Repositioning of Anthelmintic Drugs for the Treatment of Cancers of the Digestive System

 , ,
, ,
Abstract
:1. Introduction
2. Anti-Cancer Effects of Anthelmintic Drugs in Malignancies of the Digestive System
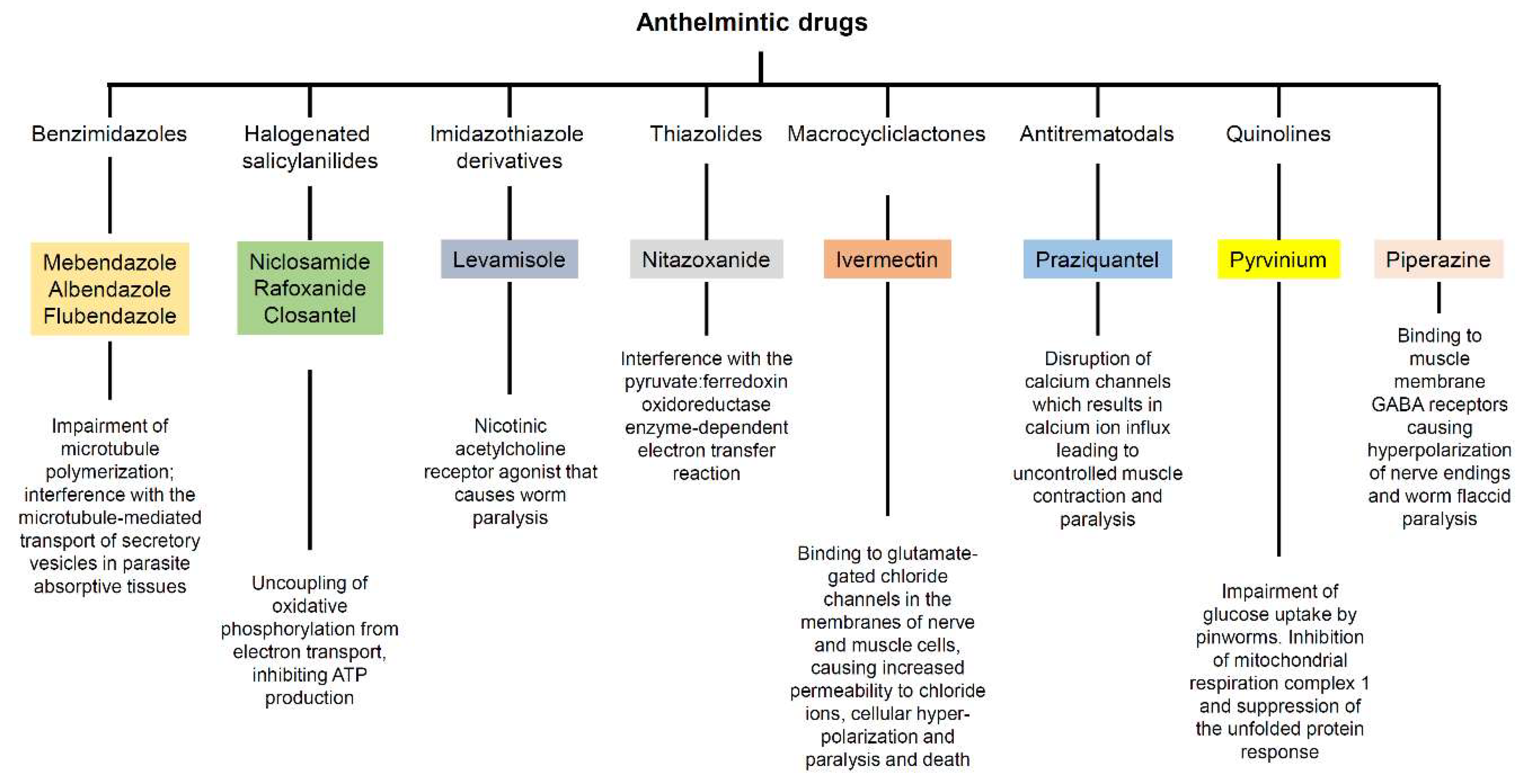
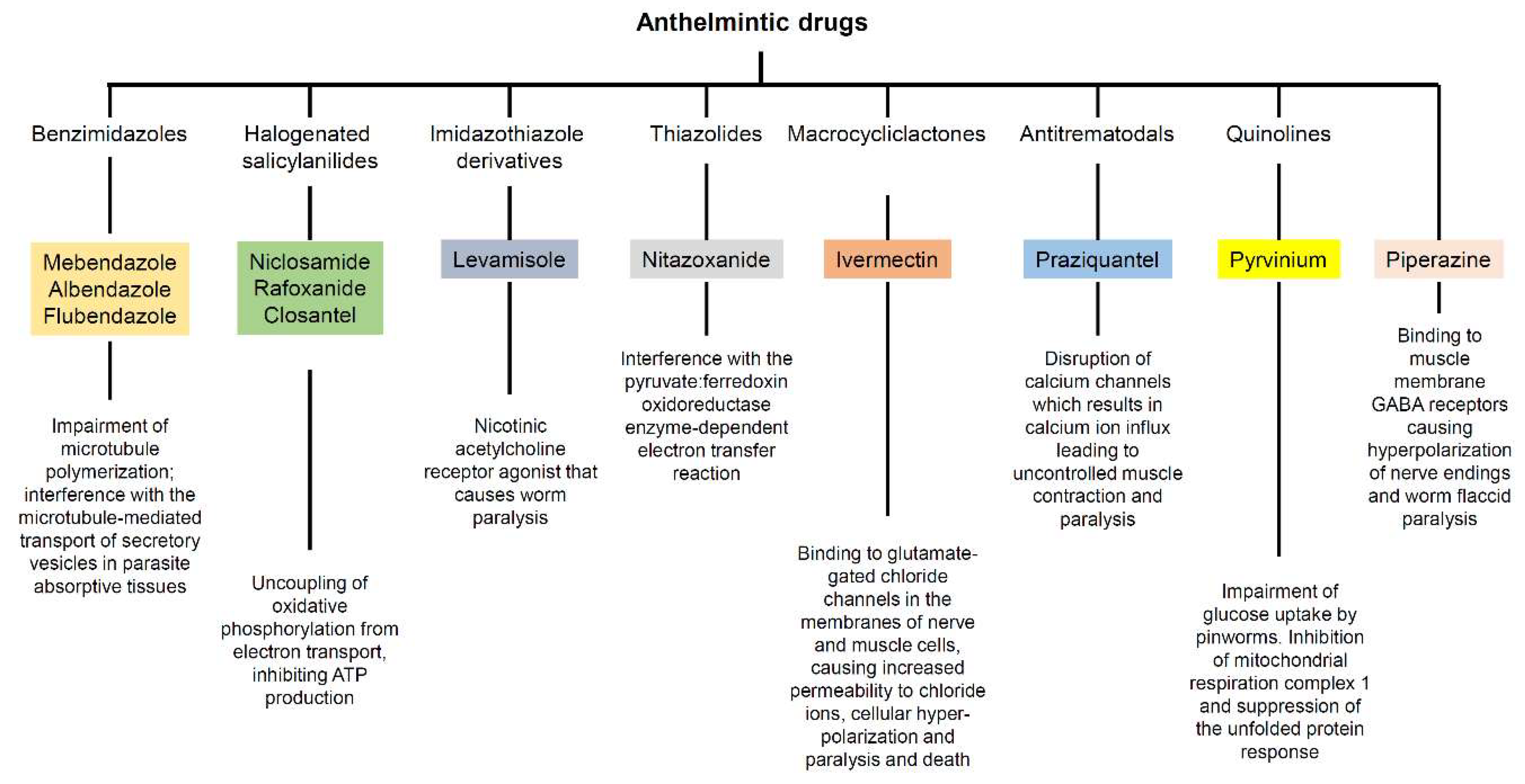
2.1. Benzimidazoles
2.1.1. Mebendazole
2.1.2. Albendazole
2.1.3. Flubendazole
2.2. Halogenated Salicylanilides
2.2.1. Niclosamide
2.2.2. Rafoxanide
2.2.3. Closantel
2.3. Other Anthelmintic Drugs
2.3.1. Levamisole
2.3.2. Nitazoxanide
2.3.3. Ivermectin
2.3.4. Praziquantel
2.3.5. Pyrvinium
2.3.6. Piperazine
3. Discussion
4. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labianca, R.; Nordlinger, B.; Beretta, G.D.; Mosconi, S.; Mandala, M.; Cervantes, A.; Arnold, D.; Group, E.G.W. Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24 (Suppl. 6), 64–72. [Google Scholar] [CrossRef] [PubMed]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sertkaya, A.; Wong, H.H.; Jessup, A.; Beleche, T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials 2016, 13, 117–126. [Google Scholar] [CrossRef]
- Yeu, Y.; Yoon, Y.; Park, S. Protein localization vector propagation: A method for improving the accuracy of drug repositioning. Mol. Biosyst. 2015, 11, 2096–2102. [Google Scholar] [CrossRef]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar]
- Collier, R. Rapidly rising clinical trial costs worry researchers. CMAJ 2009, 180, 277–278. [Google Scholar] [CrossRef] [Green Version]
- Institute of Medicine. In Drug Repurposing and Repositioning: Workshop Summary; Berger, A.C.; Olson, S.; Johnson, S.G.; Beachy, S.H. (Eds.) National Academies Press: Washington, DC, USA, 2014. [Google Scholar]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Prasad, V.; De Jesus, K.; Mailankody, S. The high price of anticancer drugs: Origins, implications, barriers, solutions. Nat. Rev. Clin. Oncol. 2017, 14, 381–390. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Front. Oncol. 2017, 7, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Zhang, Y.; Liu, K.; Liu, B.; Xu, W.; Gao, J.; Ding, L.; Tao, L. Ivermectin induces cell cycle arrest and apoptosis of HeLa cells via mitochondrial pathway. Cell Prolif. 2019, 52, e12543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangguan, F.; Liu, Y.; Ma, L.; Qu, G.; Lv, Q.; An, J.; Yang, S.; Lu, B.; Cao, Q. Niclosamide inhibits ovarian carcinoma growth by interrupting cellular bioenergetics. J. Cancer 2020, 11, 3454–3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Sun, X.; Zhang, X.; Cao, J. Targeting mitochondria by anthelmintic drug atovaquone sensitizes renal cell carcinoma to chemotherapy and immunotherapy. J. Biochem. Mol. Toxicol. 2018, 32, e22195. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Y.; Wan, H.; Hu, J. Antibiotic ivermectin selectively induces apoptosis in chronic myeloid leukemia through inducing mitochondrial dysfunction and oxidative stress. Biochem. Biophys. Res. Commun. 2018, 497, 241–247. [Google Scholar] [CrossRef]
- Zhu, M.; Li, Y.; Zhou, Z. Antibiotic ivermectin preferentially targets renal cancer through inducing mitochondrial dysfunction and oxidative damage. Biochem. Biophys. Res. Commun. 2017, 492, 373–378. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, L.; Zhou, Y.; Rajoria, P.; Wang, C. Pyrvinium selectively induces apoptosis of lymphoma cells through impairing mitochondrial functions and JAK2/STAT5. Biochem. Biophys. Res. Commun. 2016, 469, 716–722. [Google Scholar] [CrossRef]
- Xiang, W.; Cheong, J.K.; Ang, S.H.; Teo, B.; Xu, P.; Asari, K.; Sun, W.T.; Than, H.; Bunte, R.M.; Virshup, D.M.; et al. Pyrvinium selectively targets blast phase-chronic myeloid leukemia through inhibition of mitochondrial respiration. Oncotarget 2015, 6, 33769–33780. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [Green Version]
