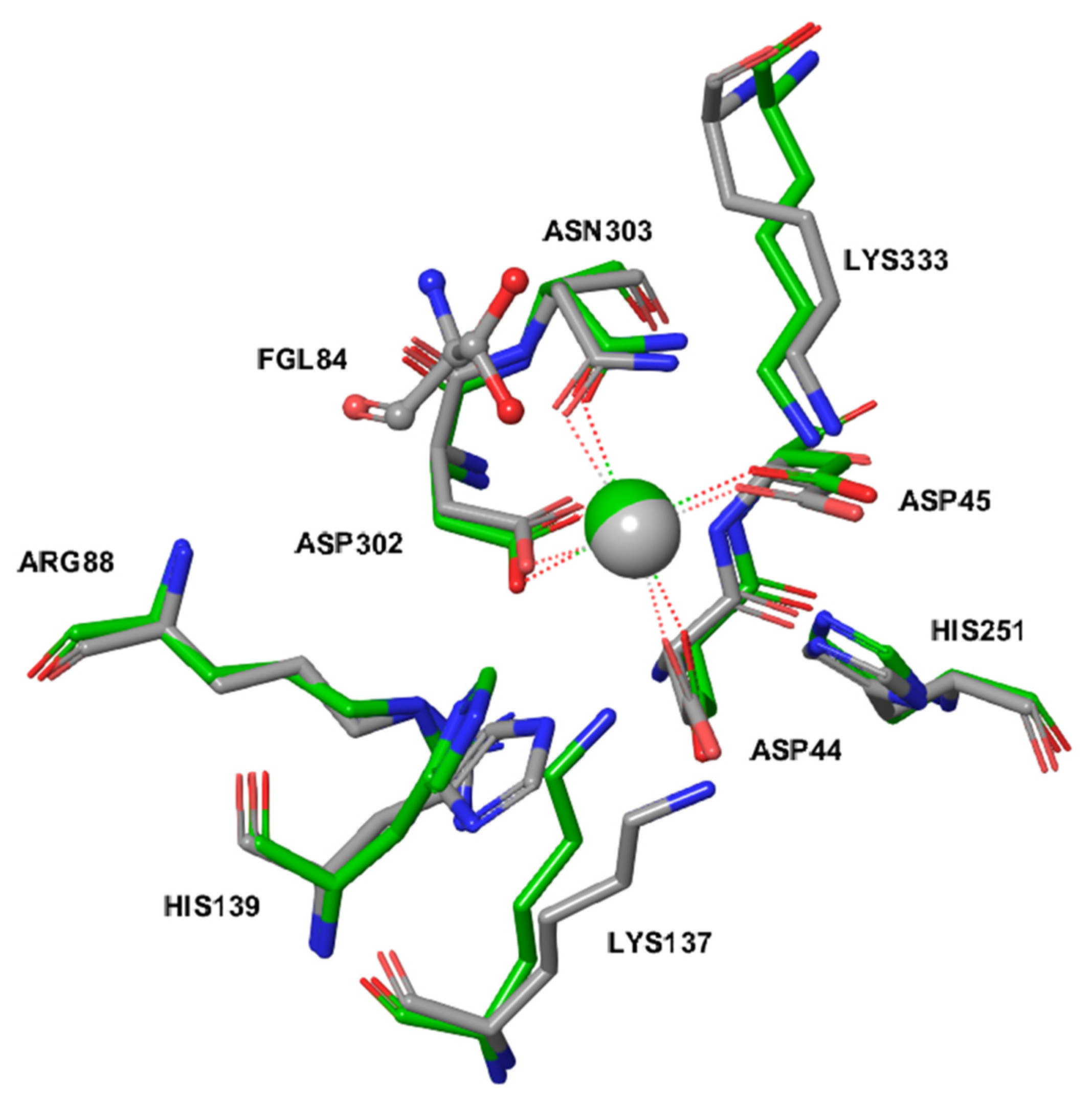
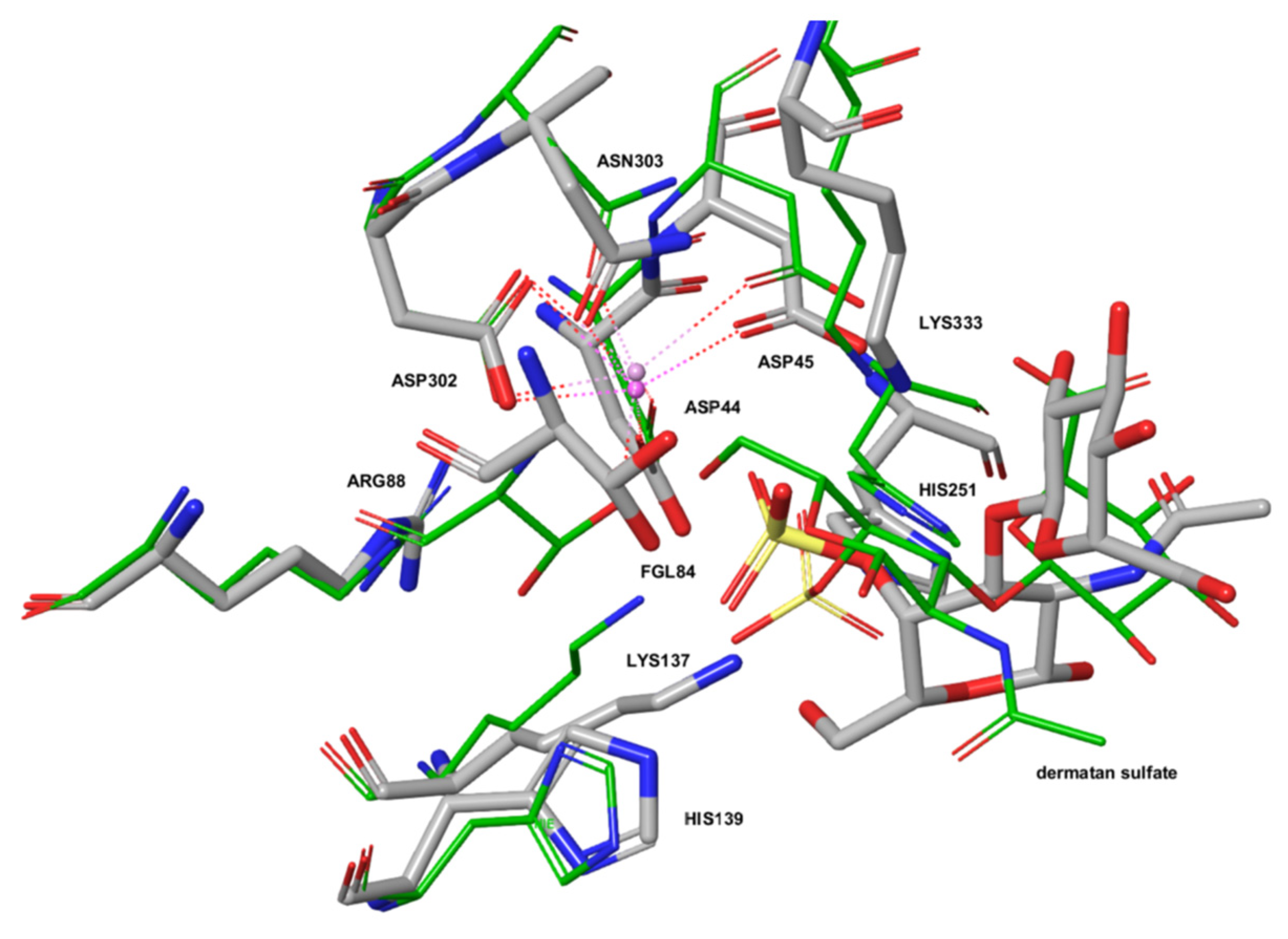
2.3. The Active Site of ARSG is Closely Similar to That of ARSB
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Gene Silencing
4.3. Cell Viability
4.4. Microarray Data Analysis and Bioinformatics Processing
4.5. RT-qPCR
4.6. Homology Modeling of Human ARSG
4.7. Ligand Docking
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARSA | Arylsulfatase A |
| ARSB | Arylsulfatase B |
| ARSG | Arylsulfatase G |
| LD | linear dichroism |
| DS | Dermatan sulfate |
| FGF-2 | Fibroblast growth factor |
| FGF-7 | Keratinocyte growth factor |
| fGly | Formylglycine |
| GAG | Glycsaminoglycans |
| GALNS | Galactosamine-6-sulfatase |
| HPAEC | Human pulmonary artery endothelial cells |
| MPS | mucopolisaccharidosis |
| PASM | Human pulmonary artery smooth muscle cells |
References
- Parenti, G.; Meroni, G.; Ballabio, A. The sulfatase gene family. Curr. Opin. Genet. Dev. 1997, 7, 386–391. [Google Scholar] [CrossRef]
- Ghosh, D. Human sulfatases: A structural perspective to catalysis. Cell. Mol. Life Sci. 2007, 64, 2013–2022. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, P.; Messali, S.; Meroni, G.; Ballabio, A. Molecular and biochemical characterisation of a novel sulphatase gene: Arylsulfatase G (ARSG). Eur. J. Hum. Genet. 2002, 10, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Frese, M.-A.; Schulz, S.; Dierks, T.; Schüpke, S. Arylsulfatase G, a Novel Lysosomal Sulfatase. J. Boil. Chem. 2008, 283, 11388–11395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalewski, B.; Lübke, T.; Kollmann, K.; Braulke, T.; Reinheckel, T.; Dierks, T.; Damme, M. Molecular characterization of arylsulfatase G expression, processing, glycosylation, transport and activity. J. Biol. Chem. 2014, 289, 27992–28005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshikawa, M.; Usami, R.; Kato, S. Characterization of the arylsulfatase I (ARSI) gene preferentially expressed in the human retinal pigment epithelium cell line ARPE-19. Mol. Vis. 2009, 15, 482–494. [Google Scholar]
- Ghosh, D. Three-Dimensional Structures of Sulfatases. Methods Enzymol. 2005, 400, 273–293. [Google Scholar] [CrossRef]
