Combination of NKT14m and Low Dose IL-12 Promotes Invariant Natural Killer T Cell IFN-γ Production and Tumor Control


Abstract
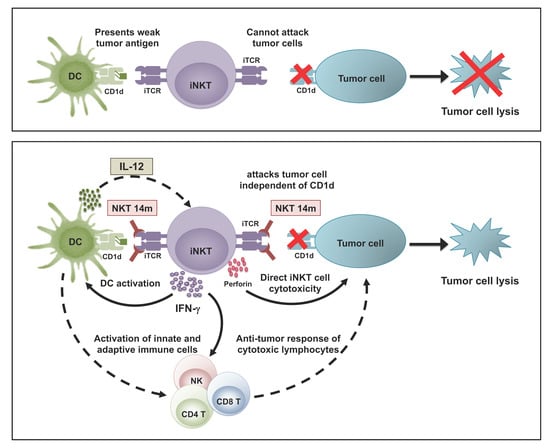
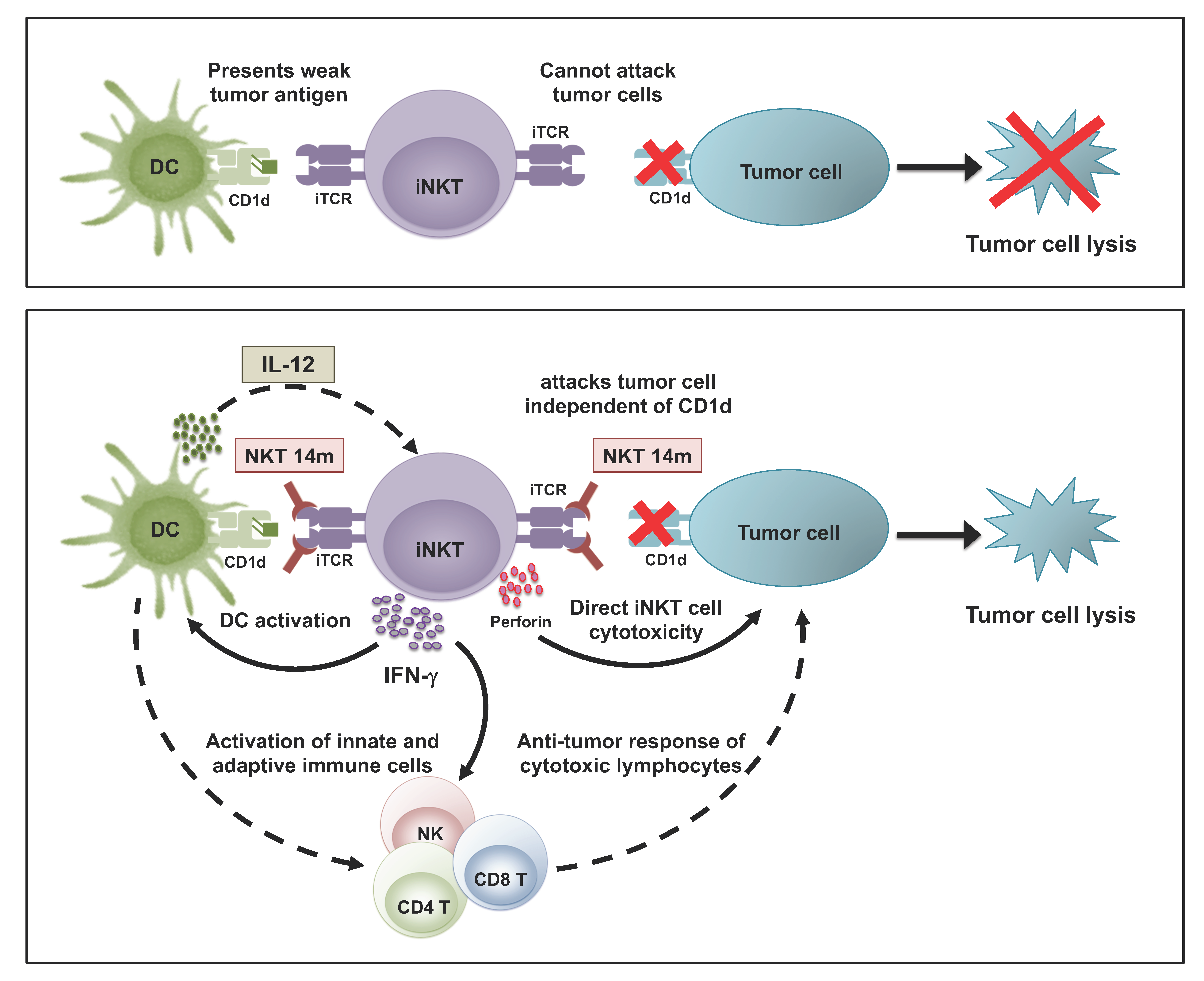
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Reagents
2.3. Antibodies and Flow Cytometry
2.4. Isolation of Purified Populations of iNKT Cell
2.5. In Vitro iNKT Cell Activation
2.6. In Vivo iNKT Cell Activation
2.7. In Vivo Tumor Model
2.8. Statistics
3. Results
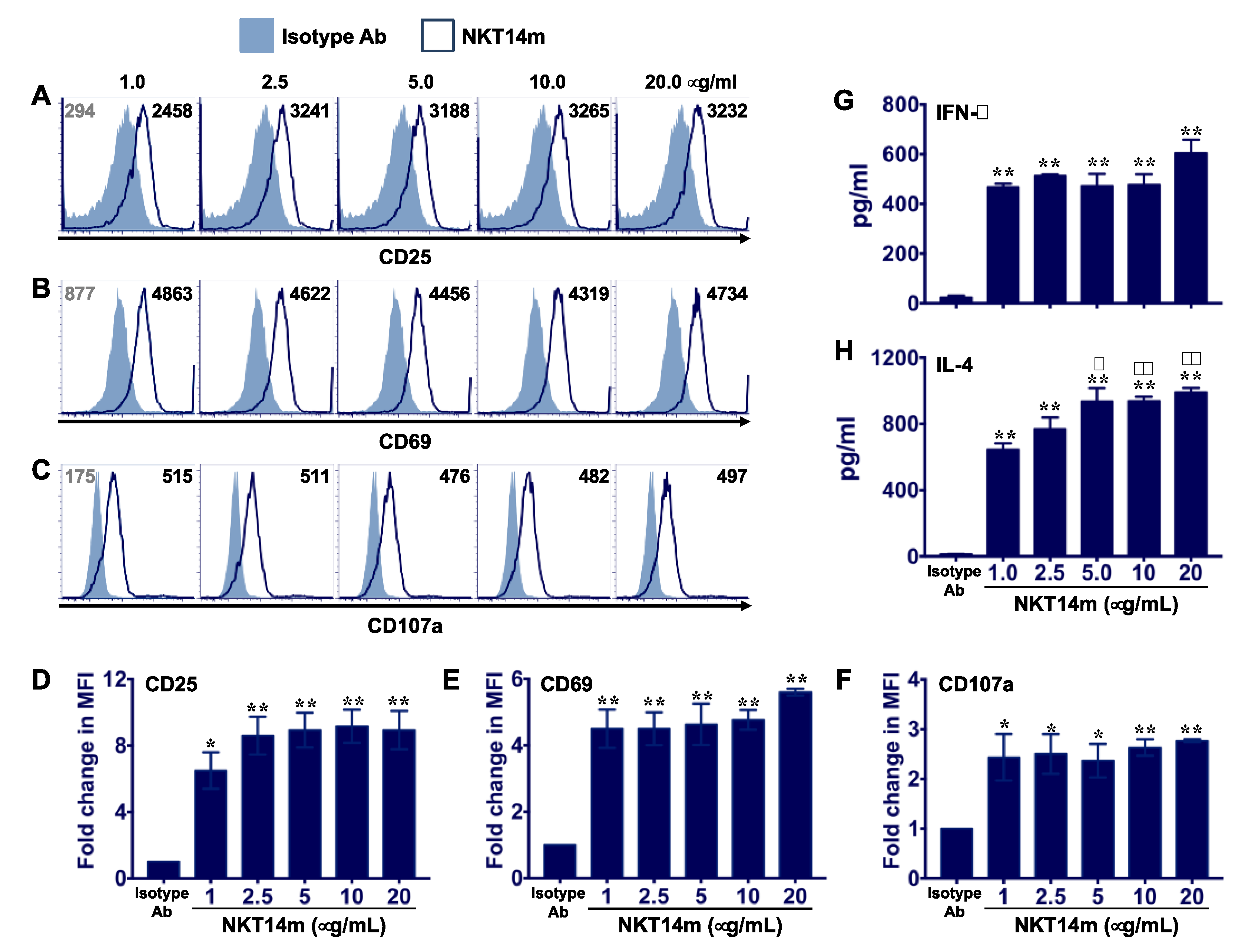
3.1. NKT14m Induces Murine iNKT Cell Activation, Degranulation and Cytokine Production In Vitro
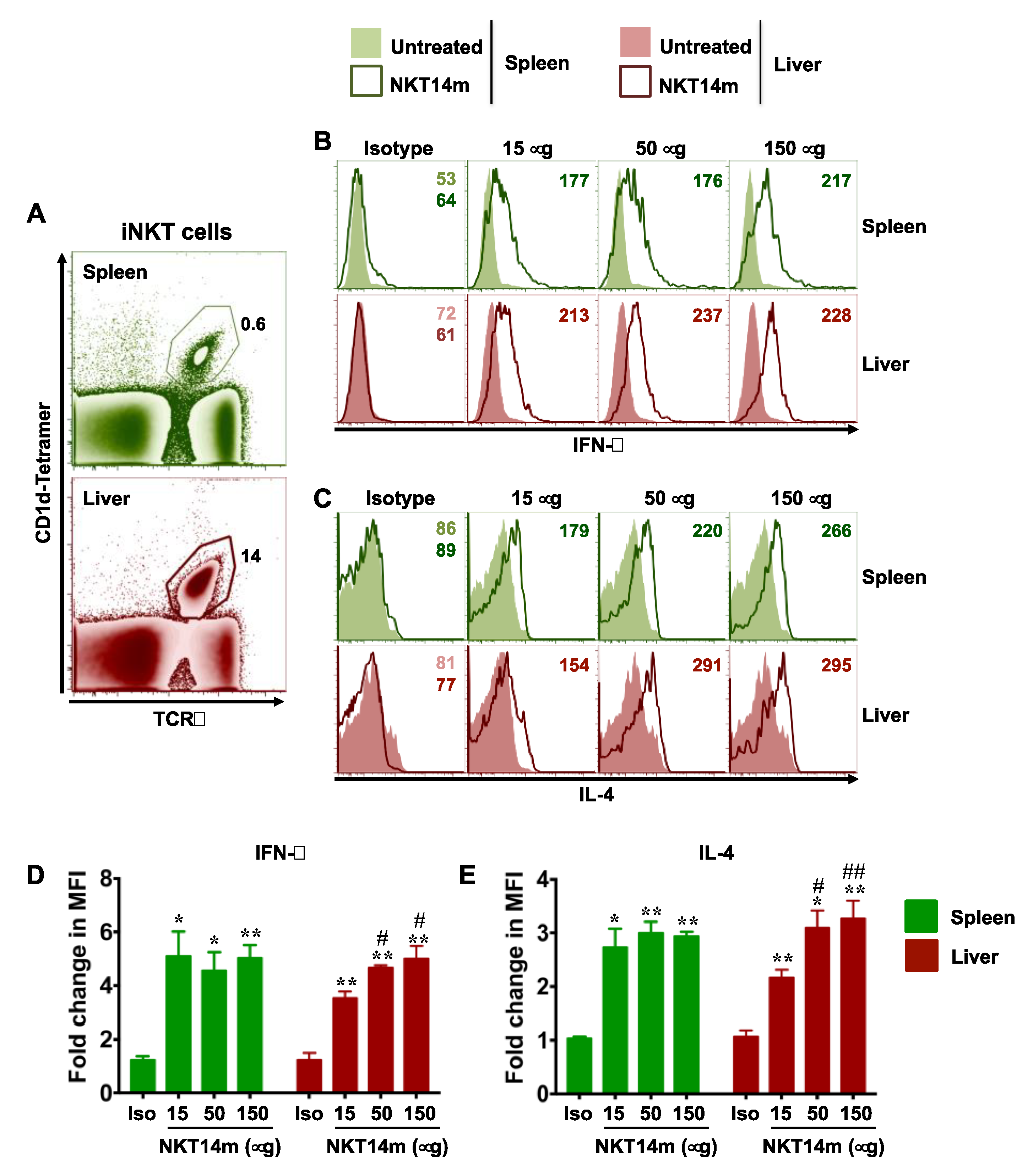
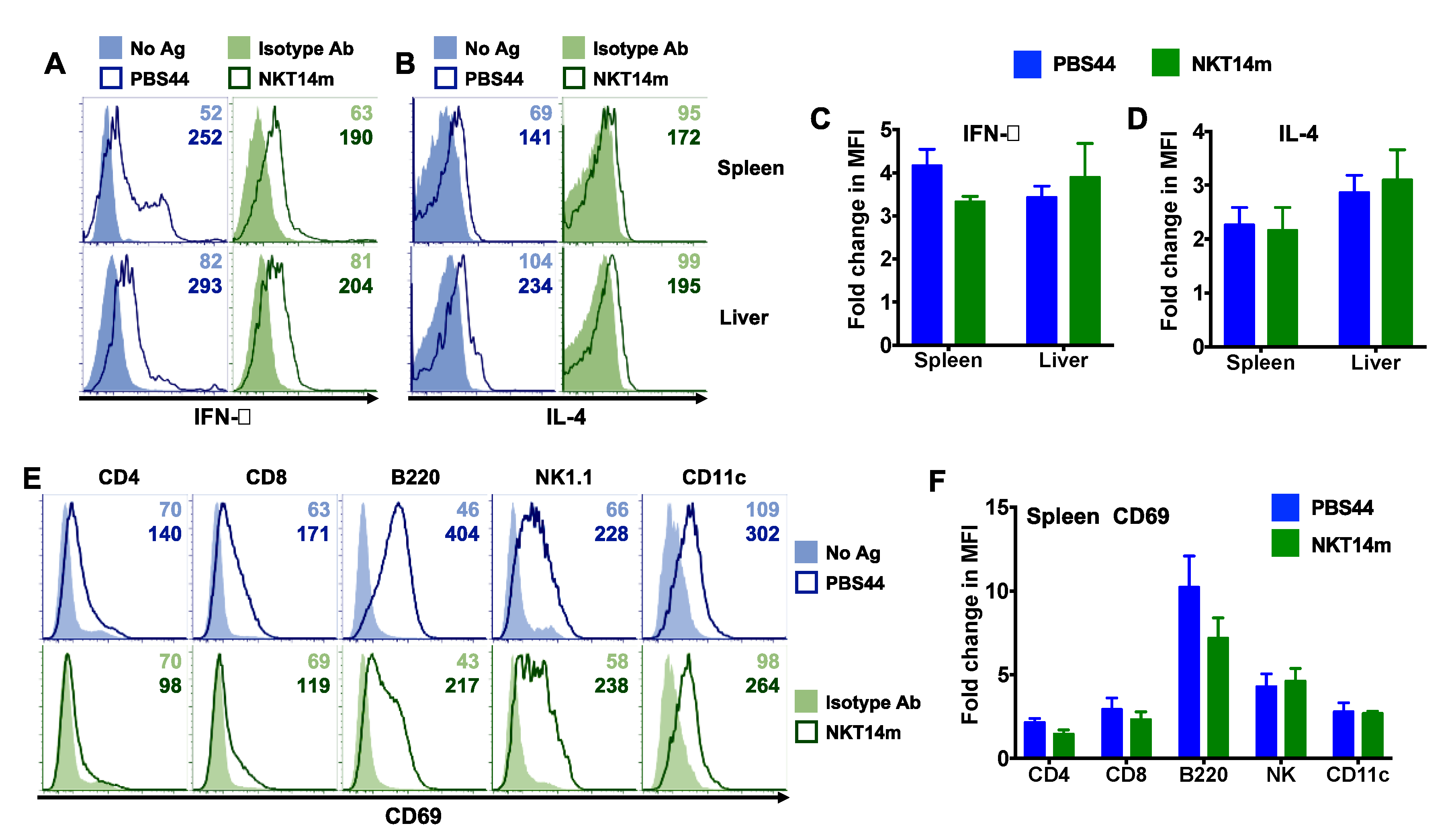
3.2. Invariant NKT Cells Readily Produce Cytokines in Response to NKT14m In Vivo
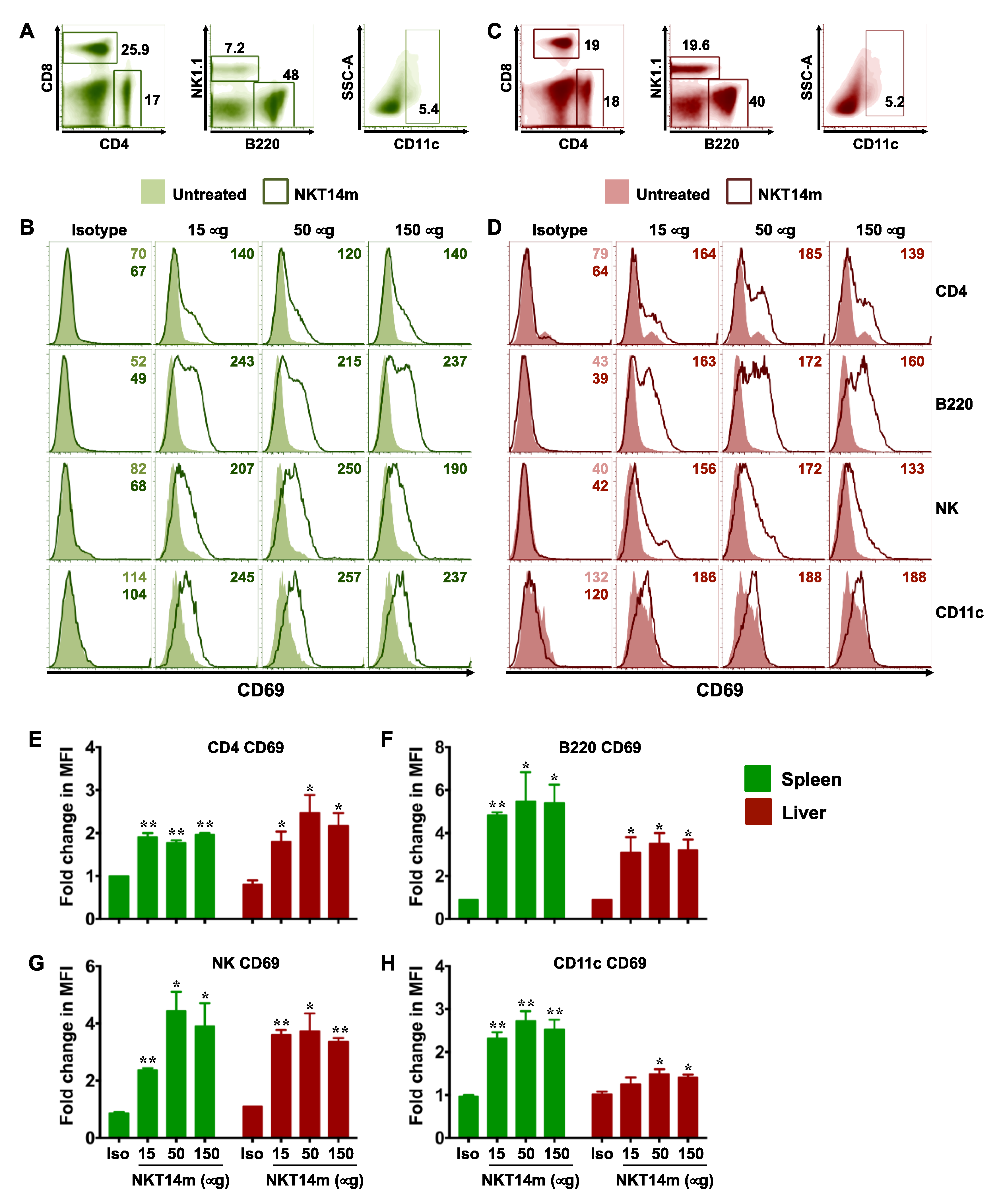
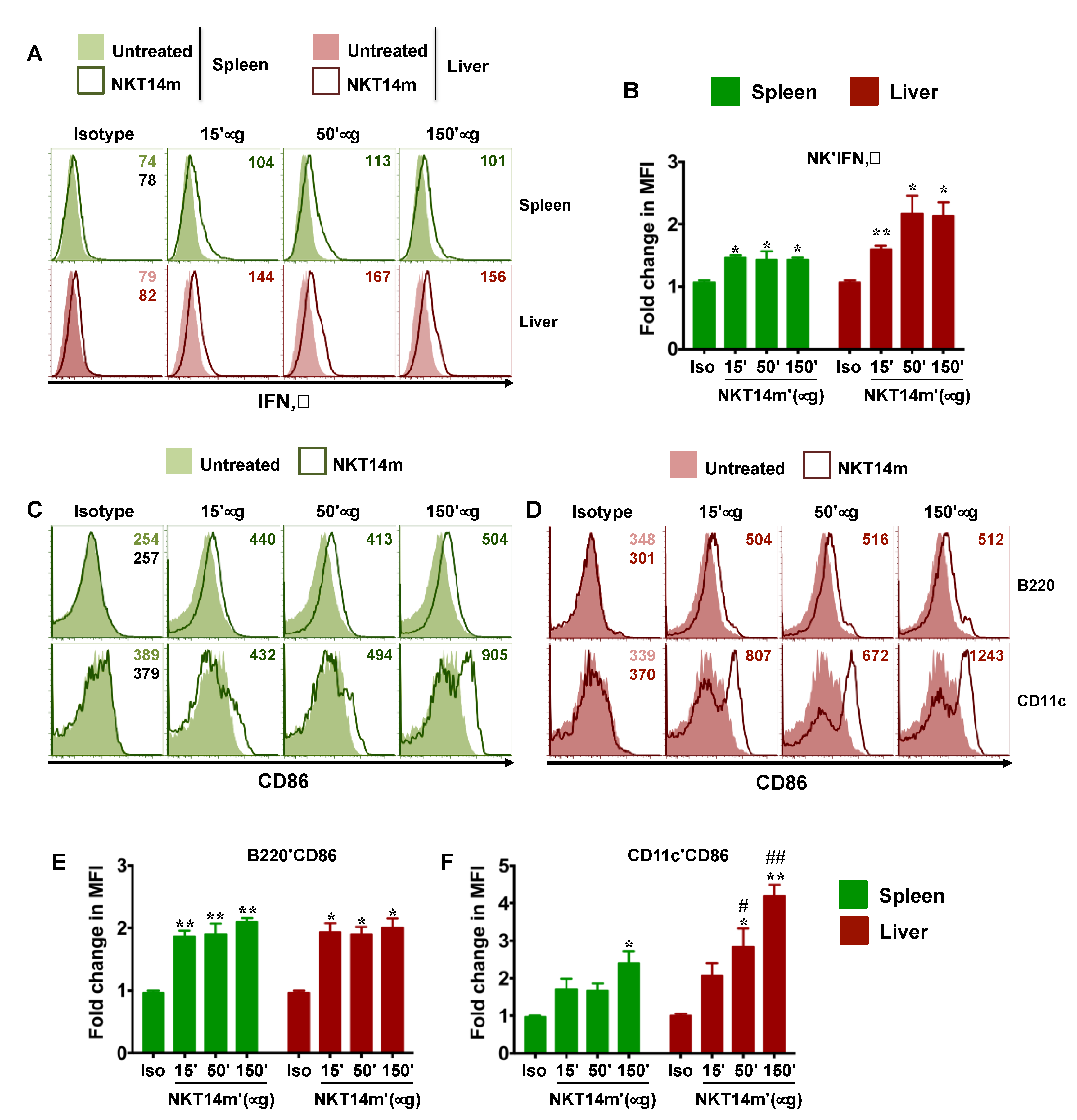
3.3. NKT14m Induces Murine iNKT Cell Activation and Immunomodulatory Functions In Vivo
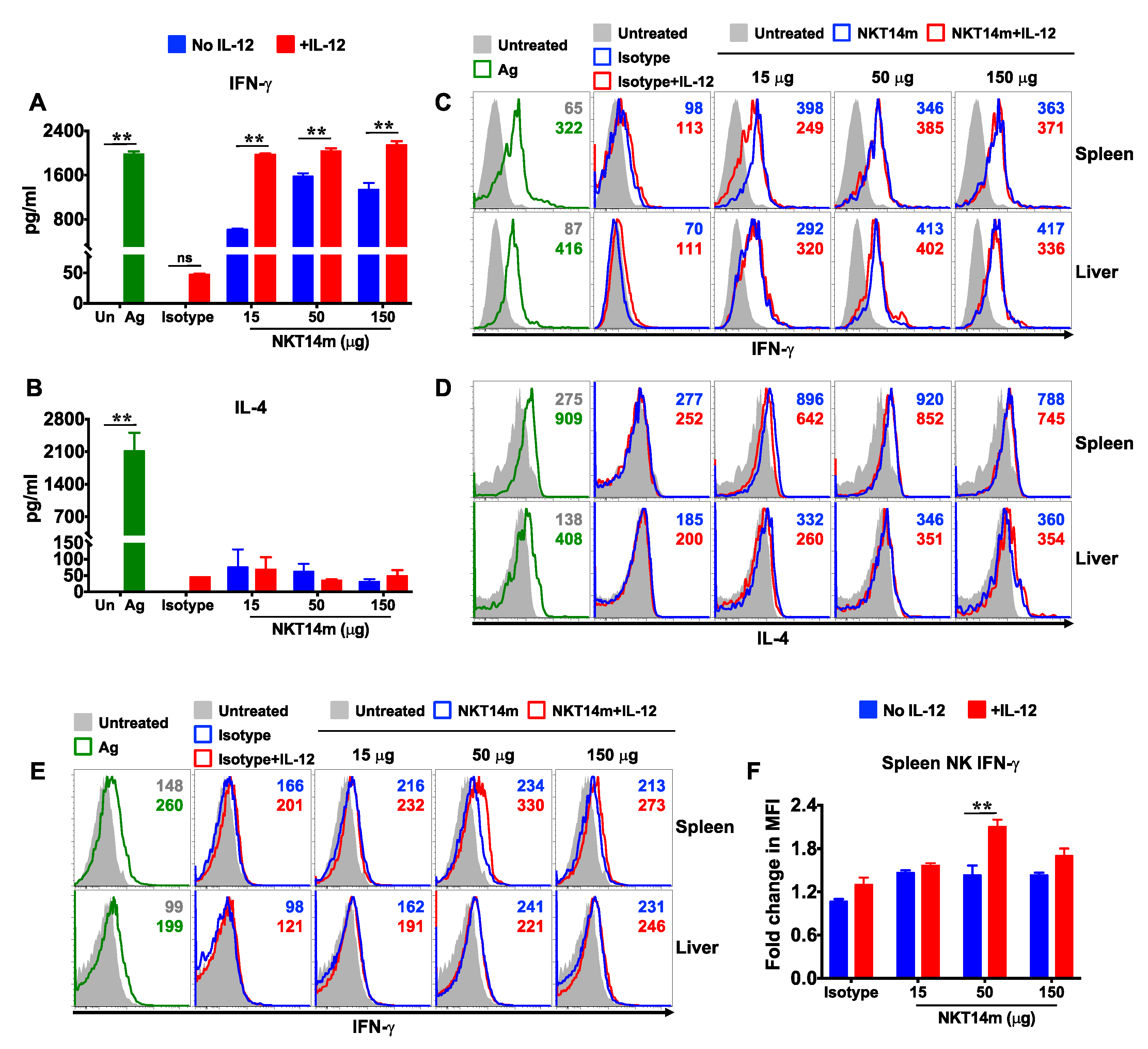
3.4. IL-12 Augments NKT14m-Induced Cytokine Production In Vivo
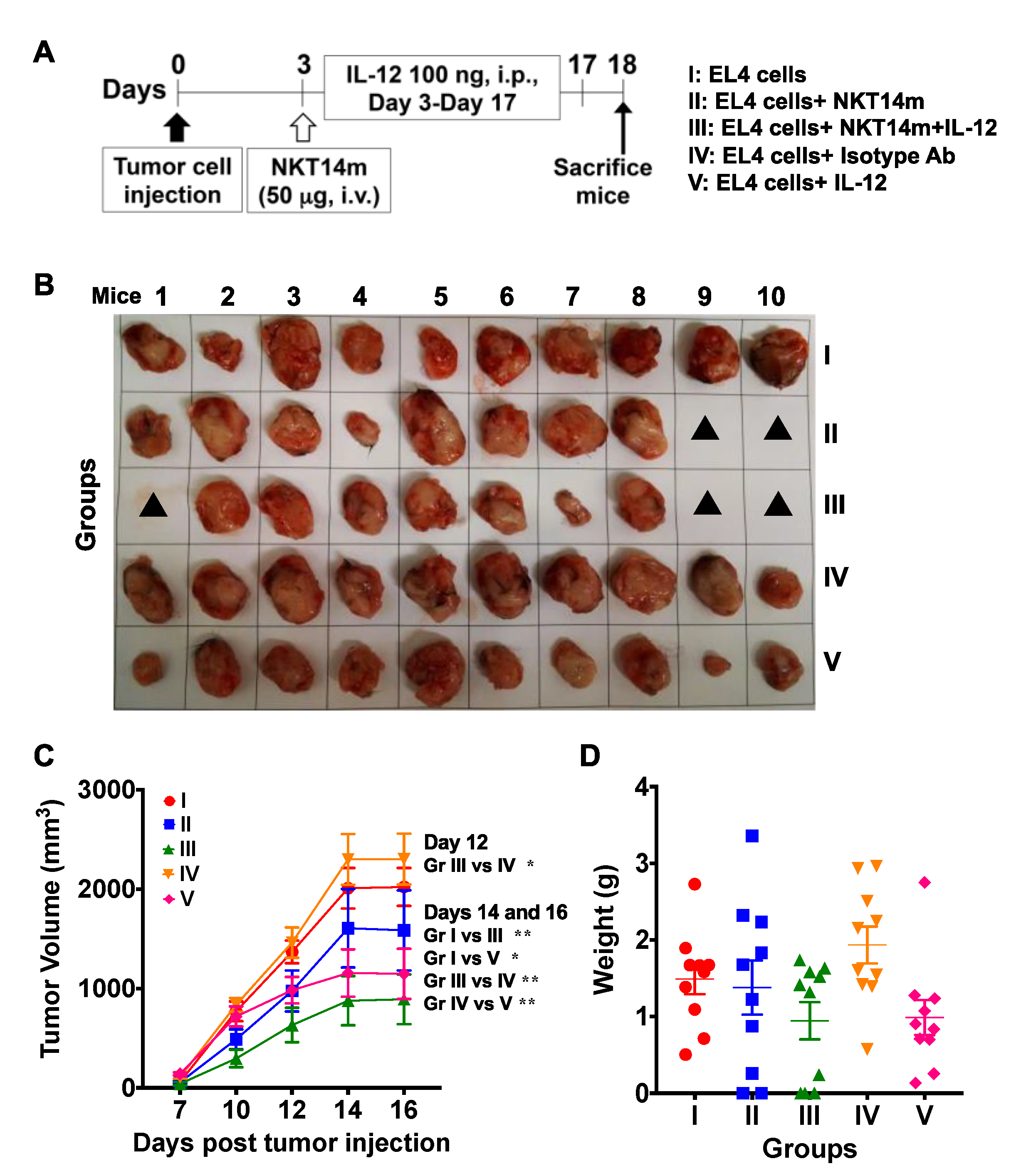
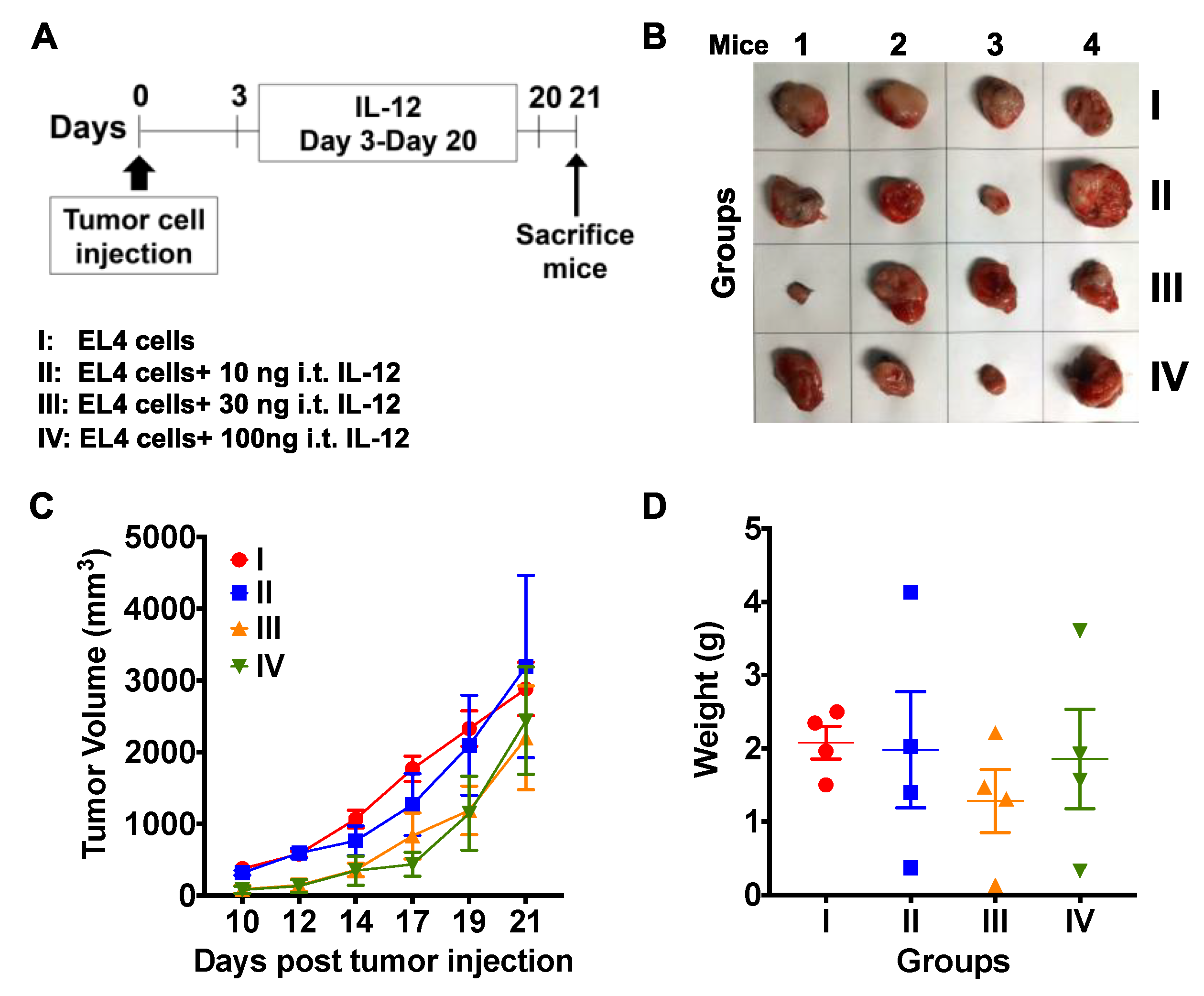
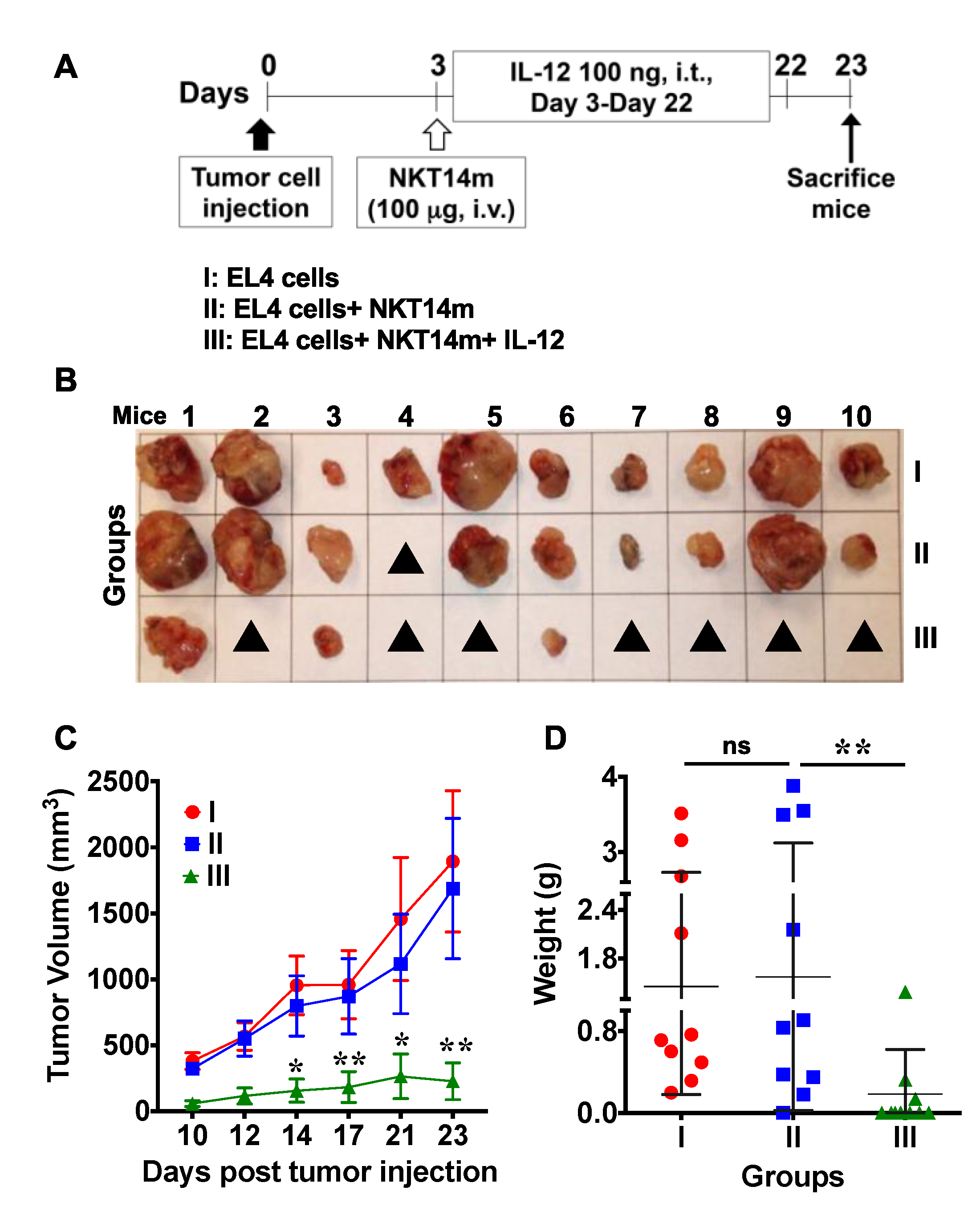
3.5. Treatment with NKT14m and IL-12 Controls Tumor Growth In Vivo.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Godfrey, D.I.; Stankovic, S.; Baxter, A.G. Raising the NKT cell family. Nat. Immunol. 2010, 11, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, D.I.; Berzins, S.P. Control points in NKT-cell development. Nat. Rev. Immunol. 2007, 7, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Motoki, K.; Ueno, H.; Nakagawa, R.; Sato, H.; Kondo, E.; et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. [Google Scholar] [CrossRef]
