ADAM-Mediated Signalling Pathways in Gastrointestinal Cancer Formation



Institute of Biochemistry, Christian-Albrechts-University, 24118 Kiel, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(14), 5133; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145133
Submission received: 27 June 2020
/
Revised: 10 July 2020
/
Accepted: 15 July 2020
/
Published: 20 July 2020
(This article belongs to the Special Issue Biological Interfaces in Gastrointestinal Cancer)
Abstract
:Tumour growth is not solely driven by tumour cell-intrinsic mechanisms, but also depends on paracrine signals provided by the tumour micro-environment. These signals comprise cytokines and growth factors that are synthesized as trans-membrane proteins and need to be liberated by limited proteolysis also termed ectodomain shedding. Members of the family of A disintegrin and metalloproteases (ADAM) are major mediators of ectodomain shedding and therefore initiators of paracrine signal transduction. In this review, we summarize the current knowledge on how ADAM proteases on tumour cells but also on cells of the tumour micro-environment contribute to the formation of gastrointestinal tumours, and discuss how these processes can be exploited pharmacologically.
1. Introduction
Gastrointestinal organs are composed of highly complex tissue and most of these tissues are constantly replenished by the proliferation and differentiation of multi-potent tissue stem cells. These processes are regulated by paracrine signals provided by cells of the stem cell niche. Moreover, signals emitted from inflammatory cells recruited to damaged or infected tissue contribute to the mobilisation of tissue stem cells, regeneration and remodelling of the damaged tissue. Several paracrine signal proteins are synthesized as membrane-bound proteins and need to be proteolytically processed to give rise to soluble factors that then can act in a paracrine or even an endocrine fashion. Additionally, receptor molecules and proteins of the extracellular matrix are subject to proteolytic remodelling. Members of the A disintegrin and metalloprotease (ADAM) family are involved in all these processes. Aberrant activation of paracrine signal transduction is key to the development of gastrointestinal tumours and it is therefore not surprising that ADAM proteases play a decisive role in gastrointestinal tumorigenesis. Here, we give an overview of ADAM protease biology and the current knowledge regarding their tumour-promoting role in gastrointestinal malignancies. We further discuss how ADAM proteases can be therapeutically targeted for the treatment of gastrointestinal cancers.
2. ADAM Proteases
2.1. Overview
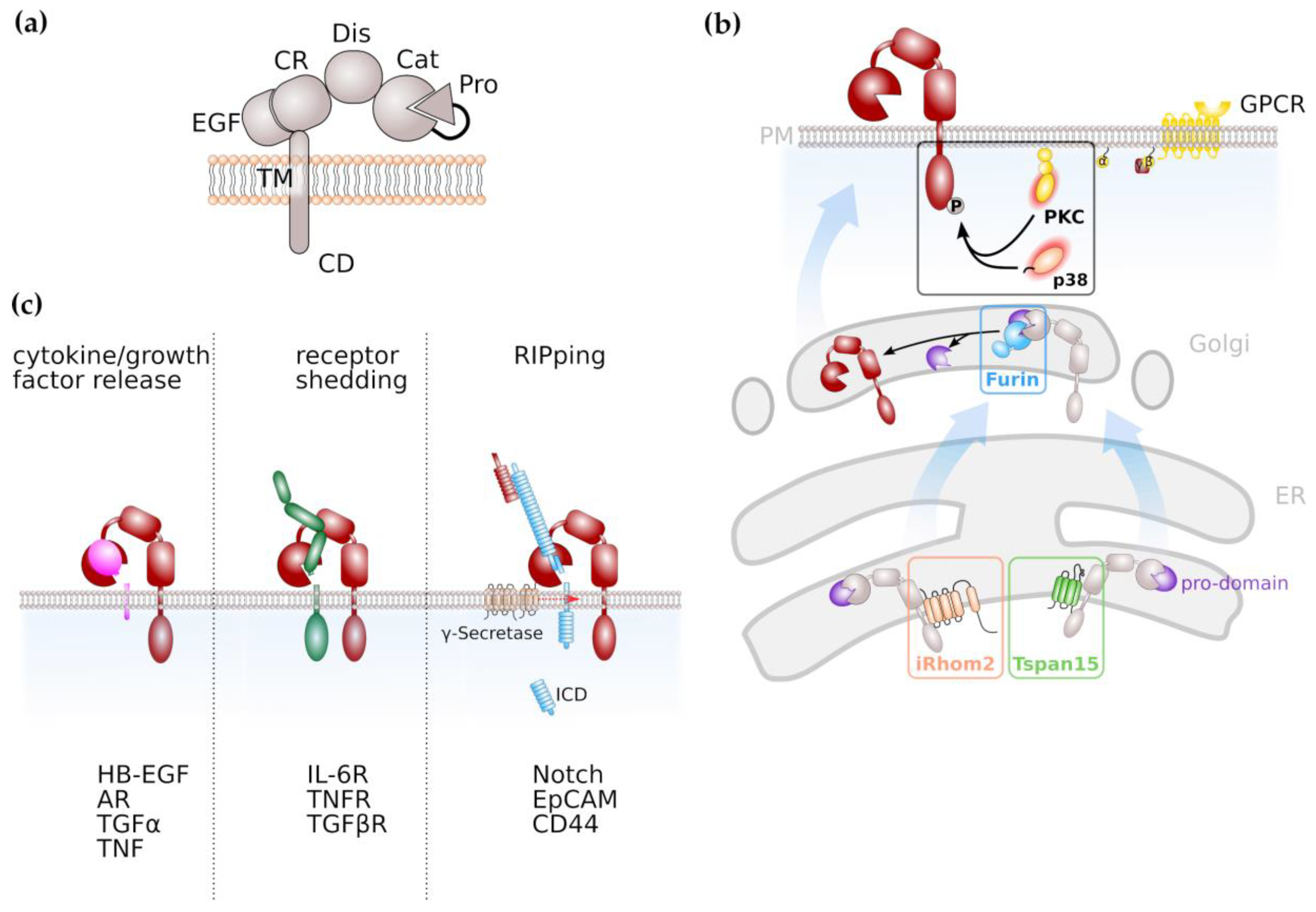