- Hanusova, V.; Skalova, L.; Kralova, V.; Matouskova, P. Potential anti-cancer drugs commonly used for other indications. Curr. Cancer Drug Targets 2015, 15, 35–52. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell. Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.C.; Soares, B.M.; Pinheiro Jde, J.; Riggins, G.J.; Assumpcao, P.P.; Burbano, R.M.; Montenegro, R.C. The anthelmintic drug mebendazole inhibits growth, migration and invasion in gastric cancer cell model. Toxicol. Vitr. 2015, 29, 2038–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygren, P.; Fryknas, M.; Agerup, B.; Larsson, R. Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 2133–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, T.; Bai, R.Y.; Staedtke, V.; Huso, D.; Riggins, G.J. Mebendazole and a non-steroidal anti-inflammatory combine to reduce tumor initiation in a colon cancer preclinical model. Oncotarget 2016, 7, 68571–68584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygren, P.; Larsson, R. Drug repositioning from bench to bedside: Tumour remission by the antihelmintic drug mebendazole in refractory metastatic colon cancer. Acta Oncol. 2014, 53, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.S.; Ghanim, A.M.H.; Saber, S. Mebendazole augments sensitivity to sorafenib by targeting MAPK and BCL-2 signalling in n-nitrosodiethylamine-induced murine hepatocellular carcinoma. Sci. Rep. 2019, 9, 19095. [Google Scholar] [CrossRef] [Green Version]
- Pourgholami, M.H.; Akhter, J.; Wang, L.; Lu, Y.; Morris, D.L. Antitumor activity of albendazole against the human colorectal cancer cell line HT-29: In vitro and in a xenograft model of peritoneal carcinomatosis. Cancer Chemother. Pharmacol. 2005, 55, 425–432. [Google Scholar] [CrossRef]
- Kralova, V.; Hanusova, V.; Stankova, P.; Knoppova, K.; Canova, K.; Skalova, L. Antiproliferative effect of benzimidazole anthelmintics albendazole, ricobendazole, and flubendazole in intestinal cancer cell lines. Anti-Cancer Drugs 2013, 24, 911–919. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Woon, L.; Almajd, R.; Akhter, J.; Bowery, P.; Morris, D.L. In vitro and in vivo suppression of growth of hepatocellular carcinoma cells by albendazole. Cancer Lett. 2001, 165, 43–49. [Google Scholar] [CrossRef]
- Chen, H.; Weng, Z.; Xu, C. Albendazole suppresses cell proliferation and migration and induces apoptosis in human pancreatic cancer cells. Anti-Cancer Drugs 2020, 31, 431–439. [Google Scholar] [CrossRef]
- Kralova, V.; Hanusova, V.; Rudolf, E.; Canova, K.; Skalova, L. Flubendazole induces mitotic catastrophe and senescence in colon cancer cells in vitro. J. Pharm. Pharmacol. 2016, 68, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Hanusova, V.; Skalova, L.; Kralova, V.; Matouskova, P. The Effect of Flubendazole on Adhesion and Migration in SW480 and SW620 Colon Cancer Cells. Anti-Cancer Agents Med. Chem. 2018, 18, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ren, X.R.; Piao, H.; Zhao, S.; Osada, T.; Premont, R.T.; Mook, R.A., Jr.; Morse, M.A.; Lyerly, H.K.; Chen, W. Niclosamide-induced Wnt signaling inhibition in colorectal cancer is mediated by autophagy. Biochem. J. 2019, 476, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Monin, M.B.; Krause, P.; Stelling, R.; Bocuk, D.; Niebert, S.; Klemm, F.; Pukrop, T.; Koenig, S. The anthelmintic niclosamide inhibits colorectal cancer cell lines via modulation of the canonical and noncanonical Wnt signaling pathway. J. Surg. Res. 2016, 203, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.; Zhou, L.; Deng, Q.; Wang, J.; Yu, Y.; Zhu, J.; Yuan, Y. Niclosamide induced cell apoptosis via upregulation of ATF3 and activation of PERK in Hepatocellular carcinoma cells. BMC Gastroenterol. 2016, 16, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhou, X.; Xu, H.; Shi, X.; Zhao, J.; Yang, M.; Zhang, L.; Jin, X.; Hu, Y.; Li, X.; et al. Niclosamide Inhibits Cell Growth and Enhances Drug Sensitivity of Hepatocellular Carcinoma Cells via STAT3 Signaling Pathway. J. Cancer 2018, 9, 4150–4155. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.C.; Chen, Y.K.; Hsu, Y.J.; Lin, B.R. Niclosamide inhibits the cell proliferation and enhances the responsiveness of esophageal cancer cells to chemotherapeutic agents. Oncol. Rep. 2020, 43, 549–561. [Google Scholar] [CrossRef]