- Selmer, T.; Hallmann, A.; Schmidt, B.; Sumper, M.; Figura, K. The Evolutionary Conservation of a Novel Protein Modification, the Conversion of Cysteine to Serinesemialdehyde in Arylsulfatase from Volvox carteri. JBIC J. Boil. Inorg. Chem. 1996, 238, 341–345. [Google Scholar] [CrossRef]
- Recksiek, M.; Selmer, T.; Dierks, T.; Schmidt, B.; Von Figura, K. Sulfatases, Trapping of the Sulfated Enzyme Intermediate by Substituting the Active Site Formylglycine. J. Boil. Chem. 1998, 273, 6096–6103. [Google Scholar] [CrossRef] [Green Version]
- Vassili, V.; Nicely, H.; Harmatz, P.; Turbeville, S. Mucopolysaccharidosis VI. Orphanet J. Rare Dis. 2010, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.; Goude, M.C.; McDevitt, T.C.; Temenoff, J.S. Molecular engineering of glycosaminoglycan chemistry for biomolecule delivery. Acta Biomater. 2013, 10, 1705–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinek, A. Vascular myocytes—The multifunctional cells. Postępy Biol. Komórki 1982, 9, 401–420. [Google Scholar]
- Lee, P.H.A.; Trowbridge, J.M.; Taylor, K.R.; Morhenn, V.B.; Gallo, R.L. Dermatan Sulfate Proteoglycan and Glycosaminoglycan Synthesis Is Induced in Fibroblasts by Transfer to a Three-dimensional Extracellular Environment. J. Boil. Chem. 2004, 279, 48640–48646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penc, S.F.; Pomahac, B.; Winkler, T.; Dorschner, R.A.; Eriksson, E.; Herndon, M.; Gallo, R.L. Dermatan Sulfate Released after Injury Is a Potent Promoter of Fibroblast Growth Factor-2 Function. J. Boil. Chem. 1998, 273, 28116–28121. [Google Scholar] [CrossRef] [Green Version]
- Trowbridge, J.M.; Rudisill, J.A.; Ron, D.; Gallo, R.L. Dermatan Sulfate Binds and Potentiates Activity of Keratinocyte Growth Factor (FGF-7). J. Boil. Chem. 2002, 277, 42815–42820. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, M.F.; Lacerda, L.; Alves, S. Glycosaminoglycan Storage Disorders: A Review. Biochem. Res. Int. 2012, 2012, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Karageorgos, L.; Brooks, D.A.; Pollard, A.; Melville, E.L.; Hein, L.K.; Clements, P.R.; Ketteridge, D.; Swiedler, S.J.; Beck, M.; Giugliani, R.; et al. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Hum. Mutat. 2007, 28, 897–903. [Google Scholar] [CrossRef]