- Stetson, D.B.; Mohrs, M.; Reinhardt, R.L.; Baron, J.L.; Wang, Z.E.; Gapin, L.; Kronenberg, M.; Locksley, R.M. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J. Exp. Med. 2003, 198, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Hammond, K.J.; Pelikan, S.B.; Crowe, N.Y.; Randle-Barrett, E.; Nakayama, T.; Taniguchi, M.; Smyth, M.J.; van Driel, I.R.; Scollay, R.; Baxter, A.G.; et al. NKT cells are phenotypically and functionally diverse. Eur. J. Immunol. 1999, 29, 3768–3781. [Google Scholar] [CrossRef]
- Swann, J.B.; Uldrich, A.P.; van Dommelen, S.; Sharkey, J.; Murray, W.K.; Godfrey, D.I.; Smyth, M.J. Type I natural killer T cells suppress tumors caused by p53 loss in mice. Blood 2009, 113, 6382–6385. [Google Scholar] [CrossRef] [Green Version]
- Bellone, M.; Ceccon, M.; Grioni, M.; Jachetti, E.; Calcinotto, A.; Napolitano, A.; Freschi, M.; Casorati, G.; Dellabona, P. iNKT cells control mouse spontaneous carcinoma independently of tumor-specific cytotoxic T cells. PLoS ONE 2010, 5, e8646. [Google Scholar] [CrossRef] [Green Version]
- Crowe, N.Y.; Smyth, M.J.; Godfrey, D.I. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J. Exp. Med. 2002, 196, 119–127. [Google Scholar] [CrossRef]
- Smyth, M.J.; Crowe, N.Y.; Godfrey, D.I. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int. Immunol. 2001, 13, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Renukaradhya, G.J.; Khan, M.A.; Vieira, M.; Du, W.; Gervay-Hague, J.; Brutkiewicz, R.R. Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood 2008, 111, 5637–5645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuling, H.; Ruijing, X.; Li, L.; Xiang, J.; Rui, Z.; Yujuan, W.; Lijun, Z.; Chunxian, D.; Xinti, T.; Wei, X.; et al. EBV-induced human CD8+ NKT cells suppress tumorigenesis by EBV-associated malignancies. Cancer Res. 2009, 69, 7935–7944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhodapkar, M.V.; Geller, M.D.; Chang, D.H.; Shimizu, K.; Fujii, S.; Dhodapkar, K.M.; Krasovsky, J. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J. Exp. Med. 2003, 197, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Metelitsa, L.S.; Wu, H.W.; Wang, H.; Yang, Y.; Warsi, Z.; Asgharzadeh, S.; Groshen, S.; Wilson, S.B.; Seeger, R.C. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J. Exp. Med. 2004, 199, 1213–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, T.; Onodera, H.; Tsuruyama, T.; Mori, A.; Nagayama, S.; Hiai, H.; Imamura, M. Increased intratumor Valpha24-positive natural killer T cells: A prognostic factor for primary colorectal carcinomas. Clin. Cancer Res. 2005, 11, 7322–7327. [Google Scholar] [CrossRef] [Green Version]
- Terabe, M.; Berzofsky, J.A. The role of NKT cells in tumor immunity. Adv. Cancer Res. 2008, 101, 277–348. [Google Scholar]
- Swann, J.; Crowe, N.Y.; Hayakawa, Y.; Godfrey, D.I.; Smyth, M.J. Regulation of antitumour immunity by CD1d-restricted NKT cells. Immunol. Cell Biol. 2004, 82, 323–331. [Google Scholar] [CrossRef]
- Fujii, S.; Shimizu, K.; Smith, C.; Bonifaz, L.; Steinman, R.M. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J. Exp. Med. 2003, 198, 267–279. [Google Scholar] [CrossRef]
- Dao, T.; Mehal, W.Z.; Crispe, I.N. IL-18 augments perforin-dependent cytotoxicity of liver NK-T cells. J. Immunol. 1998, 161, 2217–2222. [Google Scholar]
- Matsuda, J.L.; Zhang, Q.; Ndonye, R.; Richardson, S.K.; Howell, A.R.; Gapin, L. T-bet concomitantly controls migration, survival, and effector functions during the development of Valpha14i NKT cells. Blood 2006, 107, 2797–2805. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, T.; Takeda, K.; Mendiratta, S.K.; Kawamura, H.; van Kaer, L.; Yagita, H.; Abo, T.; Okumura, K. Critical role of NK1+ T cells in IL-12-induced immune responses in vivo. J. Immunol. 1998, 160, 16–19. [Google Scholar] [PubMed]
- Nieda, M.; Nicol, A.; Koezuka, Y.; Kikuchi, A.; Lapteva, N.; Tanaka, Y.; Tokunaga, K.; Suzuki, K.; Kayagaki, N.; Yagita, H.; et al. TRAIL expression by activated human CD4+ V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood 2001, 97, 2067–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingender, G.; Krebs, P.; Beutler, B.; Kronenberg, M. Antigen-specific cytotoxicity by invariant NKT cells in vivo is CD95/CD178-dependent and is correlated with antigenic potency. J. Immunol. 2010, 185, 2721–2729. [Google Scholar] [CrossRef] [Green Version]