- Alasadi, A.; Chen, M.; Swapna, G.V.T.; Tao, H.; Guo, J.; Collantes, J.; Fadhil, N.; Montelione, G.T.; Jin, S. Effect of mitochondrial uncouplers niclosamide ethanolamine (NEN) and oxyclozanide on hepatic metastasis of colon cancer. Cell Death Dis. 2018, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Laudisi, F.; Di Grazia, A.; De Simone, V.; Cherubini, F.; Colantoni, A.; Ortenzi, A.; Franze, E.; Dinallo, V.; Di Fusco, D.; Monteleone, I.; et al. Induction of endoplasmic reticulum stress and inhibition of colon carcinogenesis by the anti-helmintic drug rafoxanide. Cancer Lett. 2019, 462, 1–11. [Google Scholar] [CrossRef]
- Di Grazia, A.; Laudisi, F.; Di Fusco, D.; Franze, E.; Ortenzi, A.; Monteleone, I.; Monteleone, G.; Stolfi, C. Rafoxanide Induces Immunogenic Death of Colorectal Cancer Cells. Cancers 2020, 12, 1314. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Hu, Y.L.; Feng, Y.; Guo, Y.B.; Liu, Y.F.; Yang, J.L.; Mao, Q.S.; Xue, W.J. Rafoxanide promotes apoptosis and autophagy of gastric cancer cells by suppressing PI3K /Akt/mTOR pathway. Exp. Cell Res. 2019, 385, 111691. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.Y.; Xia, B.; Liu, H.C.; Xu, Y.Q.; Huang, C.J.; Gao, J.M.; Dong, Q.X.; Li, C.Q. Closantel Suppresses Angiogenesis and Cancer Growth in Zebrafish Models. Assay Drug Dev. Technol. 2016, 14, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, J.; Sidler, D.; Nachbur, U.; Wastling, J.; Brunner, T.; Hemphill, A. Thiazolides inhibit growth and induce glutathione-S-transferase Pi (GSTP1)-dependent cell death in human colon cancer cells. Int. J. Cancer 2008, 123, 1797–1806. [Google Scholar] [CrossRef]
- Senkowski, W.; Zhang, X.; Olofsson, M.H.; Isacson, R.; Hoglund, U.; Gustafsson, M.; Nygren, P.; Linder, S.; Larsson, R.; Fryknas, M. Three-Dimensional Cell Culture-Based Screening Identifies the Anthelmintic Drug Nitazoxanide as a Candidate for Treatment of Colorectal Cancer. Mol. Cancer Ther. 2015, 14, 1504–1516. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Olsen, J.R.; Yuan, X.; Cheng, P.F.; Levesque, M.P.; Brokstad, K.A.; Hoffman, P.S.; Oyan, A.M.; Zhang, W.; Kalland, K.H.; et al. Small molecule promotes beta-catenin citrullination and inhibits Wnt signaling in cancer. Nat. Chem. Biol. 2018, 14, 94–101. [Google Scholar] [CrossRef]
- Ripani, P.; Delp, J.; Bode, K.; Delgado, M.E.; Dietrich, L.; Betzler, V.M.; Yan, N.; von Scheven, G.; Mayer, T.U.; Leist, M.; et al. Thiazolides promote G1 cell cycle arrest in colorectal cancer cells by targeting the mitochondrial respiratory chain. Oncogene 2020, 39, 2345–2357. [Google Scholar] [CrossRef]
- Melotti, A.; Mas, C.; Kuciak, M.; Lorente-Trigos, A.; Borges, I.; Ruiz i Altaba, A. The river blindness drug Ivermectin and related macrocyclic lactones inhibit WNT-TCF pathway responses in human cancer. EMBO Mol. Med. 2014, 6, 1263–1278. [Google Scholar] [CrossRef]
- Nambara, S.; Masuda, T.; Nishio, M.; Kuramitsu, S.; Tobo, T.; Ogawa, Y.; Hu, Q.; Iguchi, T.; Kuroda, Y.; Ito, S.; et al. Antitumor effects of the antiparasitic agent ivermectin via inhibition of Yes-associated protein 1 expression in gastric cancer. Oncotarget 2017, 8, 107666–107677. [Google Scholar] [CrossRef]
- Intuyod, K.; Hahnvajanawong, C.; Pinlaor, P.; Pinlaor, S. Anti-parasitic Drug Ivermectin Exhibits Potent Anticancer Activity Against Gemcitabine-resistant Cholangiocarcinoma In Vitro. Anticancer Res. 2019, 39, 4837–4843. [Google Scholar] [CrossRef]