Limited ectodomain proteolysis is a regulatory mechanism mediated by proteases that converts membrane-bound proteins irreversibly into their soluble isoforms. The family of A disintegrin and metalloproteases (ADAMs) consists of 21 members (Table 1) with 13 of them being proteolytically active [1]. ADAM proteases belong to the superfamily of zinc-proteases that is characterised by the presence of an invariant HEXXHXXGXXH zinc-binding motif with the catalytic domain [2,3]. ADAM proteases are expressed as inactive zymogens, consisting of an N-terminal signal sequence, a pro-domain, a catalytic metalloproteinase domain, a disintegrin domain and a cysteine-rich, EGF-like domain followed by a transmembrane part and cytoplasmic tail (Figure 1a). In place of the EGF-like, cysteine-rich domain, ADAM17 and its close relative ADAM10 are characterized by a membrane-proximal domain in their extracellular region, which has also been reported to be involved in substrate recognition [4,5]. The prodomain has chaperone-like functions, inhibits enzymatic activity and is removed during maturation by the intracellular proprotein-convertase furin (Figure 1b) with additional proprotein convertase cleavage sites recently identified [6,7,8].
2.2. Regulation of ADAM Protease Activity
Proteolytic activity of ADAM proteases can either be constitutive or activated by signal transduction pathways. Regulation of ADAM protease activity is best described for the family members ADAM10 and ADAM17. Members of the membrane multi-pass families inactive rhomboids (iRhoms) [9,10] and tetraspanins (Tspans) [11,12] are involved in maturation of ADAMs throughout the secretory pathway (Figure 1b).
The rhomboid family comprises several transmembrane proteins that are evolutionarily conserved. The inactive rhomboid members lack the amino-acid residues that are indispensable for catalytic activity.
ADAM17 needs to be located at the plasma membrane in order to exert its proteolytic activity. An indispensable factor for ADAM17 activity is the inactive rhomboid protease (iRhom) 2, encoded by the rhomboid family member (RHBDF) 2 gene. iRhom2 is localized in the ER and shuttles ADAM17 from the ER to the Golgi apparatus [9]. Importantly, it is specific for ADAM17, since the transport of ADAM10 to the cell membrane is not affected by the absence of iRhom2 [10]. Whereas iRhom2 is proposed to be a myeloid-specific regulator of ADAM17 activation, its closest homologue iRhom1 is more broadly expressed [13]. The cleavage of ADAM17 substrates is seriously impaired in embryonic fibroblasts derived from iRhom2-deficient mice. However, not all ADAM17 substrates are affected by an iRhom2 deficiency, indicating a role of iRhom2 in ADAM17 substrate selection and discrimination [14].