- Harmatz, P.; Shediac, R. Mucopolysaccharidosis VI: Patophysiology, diagnosis and treatmnt. Front. Biosci.-Landmrk 2017, 22, 385–406. [Google Scholar]
- Golda, A.; Jurecka, A.; Gajda, K.; Tylki-Szymańska, A.; Lalik, A. Human pulmonary artery endothelial cells in the model of mucopolysaccharidosis VI present a prohypertensive phenotype. Mol. Genet. Metab. Rep. 2015, 3, 11–17. [Google Scholar] [CrossRef]
- Scott, R.A.; Panitch, A. Decorin Mimic Regulates Platelet-Derived Growth Factor and Interferon-γ Stimulation of Vascular Smooth Muscle Cells. Biomacromolecules 2014, 15, 2090–2103. [Google Scholar] [CrossRef] [Green Version]
- Jufri, N.F.; Mohamedali, A.; Avolio, A.P.; Baker, M.S. Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vasc. Cell 2015, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovar, A.M.; Teixeira, L.A.; Marinho, A.C.; Pinho, D.A.; Silva, L.-F.; Mourão, P.A.D.S. The dermatan sulfate-dependent anticoagulant pathway is mostly preserved in aneurysm and in severe atherosclerotic lesions while the heparan sulfate pathway is disrupted. Clin. Chim. Acta 2011, 412, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Ramaswamy, A.K.; Park, K.; Panitch, A. Decorin mimic promotes endothelial cell health in endothelial monolayers and endothelial-smooth muscle co-cultures. J. Tissue Eng. Regen. Med. 2015, 11, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Paderi, J.E.; Sturek, M.; Panitch, A. Decorin Mimic Inhibits Vascular Smooth Muscle Proliferation and Migration. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Rasente, R.Y.; Egitto, P.; Calabrese, G.C. Low molecular mass dermatan sulfate modulates endothelial cells proliferation and migration. Carbohydr. Res. 2012, 356, 233–237. [Google Scholar] [CrossRef]
- Appel, M.; Bertozzi, C.R. Formylglycine, a Post-Translationally Generated Residue with Unique Catalytic Capabilities and Biotechnology Applications. ACS Chem. Boil. 2014, 10, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer Science and Business Media LLC: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Gentleman, R.C.; Carey, V.; Bates, D.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Boil. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Thorne, N.P. Normalization for two-color cDNA microarray data. Lect. Notes-Monograph Series 2003, 40, 403–418. [Google Scholar] [CrossRef]