- Wu, Z.H.; Lu, M.K.; Hu, L.Y.; Li, X. Praziquantel synergistically enhances paclitaxel efficacy to inhibit cancer cell growth. PLoS ONE 2012, 7, e51721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esumi, H.; Lu, J.; Kurashima, Y.; Hanaoka, T. Antitumor activity of pyrvinium pamoate, 6-(dimethylamino)-2-[2-(2,5-dimethyl-1-phenyl-1H-pyrrol-3-yl)ethenyl]-1-methyl-qu inolinium pamoate salt, showing preferential cytotoxicity during glucose starvation. Cancer Sci. 2004, 95, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.H.; Macdonald, J.; Liu, G.; Lee, A.S.; Ly, M.; Davis, T.; Ke, N.; Zhou, D.; Wong-Staal, F.; Li, Q.X. Pyrvinium targets the unfolded protein response to hypoglycemia and its anti-tumor activity is enhanced by combination therapy. PLoS ONE 2008, 3, e3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Flaveny, C.A.; Giambelli, C.; Fei, D.L.; Han, L.; Hang, B.I.; Bai, F.; Pei, X.H.; Nose, V.; Burlingame, O.; et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS ONE 2014, 9, e101969. [Google Scholar] [CrossRef] [Green Version]
- Wiegering, A.; Uthe, F.W.; Huttenrauch, M.; Muhling, B.; Linnebacher, M.; Krummenast, F.; Germer, C.T.; Thalheimer, A.; Otto, C. The impact of pyrvinium pamoate on colon cancer cell viability. Int. J. Color. Dis. 2014, 29, 1189–1198. [Google Scholar] [CrossRef]
- Chopra, A.; Anderson, A.; Giardina, C. Novel piperazine-based compounds inhibit microtubule dynamics and sensitize colon cancer cells to tumor necrosis factor-induced apoptosis. J. Biol. Chem. 2014, 289, 2978–2991. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kwon, H.Y.; Ryu, D.H.; Nam, M.H.; Shim, B.S.; Kim, J.H.; Lee, J.Y.; Kim, S.H. Inhibition of CUG-binding protein 1 and activation of caspases are critically involved in piperazine derivative BK10007S induced apoptosis in hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0186490. [Google Scholar] [CrossRef]
- Moertel, C.G.; Fleming, T.R.; Macdonald, J.S.; Haller, D.G.; Laurie, J.A.; Goodman, P.J.; Ungerleider, J.S.; Emerson, W.A.; Tormey, D.C.; Glick, J.H.; et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N. Engl. J. Med. 1990, 322, 352–358. [Google Scholar] [CrossRef]
- O’Connell, M.J.; Laurie, J.A.; Kahn, M.; Fitzgibbons, R.J., Jr.; Erlichman, C.; Shepherd, L.; Moertel, C.G.; Kocha, W.I.; Pazdur, R.; Wieand, H.S.; et al. Prospectively randomized trial of postoperative adjuvant chemotherapy in patients with high-risk colon cancer. J. Clin. Oncol. 1998, 16, 295–300. [Google Scholar] [CrossRef]
- Wolmark, N.; Rockette, H.; Mamounas, E.; Jones, J.; Wieand, S.; Wickerham, D.L.; Bear, H.D.; Atkins, J.N.; Dimitrov, N.V.; Glass, A.G.; et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: Results from National Surgical Adjuvant Breast and Bowel Project C-04. J. Clin. Oncol. 1999, 17, 3553–3559. [Google Scholar] [PubMed]
- Morris, D.L.; Jourdan, J.L.; Pourgholami, M.H. Pilot study of albendazole in patients with advanced malignancy. Effect on serum tumor markers/high incidence of neutropenia. Oncology 2001, 61, 42–46. [Google Scholar] [CrossRef] [PubMed]
- De Simone, V.; Franze, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [Green Version]
- Armour, J.; Corba, J. The anthelmintic activity of rafoxanide against immature Fasciola hepatica in sheep. Vet. Rec. 1970, 87, 213–214. [Google Scholar] [CrossRef]
- Knapp, S.E.; Presidente, P.J. Efficacy of rafoxanide against natural Fasciola hepatica infections in cattle. Am. J. Vet. Res. 1971, 32, 1289–1291. [Google Scholar]
- Swan, G.E. The pharmacology of halogenated salicylanilides and their anthelmintic use in animals. J. S. Afr. Vet. Assoc. 1999, 70, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Yurdakok, M. [Rafanoxide therapy in a child with fascioliasis]. Mikrobiyol. Bul. 1985, 19, 38–40. [Google Scholar]