As the name indicates, tetraspanins span the membrane four times, resulting in the formation of a short and a large extracellular loop. Tspans have the capacity to form large interacting networks, the so-called tetraspanin-enrichted microdomains (TEMs) which regulate signal transduction pathways similar to lipid rafts [15,16]. ADAM10 was previously identified to interact with members of the TspanC8 family within the ER which promoted maturation of ADAM10 to the plasma membrane [11,12]. Therefore, Tspans might emerge in general as regulators and facilitators of ADAM protease activity.
The intracellular region of ADAM17 contains numerous phosphorylation sites with phosphorylation of threonine 735 leading to rapid translocation of ADAM17 to the cell surface, resulting in increased ADAM17 activity [17,18]. It is described that the mitogen-activated protein (MAP) kinases ERK and p38 are involved in ADAM17 phosphorylation thus regulating its activity [19,20], as well as family members of the protein kinase C (PKC) family (Figure 1b). Activation of PKCs can result from engagement of G-protein coupled receptors (GPCRs) that are coupled to an αq/11 subunit resulting in phospholipase C (PLC)-mediated increase in second messengers diacylglycerol (DAG), inositol triphosphate (IP3) and Ca2+ [21,22].
Proteolytic activity of ADAM10 and ADAM17 is also triggered by exposure to negatively charged phosphatidylserine [23,24]. Phosphatidylserine in the outer leaflet of the cell membrane usually indicates cells which undergo apoptosis and cell death was indeed identified as a potent activator of ADAM proteases [25]. Moreover, ADAM protease activity can be regulated by limited proteolysis and ADAM proteases were suggested to be cleaved by other ADAM family members and the astacin family member meprin β [26,27,28,29].
The regulation of other ADAM family members is less well understood. The key to the activation of all ADAM family members is the removal of the pro-domain by proprotein convertases, as described above. However, the pro-domain of the family member ADAM8 is removed in an autocatalytic process [30]. Expression of ADAM8 itself is regulated by inflammatory cytokines such as TNFα, IL-1β, IL-4 and IL-13 [30]. ADAM12 is stored as active protease intracellularly and substrate exposure occurs via trafficking to the plasma membrane [31].
2.3. Signalling Pathways Regulated by ADAM Proteases
ADAM proteases play a decisive role in inflammation and cancer due to their variety of substrates. Among these substrates are cytokines, growth factors and cell surface receptors (Figure 1c). The most prominent substrate for ADAM17 is Tumor Necrosis Factor α (TNFα), a cytokine which binds to TNFα receptors (TNFRs) 1 and 2 which both are ADAM17 substrates as well [32]. The resulting soluble TNFR ectodomains can still bind TNFα thus act as antagonistic decoy receptors which has been used therapeutically. In contrast to the antagonistic activity of soluble TNFRs in TNFα signalling, the proteolytically cleaved receptor for the pleiotropic cytokine Interleukin-6 (IL-6) acts in an agonistic fashion. IL-6 signals via a membrane-bound alpha-receptor (IL-6R) and a homodimer of the signal-transducing beta-receptor subunit glycoprotein 130 (gp130), which is ubiquitously expressed, while expression of IL-6R is limited to selected cell types, e.g., hepatocytes and leukocytes. This process is termed IL-6 classic signalling. However, cells that do not express the membrane-bound IL-6R are still responsive to IL-6 via IL-6 trans-signalling. In this process the complex of IL-6 and soluble IL-6R (sIL-6R) which can be generated via limited proteolysis mediated by ADAM10 and ADAM17, is able to engage gp130 homodimers on target cells [33,34,35].
ADAM protease activity was also demonstrated to increase bioavailability of epidermal growth factor receptor (EGFR) ligands like transforming growth factor α (TGFα), heparin-bound EGF (HB-EGF) and amphiregulin (AR) [36,37,38]. EGFR signalling is crucial for regenerative processes as it was, e.g., demonstrated that both EGFR-hypomorphic mice (Waved-2) and ADAM17-hypomorphic mice (ADAM17ex/ex) display impaired intestinal epithelial regeneration [39,40]. The enhanced release of EGFR ligands via ADAM17 was shown to drive cancer progression and drug resistance in cell lines and experimental mouse models [41,42,43] (see also below).