- Bassiri, H.; Das, R.; Guan, P.; Barrett, D.M.; Brennan, P.J.; Banerjee, P.P.; Wiener, S.J.; Orange, J.S.; Brenner, M.B.; Grupp, S.A.; et al. iNKT cell cytotoxic responses control T-lymphoma growth in vitro and in vivo. Cancer Immunol. Res. 2014, 2, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Sato, H.; Kondo, E.; Harada, M.; Koseki, H.; Nakayama, T.; et al. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Valpha14 NKT cells. Proc. Natl. Acad. Sci. USA 1998, 95, 5690–5693. [Google Scholar] [CrossRef] [Green Version]
- Kawano, T.; Nakayama, T.; Kamada, N.; Kaneko, Y.; Harada, M.; Ogura, N.; Akutsu, Y.; Motohashi, S.; Iizasa, T.; Endo, H.; et al. Antitumor cytotoxicity mediated by ligand-activated human V alpha24 NKT cells. Cancer Res. 1999, 59, 5102–5105. [Google Scholar]
- Das, R.; Bassiri, H.; Guan, P.; Wiener, S.; Banerjee, P.P.; Zhong, M.C.; Veillette, A.; Orange, J.S.; Nichols, K.E. The adaptor molecule SAP plays essential roles during invariant NKT cell cytotoxicity and lytic synapse formation. Blood 2013, 121, 3386–3395. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.P.; Guan, P.; Bahal, D.; Hashem, T.; Scheuplein, F.; Schaub, R.; Nichols, K.E.; Das, R. Cancer Immunotherapeutic Potential of NKTT320, a Novel, Invariant, Natural Killer T Cell-Activating, Humanized Monoclonal Antibody. Int. J. Mol. Sci. 2020, 21, 4317. [Google Scholar] [CrossRef]
- Scheuplein, F.; Lamont, D.J.; Poynter, M.E.; Boyson, J.E.; Serreze, D.; Lundblad, L.K.; Mashal, R.; Schaub, R. Mouse Invariant Monoclonal Antibody NKT14: A Novel Tool to Manipulate iNKT Cell Function In Vivo. PLoS ONE 2015, 10, e0140729. [Google Scholar] [CrossRef]
- Escriba-Garcia, L.; Alvarez-Fernandez, C.; Caballero, A.C.; Schaub, R.; Sierra, J.; Briones, J. The novel agonistic iNKT-cell antibody NKT14m induces a therapeutic antitumor response against B-cell lymphoma. Oncoimmunology 2019, 8, e1546543. [Google Scholar] [CrossRef] [Green Version]