- Gooyit, M.; Janda, K.D. Reprofiled anthelmintics abate hypervirulent stationary-phase Clostridium difficile. Sci. Rep. 2016, 6, 33642. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guo, B.; Xu, Z.; Li, B.; Cai, T.; Zhang, X.; Yu, Y.; Wang, H.; Shi, J.; Zhu, W. Repositioning organohalogen drugs: A case study for identification of potent B-Raf V600E inhibitors via docking and bioassay. Sci. Rep. 2016, 6, 31074. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.J.; Robertson, A.P.; Buxton, S.K.; Beech, R.N.; Charvet, C.L.; Neveu, C. Levamisole receptors: A second awakening. Trends Parasitol. 2012, 28, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.Y.; Lin, Y.L.; Chiang, B.L. Levamisole enhances immune response by affecting the activation and maturation of human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2008, 151, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Blagburn, B.L.; Drain, K.L.; Land, T.M.; Kinard, R.G.; Moore, P.H.; Lindsay, D.S.; Patrick, D.A.; Boykin, D.W.; Tidwell, R.R. Comparative efficacy evaluation of dicationic carbazole compounds, nitazoxanide, and paromomycin against Cryptosporidium parvum infections in a neonatal mouse model. Antimicrob. Agents Chemother. 1998, 42, 2877–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodos, C.M.; Griffiths, J.K.; D’Onfro, J.; Fairfield, A.; Tzipori, S. Efficacy of nitazoxanide against Cryptosporidium parvum in cell culture and in animal models. Antimicrob. Agents Chemother. 1998, 42, 1959–1965. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, J.J.; Ayoub, A.; Gargala, G.; Chegne, N.L.; Favennec, L. Randomized clinical study of nitazoxanide compared to metronidazole in the treatment of symptomatic giardiasis in children from Northern Peru. Aliment. Pharmacol. Ther. 2001, 15, 1409–1415. [Google Scholar] [CrossRef]
- Musher, D.M.; Logan, N.; Hamill, R.J.; Dupont, H.L.; Lentnek, A.; Gupta, A.; Rossignol, J.F. Nitazoxanide for the treatment of Clostridium difficile colitis. Clin. Infect. Dis. 2006, 43, 421–427. [Google Scholar] [CrossRef]
- Rossignol, J.F. Nitazoxanide: A first-in-class broad-spectrum antiviral agent. Antivir. Res. 2014, 110, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Campbell, W.C.; Fisher, M.H.; Stapley, E.O.; Albers-Schonberg, G.; Jacob, T.A. Ivermectin: A potent new antiparasitic agent. Science 1983, 221, 823–828. [Google Scholar] [CrossRef]
- Juarez, M.; Schcolnik-Cabrera, A.; Duenas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar]
- Choi, W.; Kim, J.; Park, J.; Lee, D.H.; Hwang, D.; Kim, J.H.; Ashktorab, H.; Smoot, D.; Kim, S.Y.; Choi, C.; et al. YAP/TAZ Initiates Gastric Tumorigenesis via Upregulation of MYC. Cancer Res. 2018, 78, 3306–3320. [Google Scholar] [CrossRef] [Green Version]
- Vale, N.; Gouveia, M.J.; Rinaldi, G.; Brindley, P.J.; Gartner, F.; Correia da Costa, J.M. Praziquantel for Schistosomiasis: Single-Drug Metabolism Revisited, Mode of Action, and Resistance. Antimicrob. Agents Chemother. 2017, 61, e02582-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamsa-Ard, S.; Luvira, V.; Pugkhem, A.; Luvira, V.; Thinkhamrop, B.; Suwanrungruang, K.; Bhudhisawasdi, V. Association between praziquantel treatment and cholangiocarcinoma: A hospital-based matched case-control study. BMC Cancer 2015, 15, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momtazi-Borojeni, A.A.; Abdollahi, E.; Ghasemi, F.; Caraglia, M.; Sahebkar, A. The novel role of pyrvinium in cancer therapy. J. Cell. Physiol. 2018, 233, 2871–2881. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.J. gamma-Aminobutyric acid- and piperazine-activated single-channel currents from Ascaris suum body muscle. Br. J. Pharmacol. 1985, 84, 445–461. [Google Scholar] [CrossRef]