Signal transduction via the Notch receptor is an evolutionary conserved pathway in which the Notch receptor undergoes a series of proteolytic events. Notch signalling was demonstrated to be essentially involved in stem cell maintenance and differentiation, but also in tumour formation [44,45]. In the majority of tissues analysed, ADAM10 was identified as the major extracellular protease for Notch [46,47]. This cleavage is followed by regulated intramembrane proteolysis (RIP) by the γ-secretase complex in order to release an intracellular domain (Figure 1c) that translocates to the nucleus and induces expression of Notch target genes [44,45]. Next to ADAM10, ADAM17 is discussed to promote Notch signalling [48,49].
Additionally, ADAM17 is described to be responsible for the proteolysis of Notch1 receptor, which increases EGFR expression [49]. The Notch receptor undergoes a series of proteolytic events in order to release an intracellular domain which is necessary for signal transduction. However, in this process the involvement of both proteases, ADAM10 and ADAM17, is discussed and seems to be context dependent [48].
3. ADAM-Mediated Pathways in Gastrointestinal Tumour Formation
3.1. ADAM Proteases in Gastric Cancer
Gastric cancer is the fourth most frequently occurring but the second most deadly cancer worldwide. One major risk for the development of gastric cancer is infection by Helicobacter pylori which is reflected by the fact that gastric cancer incidence is increased in areas with high H. pylori prevalence. Transactivation of EGFR family members and chronic inflammation are potential mechanisms how H. pylori infection contribute to the development of gastric cancer [50]. The vast majority of gastric cancers are adenocarcinoma arising from glandular epithelial cells of the gastric mucosa. Gastric atrophy, i.e., the loss of glandular tissue, including parietal cells, can progress to metaplasia due to the absence of signals regulating stem cell proliferation and differentiation provided by parietal cells [51]. Chronic inflammation of the gastric mucosa, e.g., triggered by bacterial infections such as H.pylori often results in the formation of pre-malignant lesions. Accordingly, mice with hyperactive gp130, the signal transducing subunit of the interleukin 6 (IL-6) receptor complex resulted in an excessive inflammatory response of the gastric epithelium and the formation of gastric metaplasia and spontaneous adenoma [52].
The most common genetic alterations in gastric cancer comprise among others, mutations in genes encoding for the tumour suppressor p53, the small GTPase KRAS, phosphoinositide-3 kinase (PI3K), but also members of the epidermal growth factor receptor (EGFR) family, including EGFR and ErbB2 [53,54]. KRAS, PI3K and EGFR proteins are connected within a common signal transduction pathway, highlight the importance of the EGFR/MAPK pathway for the development of gastric cancer.
Several ADAM proteases including ADAM9, 10, 12, 15, 17 and 33 were shown to be overexpressed in gastric cancer [55,56,57,58,59,60], while ADAM23 was shown to be epigenetically silenced by promoter CpG methylation in gastric tumour tissue [61]. Expression of ADAM10 and ADAM17 was demonstrated to be induced by H. pylori infection [55,62] and expression of ADAM17 correlated with the forkhead transcription factor FoxM1 which is activated by PI3K/AKT [60]. ADAM17 activity is not solely dependent on its transcription level but needs additional stimuli. It is therefore not surprising that the enhanced release of EGFR ligands amphiregulin and HB-EGF upon H. pylori infection was dependent on the phosphorylation of ADAM17 C-terminus [62].