- Van Kaer, L.; Parekh, V.V.; Wu, L. Invariant natural killer T cells: Bridging innate and adaptive immunity. Cell Tissue Res. 2011, 343, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.P.; Trinchieri, G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 155–168. [Google Scholar] [CrossRef]
- Cui, J.; Shin, T.; Kawano, T.; Sato, H.; Kondo, E.; Toura, I.; Kaneko, Y.; Koseki, H.; Kanno, M.; Taniguchi, M. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science 1997, 278, 1623–1626. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Gritzapis, A.D.; Papamichail, M. In vivo antitumor activity of NKT cells activated by the combination of IL-12 and IL-18. J. Immunol. 2003, 171, 2953–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Kerkar, S.P.; Yu, Z.; Zheng, Z.; Yang, S.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther. 2011, 19, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, P.; Bassiri, H.; Patel, N.P.; Nichols, K.E.; Das, R. Invariant natural killer T cells in hematopoietic stem cell transplantation: Killer choice for natural suppression. Bone Marrow Transplant. 2016, 51, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Ngai, H.; Tian, G.; Courtney, A.N.; Ravari, S.B.; Guo, L.; Liu, B.; Jin, J.; Shen, E.T.; Di Pierro, E.J.; Metelitsa, L.S. IL-21 Selectively Protects CD62L(+) NKT Cells and Enhances Their Effector Functions for Adoptive Immunotherapy. J. Immunol. 2018, 201, 2141–2153. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Investig. 2016, 126, 2341–2355. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Berg, J.V.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Mizoguchi, I.; Morishima, N.; Chiba, Y.; Mizuguchi, J.; Yoshimoto, T. Regulation of antitumor immune responses by the IL-12 family cytokines, IL-12, IL-23, and IL-27. Clin. Dev. Immunol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, H.; Iwakabe, K.; Yahata, T.; Nishimura, S.; Ohta, A.; Ohmi, Y.; Sato, M.; Takeda, K.; Okumura, K.; van Kaer, L.; et al. The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J. Exp. Med. 1999, 189, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberl, G.; MacDonald, H.R. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur. J. Immunol. 2000, 30, 985–992. [Google Scholar] [CrossRef]
- Chiodoni, C.; Stoppacciaro, A.; Sangaletti, S.; Gri, G.; Cappetti, B.; Koezuka, Y.; Colombo, M.P. Different requirements for alpha-galactosylceramide and recombinant IL-12 antitumor activity in the treatment of C-26 colon carcinoma hepatic metastases. Eur. J. Immunol. 2001, 31, 3101–3110. [Google Scholar] [CrossRef]
- Nagai, H.; Hara, I.; Horikawa, T.; Fujii, M.; Kurimoto, M.; Kamidono, S.; Ichihashi, M. Antitumor effects on mouse melanoma elicited by local secretion of interleukin-12 and their enhancement by treatment with interleukin-18. Cancer Investig. 2000, 18, 206–213. [Google Scholar] [CrossRef]
- Osaki, T.; Peron, J.M.; Cai, Q.; Okamura, H.; Robbins, P.D.; Kurimoto, M.; Lotze, M.T.; Tahara, H. IFN-gamma-inducing factor/IL-18 administration mediates IFN-gamma- and IL-12-independent antitumor effects. J. Immunol. 1998, 160, 1742–1749. [Google Scholar]
- Micallef, M.J.; Yoshida, K.; Kawai, S.; Hanaya, T.; Kohno, K.; Arai, S.; Tanimoto, T.; Torigoe, K.; Fujii, M.; Ikeda, M.; et al. In vivo antitumor effects of murine interferon-gamma-inducing factor/interleukin-18 in mice bearing syngeneic Meth A sarcoma malignant ascites. Cancer Immunol. Immunother. 1997, 43, 361–367. [Google Scholar] [CrossRef]
- Takeda, K.; Tsutsui, H.; Yoshimoto, T.; Adachi, O.; Yoshida, N.; Kishimoto, T.; Okamura, H.; Nakanishi, K.; Akira, S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 1998, 8, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Akamatsu, S.; Arai, N.; Hanaya, T.; Arai, S.; Tanimoto, T.; Fujii, M.; Kohno, K.; Micallef, M.J.; Ikeda, M.; Kurimoto, M. Antitumor activity of interleukin-18 against the murine T-cell leukemia/lymphoma EL-4 in syngeneic mice. J. Immunother. 