- McNair, T.J.; Wibin, F.A.; Hoppe, E.T.; Schmidt, J.L.; Depeyster, J.A. Antitumor action of several new piperazine derivatives compared to certain standard anticancer agents. J. Surg. Res. 1963, 3, 130–136. [Google Scholar] [CrossRef]
- Sleire, L.; Forde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.O. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Scapozza, L.; Ruiz, I.A.A. Drug repurposing in oncology: Compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 434–454. [Google Scholar] [CrossRef]
- Fearon, E.R.; Carethers, J.M. Molecular subtyping of colorectal cancer: Time to explore both intertumoral and intratumoral heterogeneity to evaluate patient outcome. Gastroenterology 2015, 148, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Ulintz, P.J.; Greenson, J.K.; Wu, R.; Fearon, E.R.; Hardiman, K.M. Lymph Node Metastases in Colon Cancer Are Polyclonal. Clin. Cancer Res. 2018, 24, 2214–2224. [Google Scholar] [CrossRef] [Green Version]
- Edwards, G.; Breckenridge, A.M. Clinical pharmacokinetics of anthelmintic drugs. Clin. Pharmacokinet. 1988, 15, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, E.J.; Lobenberg, R.; de Araujo, G.L.B.; Bou-Chacra, N.A. Niclosamide repositioning for treating cancer: Challenges and nano-based drug delivery opportunities. Eur. J. Pharm. Biopharm. 2019, 141, 58–69. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Drug | Cancer Type | Observation | Ref. |
|---|---|---|---|
| Mebendazole | Gastric cancer | Inhibition of cell growth and invasion of a human malignant cell line derived from a primary gastric tumor, whether used alone or in combination 5-FU | [23] |
| Mebendazole | CRC | Cytotoxic activity against CRC cell lines (HCT-116, RKO, HT29, HT-8 and SW626) | [24] |
| Mebendazole | CRC | Reduction of the number and size of intestinal microadenomas in Apcmin/+ mice, once used in combination with sulindac, through the inhibition of MYC and COX2 pathways, cytokine release and angiogenesis | [25] |
| Mebendazole | CRC | Induction of lung and lymph node metastases remission as well as partial liver metastases remission in a patient with refractory metastatic colon cancer | [26] |
| Mebendazole | HCC | Inhibition of the MAPK pathway in vitro and in vivo, whether used alone or in combination with Sorafenib, and improvement of liver function in an animal model of HCC | [27] |
| Albendazole | CRC | Cytostatic effect on HT29 cells in vitro and reduction of cancer growth in nude mice bearing intraperitoneal HT29-derived tumors | [28] |
| Albendazole | CRC | Anti-proliferative effects on SW480, SW620, HCT8 and Caco2 cells, especially when used in combination with paclitaxel | [29] |
| Albendazole | HCC | Cytostatic effects on rat, mice and human HCC cells. Reduction of tumor growth in nude mice inoculated with SK-HEP1 cells | [30] |
| Albendazole | Pancreatic cancer | Cytostatic and pro-apoptotic effects on PANC-1 and SW1990 cells in vitro and in a mouse xenograft model | [31] |
| Flubendazole | CRC | Anti-proliferative effect on SW480, SW620, HCT8, and Caco2 cell lines, also in combination with paclitaxel. Such effect was associated with cyclin B1 and cyclin D1 down-regulation | [29,32] |
| Flubendazole | CRC | Impairment of phosphorylation/activity of NF-kB in SW480 and SW620 cell lines, suppression of the expression of metastatic markers as well as cell migration | [33] |
| Niclosamide | CRC | Inhibition of the Wnt/β-catenin signaling pathway in the human CRC cell lines HCT-116, HT-29 and Caco2 by down-regulation of Dishevelled-2. Anti-cancer effects in CRC cells isolated by surgical resection of metastatic disease as well as in NOD/SCID mice implanted with human CRC cell-derived xenografts | [34] |
| Niclosamide | CRC | Anti-cancer effects on CRC mediated by the induction of autophagy | [35] |
| Niclosamide | CRC | Anti-proliferative and pro-apoptotic effects on SW480, SW620 and CC531 cells by affecting the formation of β-catenin-Bcl9-LEF/TCF triple-complex and inducing c-jun expression | [36] |