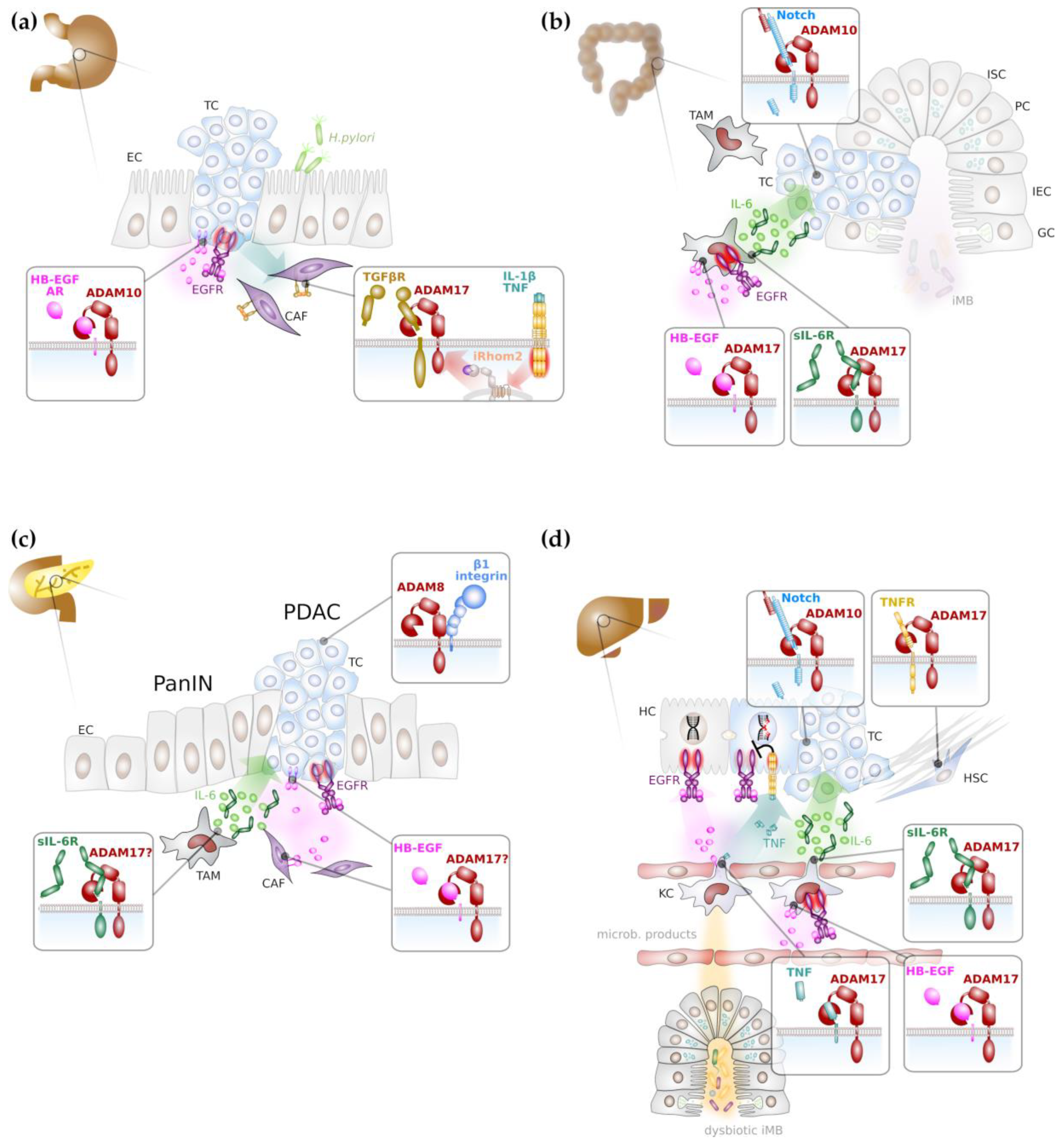
As described above, ligands for the EGFR can be liberated by limited proteolysis mainly via ADAM10 or ADAM17 activity. It is therefore plausible that ADAM proteases provide soluble ligands for the activation of EGFR family members on gastric tumour cells. It was indeed demonstrated that the inflammatory cytokines IL-1β and IL-8 [63], but TGFβ [64] also induced EGFR trans-activation in gastric cancer cells, particularly via soluble amphiregulin and HB-EGF (Figure 2a). Depletion of ADAM10 but not ADAM12 or ADAM17 abrogated the release of soluble EGFR ligands and hence EGFR trans-activation [65]. Upon proteolytic release of HB-EGF ectodomain, a C-terminal fragment (CTF) remains in the signal-sending cell and it was suggested that nuclear translocation of HB-EGF CTF contributes to the pathogenicity of gastric cancer [63].