- Bolstad, B.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Expression (log2FC) | |
|---|---|---|
| HPAEC | PASM | |
| Enzymes Involved in Dermatan Sulfate Metabolism | ||
| Iduronate 2-sulfatase | 0.18 | −0.13 |
| A-L-iduronidase | 0.11 | −0.03 |
| Arylsulfatase B | −1.78 * | −1.63 * |
| β-Hexoaminidase A | −0.15 | 0.04 |
| β-Hexoaminidase B | 0.05 | −0.05 |
| β-glucuronidase | 0.09 | −0.04 |
| Gene | Expression [log2FC] | |
|---|---|---|
| HPAEC | PASM | |
| Arylsulfatases | ||
| Arylsulfatase A | 0.08 | −0.10 |
| Arylsulfatase B | −1.78 * | −1.63 * |
| Arylsulfatase D | −0.22 | 0.04 |
| Arylsulfatase E | 0.18 | −0.23 |
| Arylsulfatase F | 0.22 | 0.08 |
| Arylsulfatase G | 0.004 | 0.67 * |
| Arylsulfatase H | −0.08 | −0.03 |
| Arylsulfatase I | −0.02 | 0.06 |
| Arylsulfatase J | −0.43 | −0.02 |
| Arylsulfatase K | 0.12 | −0.12 |
| ARSG | ARSB | GALNS | ARSA |
|---|---|---|---|
| Asp44 | Asp53 | Asp39 | Asp29 |
| Asp45 | Asp54 | Asp40 | Asp30 |
| FGly84 | FGly91 | FGly79 | FGly69 |
| Arg88 | Arg95 | Arg83 | Arg73 |
| Lys137 | Lys145 | Lys140 | Lys123 |
| His139 | His147 | His142 | His125 |
| His251 | His242 | His236 | His229 |
| Asp302 | Asp300 | Asp288 | Asp281 |
| Asn303 | Asn301 | Asn289 | Asn282 |
| Lys333 | Lys318 | Lys310 | Lys302 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poterala-Hejmo, A.; Golda, A.; Pacholczyk, M.; Student, S.; Tylki-Szymańska, A.; Lalik, A. A Possible Role for Arylsulfatase G in Dermatan Sulfate Metabolism. Int. J. Mol. Sci. 2020, 21, 4913. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144913
Poterala-Hejmo A, Golda A, Pacholczyk M, Student S, Tylki-Szymańska A, Lalik A. A Possible Role for Arylsulfatase G in Dermatan Sulfate Metabolism. International Journal of Molecular Sciences. 2020; 21(14):4913. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144913
Chicago/Turabian StylePoterala-Hejmo, Aleksandra, Adam Golda, Marcin Pacholczyk, Sebastian Student, Anna Tylki-Szymańska, and Anna Lalik. 2020. "A Possible Role for Arylsulfatase G in Dermatan Sulfate Metabolism" International Journal of Molecular Sciences 21, no. 14: 4913. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144913