| Niclosamide | HCC | Cell growth inhibition and induction of apoptotic cell death in HepG2 and QGY7701 cell lines by eliciting ER stress | [37] |
| Niclosamide | HCC | Impairment of proliferation and induction of apoptosis in HepG2, QGY-7703 and SMMC-7721 cell lines by negatively affecting the phosphorylation/activity of the oncogenic transcription factor STAT3 | [38] |
| Niclosamide | Esophageal cancer | Suppression of STAT3 signaling pathway resulting in the arrest of esophageal adenocarcinoma cells (BE3) and esophageal squamous cell carcinoma cells (CE48T and CE81T) in the G0/G1 phase of the cell cycle | [39] |
| NiclosamideEthanolamine | HCC | Cytostatic effect on HCC cells and impairment of cell migration, whether used alone or in combination with oxyclozanide | [40] |
| Niclosamide Ethanolamine | CRC | Reduction of intestinal polyp formation in Apcmin/+ mice. Decrease of hepatic metastasis in a mouse model of CRC metastasis | [40] |
| Rafoxanide | CRC | Selective induction of ER stress in HCT-116 and DLD1 cells associated with cyclin D1 protein down-regulation, accumulation of cells in the G0/G1 phase and subsequent caspase-dependent apoptosis. Ani-mitogenic effect in human CRC explants. Reduction of both number and size of neoplastic lesions in Apcmin/+ mice | [41] |
| Rafoxanide | CRC | Induction of autophagy and DAMPs (i.e., ecto-calreticulin exposure, ATP/HMGB1 release) in HCT-116 and DLD1 cells, resulting in immunogenic cell death. Reduction of tumor growth in vaccination experiments in vivo using immunocompetent mice and syngeneic cancer cells | [42] |
| Rafoxanide | Gastric cancer | Arrest of gastric cancer cells in the G0/G1 phase of the cell cycle, induction of autophagy and apoptosis through the inhibition of the PI3K/Akt pathway both in vitro and in vivo | [43] |
| Closantel | Liver cancer, pancreatic cancer | Cytostatic effect in zebrafishes xenotransplanted with human liver and pancreatic cancer cells | [44] |
| Nitazoxanide | CRC | Induction of apoptosis in CRC cell lines in a GSTP1-dependent manner | [45] |
| Nitazoxanide | CRC | Inhibition of mitochondrial respiration and mTOR pathway in HCT-116- and HT-29-derived spheroids. Suppression of tumor growth in combination with Irinotecan in a mouse xenograft model | [46] |
| Nitazoxanide | CRC | Anti-cancer activity on CRC cells via the impairment of Wnt/β-catenin signaling pathway | [47] |
| Nitazoxanide derivative(RM4819) | CRC | Cell cycle arrest and suppression of mitochondrial complex III activity. Inhibition of the proliferation of intestinal tumoroids | [48] |
| Ivermectin | CRC | Inhibition of Wnt-TCF pathway in DLD1 and Ls174T cells. Blockade of colon cancer stem cell self-renewal. Impairment of the growth of DLD1- and HT-29-derived xenografts in nude mice in a TCF-dependent fashion | [49] |
| Ivermectin | Gastric cancer | Suppression of MKN1 cell growth in vitro and in vivo through the inhibition of the nuclear expression of YAP1 | [50] |
| Ivermectin | CCA | Induction of S-phase cell cycle arrest and apoptotic cell death in both gemcitabine-sensitive (KKU214) and gemcitabine-resistant (KKU214GemR) CCA cell lines | [51] |
| Praziquantel | CRC | Synergistic negative effect on DLD1 cell growth and viability, associated with XIAP down-regulation, in combination with paclitaxel | [52] |
| Pyrvinium pamoate | Pancreatic cancer | Cytotoxic effect on PANC-1 cells cultured under glucose starvation associated with the inhibition of Akt phosphorylation. Anti-tumor activity in vivo in a hypovascular pancreatic cancer model where immunocompromised mice were xenografted with PANC-1 cells | [53] |