Mouse models with activating mutations in KRAS in gastric epithelial cells but not Helicobacter infections resulted in an increase in AR and HB-EGF secretion, supporting the existence of a RTK/MAPK feed-forward loop in gastric cancer. However, while aberrant KRAS activation alone induced gastric metaplasia [66], the formation of adenoma was dependent on the additional activation of gp130 [54], mainly through interleukin 11 (IL-11) [67]. While in vitro experiments showed that IL-6 trans-signalling can overcome the repressive effects of trefoil factor 1 (TFF1) on IL-6 classic-signalling [68], gastric metaplasia induced by a hyperactive gp130 variant was not abrogated by a dimerised form of soluble gp130 (sgp130Fc) [69]. These data suggest that in gastric cancer ADAM10 and ADAM17 rather have a major role in EGFR trans-activation but not in boosting signal transduction via IL-6 family cytokines as in other gastrointestinal tumours (see below).
ADAM17 was also shown to be overexpressed in gastrointestinal stroma tumours (GIST) where it co-localised with EGF and EGFR [70]. Gastrointestinal stroma tumours are rare malignant tumours of non-epithelial origin and the most common mesenchymal neoplasms of the gastrointestinal tract [71]. The majority of GISTs localise to the stomach and they are thought to originate from interstitial cells of Cajal [71], a cell type that under physiological conditions controls gastric smooth muscle cell contraction. GISTs are characterised by the occurrence of activating mutations in genes coding for the platelet derived growth factor receptor alpha (PDGFRA) or the close homologue c-Kit [Schmidt-Arras and Böhmer Trends Mol Med, in press]. PDGFRβ was previously shown to induce ADAM17 activation [72] and it is likely that ADAM17 activity in cells is elevated by mutant PDGFRα or c-Kit, resulting in the additional activation of EGFR. However, experimental evidence is still lacking.
The progression of gastric cancer correlates with the expression of ADAM10 and ADAM17 and high ADAM10/ADAM17 expression levels were associated with patient’s poor prognosis [58,59] and the establishment of lymph node metastasis [73]. A recent report demonstrated that inflammatory cytokines IL-1α, IL-1β or TNFα secreted by diffuse-type gastric cancer cells induced upregulation of rhomboid 5 homolog 2 (RHBDF2), also termed iRhom2 in cancer-associated fibroblasts (CAFs). iRhom2 promoted ADAM17 activity and TGFβ receptor cleavage (Figure 2a) and as a consequence CAF motility to invade extracellular matrix and lymphatic vessels [74]. Hence, progression of gastric cancer is not only promoted by ADAM17 on tumour cells, but also on cells of the tumour micro-environment.
3.2. ADAM Proteases in Colorectal Cancer
The intestine is a complex organ comprising multiple types of differentiated and specialized cell types. The intestinal lumen is lined by a single layer of columnar epithelial cels which are constantly replenished by proliferative crypt cells. In the adult, Lgr5+ columnar base crypt (CBC) stem cells locate to the crypt base and give rise to the multiple intestinal cell types. The stem cell niche is formed and maintained by Paneth cells that intersperse the CBCs and provide Wnt ligands, the Notch ligands delta like (Dll) 1 and 4 and EGF. Upon exiting the stem cell niche, CBCs give rise to rapidly proliferating transit-amplifying (TA) progenitors and, subsequently, to either absorptive progenitors or secretory progenitors, dependent on Notch receptor activity. High Notch activity results in absorptive cells while low Notch activity induces secretory differentiation [75]. As outlined above, ADAM10 is the major sheddase for the Notch receptor in multiples tissues and genetic deficiency of ADAM10 in villin-expressing intestinal cells demonstrated its importance for intestinal stem cell fitness [47]. In contrast, while complete loss of ADAM10 in Paneth cells did not alter crypt homeostasis [76], overexpression of a catalytic inactive ADAM10 variant resulted in Paneth cell mislocalisation, partially phenocopying EphB3-/- mice [77].
Proliferation of CBCs and TA progenitor cells is driven by EGFR activation [78]. Surprisingly, albeit ADAM17 plays a major role for the release of EGFR ligands, loss of ADAM17 did not influence intestinal crypt homeostasis. However, under conditions of epithelial regeneration, loss of ADAM17 in intestinal epithelial cells but not in myeloid cells impaired intestinal regeneration which was associated with impaired EGFR activation [40,79].