2002, 25, S28–S34. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, H.; Ju, D.W.; He, L.; Pan, J.P.; Xia, D.J.; Zhang, L.H.; Cao, X. Intratumoral IL-18 gene transfer improves therapeutic efficacy of antibody-targeted superantigen in established murine melanoma. Gene Ther. 2001, 8, 542–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, D.W.; Yang, Y.; Tao, Q.; Song, W.G.; He, L.; Chen, G.; Gu, S.; Ting, C.C.; Cao, X. Interleukin-18 gene transfer increases antitumor effects of suicide gene therapy through efficient induction of antitumor immunity. Gene Ther. 2000, 7, 1672–1679. [Google Scholar] [CrossRef] [Green Version]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef]
- Lauwerys, B.R.; Garot, N.; Renauld, J.C.; Houssiau, F.A. Cytokine production and killer activity of NK/T-NK cells derived with IL-2, IL-15, or the combination of IL-12 and IL-18. J. Immunol. 2000, 165, 1847–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite-De-Moraes, M.C.; Hameg, A.; Arnould, A.; Machavoine, F.; Koezuka, Y.; Schneider, E.; Herbelin, A.; Dy, M. A distinct IL-18-induced pathway to fully activate NK T lymphocytes independently from TCR engagement. J. Immunol. 1999, 163, 5871–5876. [Google Scholar] [PubMed]
- Hyodo, Y.; Matsui, K.; Hayashi, N.; Tsutsui, H.; Kashiwamura, S.; Yamauchi, H.; Hiroishi, K.; Takeda, K.; Tagawa, Y.; Iwakura, Y.; et al. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 1999, 162, 1662–1668. [Google Scholar] [PubMed]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Guan, P.; Wiener, S.J.; Patel, N.P.; Gohl, T.G.; Evans, E.; Zauderer, M.; Nichols, K.E. Enhancing the antitumor functions of invariant natural killer T cells using a soluble CD1d-CD19 fusion protein. Blood Adv. 2019, 3, 813–824. [Google Scholar] [CrossRef]