| Pyrvinium pamoate | Pancreatic cancer, CRC | Impairment of glucose starvation-driven transcriptional activation of UPR-related genes (e.g., GRP78, GRP94, XBP-1, ATF4). Anti-tumor activity in nude mice transplanted with either HCT-116 or the pancreatic cancer cell line AsPC-1 in combination with doxorubicin | [54] |
| Pyrvinium pamoate | CRC | Inhibition of Wnt/β-catenin signaling in HCT-116 and SW480 cells via interaction with CK1α and pygopus down-regulation | [55] |
| Pyrvinium pamoate | CRC | Reduction of intestinal adenoma formation in Apcmin/+ mice through the inhibition of the Wnt signaling | [56] |
| Pyrvinium pamoate | CRC | Synergistic anti-cancer effect on HCT-116 and SW620 cell lines in combination with 5-FU. Inhibition of the Wnt signaling in HCT-116 and SW620 cells as well as in human CRC explants. Impairment of liver metastases formation in nude mice injected with HCT-116 in the portal vein | [57] |
| Piperazine derivative(AK301) | CRC | Impairment of tubulin polymerization and induction of mitotic arrest in HT-29 and HCT116 cells. Increase of the susceptibility to TNF-α-mediated apoptosis | [58] |
| Piperazine derivative(BK1000S7) | HCC | Blockade of HepG2 and SK-Hep1 cell growth upon cyclin D1 down-regulation. Induction of apoptosis via caspase-3 and PARP-1 protein cleavage, impairment of AKT/ERK kinase phosphorylation and survivin expression | [59] |
| Drug | Cancer Type | Title of the Study | Phase | Identifier/Ref. |
|---|---|---|---|---|
| Mebendazole | CRC | Clinical study evaluating Mebendazole as adjuvant therapy in patients with colorectal cancer | 2 | NCT03925662 |
| Mebendazole | Gastrointestinal cancer | Cytotoxic activity against five CRC cell lines | Terminated (lack of effect) | NCT03628079 |
| Niclosamide | CRC | A Phase I study of Niclosamide in patients with resectable colon cancer | Terminated (low accrual) | NCT02687009 |
| Niclosamide | CRC | Phase II Trial to investigate the safety and efficacy of orally applied Niclosamide in patients with metachronous or synchronous metastases of colorectal cancer progressing after therapy | 2 | NCT02519582 |
| Niclosamide | FAP | The chemopreventive effect of Niclosamide in patients with familial adenomatous polyposis: double blinded randomized controlled study | 2 | NCT04296851 |
| Levamisole | CRC | Levamisole and Fluorouracil for adjuvant therapy of resected colon carcinoma | Terminated | [60] |
| Levamisole | CRC | Prospectively randomized trial of postoperative adjuvant chemotherapy in patients with high-risk colon cancer | Terminated (lack of effect) | [61] |
| Levamisole | CRC | Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04 | Terminated (lack of effect) | [62] |
| Levamisole | HCC | Multicenter, randomized, open, parallel, prospective, exploratory clinical study of Arginine Hydrochloride and levamisole in the treatment of advanced HCC | 3 | NCT03950518 |
| Levamisole | Intrahepatic CCA | The efficacy of Levamisole Hcl in advanced intrahepatic cholangiocarcinoma. A multicenter, open, randomized, prospective study | 3 | NCT03940378 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laudisi, F.; Marônek, M.; Di Grazia, A.; Monteleone, G.; Stolfi, C. Repositioning of Anthelmintic Drugs for the Treatment of Cancers of the Digestive System. Int. J. Mol. Sci. 2020, 21, 4957. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144957
Laudisi F, Marônek M, Di Grazia A, Monteleone G, Stolfi C. Repositioning of Anthelmintic Drugs for the Treatment of Cancers of the Digestive System. International Journal of Molecular Sciences. 2020; 21(14):4957. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144957
Chicago/Turabian StyleLaudisi, Federica, Martin Marônek, Antonio Di Grazia, Giovanni Monteleone, and Carmine Stolfi. 2020. "Repositioning of Anthelmintic Drugs for the Treatment of Cancers of the Digestive System" International Journal of Molecular Sciences 21, no. 14: 4957. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144957