- Corgnac, S.; Perret, R.; Derre, L.; Zhang, L.; Stirnemann, K.; Zauderer, M.; Speiser, D.E.; Mach, J.P.; Romero, P.; Donda, A. CD1d-antibody fusion proteins target iNKT cells to the tumor and trigger long-term therapeutic responses. Cancer Immunol. Immunother. 2013, 62, 747–760. [Google Scholar] [CrossRef] [Green Version]
- Stirnemann, K.; Romero, J.F.; Baldi, L.; Robert, B.; Cesson, V.; Besra, G.S.; Zauderer, M.; Wurm, F.; Corradin, G.; Mach, J.P.; et al. Sustained activation and tumor targeting of NKT cells using a CD1d-anti-HER2-scFv fusion protein induce antitumor effects in mice. J. Clin. Investig. 2008, 118, 994–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, J.J.; Majerus, E.; Ataga, K.I.; Vichinsky, E.P.; Schaub, R.; Mashal, R.; Nathan, D.G. NNKTT120, an anti-iNKT cell monoclonal antibody, produces rapid and sustained iNKT cell depletion in adults with sickle cell disease. PLoS ONE 2017, 12, e0171067. [Google Scholar] [CrossRef] [PubMed]
- Scheuplein, F.; Thariath, A.; Macdonald, S.; Truneh, A.; Mashal, R.; Schaub, R. A humanized monoclonal antibody specific for invariant Natural Killer T (iNKT) cells for in vivo depletion. PLoS ONE 2013, 8, e76692. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, P.; Schaub, R.; Nichols, K.E.; Das, R. Combination of NKT14m and Low Dose IL-12 Promotes Invariant Natural Killer T Cell IFN-γ Production and Tumor Control. Int. J. Mol. Sci. 2020, 21, 5085. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145085
Guan P, Schaub R, Nichols KE, Das R. Combination of NKT14m and Low Dose IL-12 Promotes Invariant Natural Killer T Cell IFN-γ Production and Tumor Control. International Journal of Molecular Sciences. 2020; 21(14):5085. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145085
Chicago/Turabian StyleGuan, Peng, Robert Schaub, Kim E. Nichols, and Rupali Das. 2020. "Combination of NKT14m and Low Dose IL-12 Promotes Invariant Natural Killer T Cell IFN-γ Production and Tumor Control" International Journal of Molecular Sciences 21, no. 14: 5085. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145085