Intestinal tumorigenesis follows a typical adenoma–carcinoma sequence. The persistent activation of the Wnt signalling pathway is a major hallmark in intestinal cancer formation. Wnt ligands bind to its receptor Frizzled which results in stabilisation and activation of β-catenin which translocates to the nucleus to induce gene expression. In the unliganded state, β-catenin is bound in a protein complex also containing the protein adenomatous polyposis coli (APC) which is essential to induce phosphorylation of β-catenin by glycogen synthase kinase 3 (GSK3) β and subsequent proteasomal degradation of β-catenin. Loss of APC occurs often, particularly in colon cancer, and results in ligand-independent β-catenin activation. The loss of APC alleles in mice is commonly used as a model for intestinal adenoma formation. Other frequently found and early arising mutations in colorectal cancer are loss of the tumour suppressor p53 and aberrant KRAS activation [80].
Genetic deficiency of ADAM10 in intestinal stem cells drastically reduced colonic and small intestinal adenoma formation in mice with biallelic loss of APC which was linked to the absence of Notch signalling [75], indicating that ADAM10 is a potential attractive target for intestinal cancer therapy (Figure 2b).
ADAM17 has a dual role during intestinal cancer formation. On one hand, ADAM17 releases EGFR ligands on intestinal epithelial cells and contributes to autocrine EGFR trans-activation [81]. In addition, there is evidence that the release of EGFR ligands from cancer associated macrophages depends on ADAM17 and it was demonstrated that myeloid EGFR activation (Figure 2b) is a prerequisite for the release of tumour-promoting IL-6 [82]. On the other hand, ADAM17 provides the soluble IL-6 receptor (sIL-6R) from myeloid cells that in consequence induces IL-6 trans-signalling on intestinal epithelial cells (Figure 2b) that was demonstrated to be essential for intestinal tumorigenesis [43,83]. Furthermore, there is evidence that ADAM17 on intestinal tumour cells promotes tumour angiogenesis through enhanced vascular endothelial growth factor (VEGF)-A secretion [84].
Although there is strong experimental evidence that ADAM proteases, in particular ADAM10 and ADAM17 are involved in intestinal tumorigenesis, further work is warranted to translate these findings to the human situation.
3.3. ADAM Proteases in Pancreatic Cancer
The pancreas is an organ with exo- and endocrine functions which synthesizes digestive enzymes such as tryptic proteases and hormones like insulin and glucagon. The most frequently occurring type of pancreatic cancer is the pancreatic adenocarcinoma within the exocrine tissue. Almost all of these cancers arise from the pancreatic ductal epithelium and are therefore termed pancreatic ductal adenocarcinoma (PDAC), while the second most common type arises from acinar cells. The head of the pancreas, which lies close to the duodenum, is the most common localisation of pancreatic cancer, and tumours in this region can result in obstructions of the pancreatic or biliary tract which is the reason why patients at diagnosis present signs of jaundice.
Pancreatic cancer mainly occurs at patients aged over 40 years and common risk factors are smoking, obesity, chronic pancreatitis, which is often linked to excessive alcohol consumption, and genetic predisposition.
PDAC is thought to arise from pre-cancerous lesions, that, depending on their localisation, can be termed pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasia (IPMN), pancreatic mucinous cystic neoplasia (MCN) or intraductal tubulopapillary neoplasia. Although the pre-cancerous lesions differ in their occurrence of mutations, most common mutations in pancreatic cancer are activating mutations in KRAS, TP53-deficiency and mutations in SMAD4 [85].
Experimental models have demonstrated the importance of EGFR activation in KRAS-mutant pancreatic cancer [86,87], indicating that RAS/MAPK pathway activation is not sufficient to drive pancreatic tumorigenesis. This also suggests a potential critical role of ADAM17 on tumour or stroma cells to provide EGFR ligands (Figure 2c). Progression of pancreatic cancer is indeed associated with increased expression of ADAM17 [88], and ADAM17 expression on PDAC cells was shown to be induced by deoxycholic acid, resulting in enhanced release of the EGFR ligands AR and transforming growth factor (TGF) α [89]. Consequently, PDAC formation in a KRAS and TP53-driven mouse model of pancreatic cancer was reduced by the use of an ADAM17-directed antibody [90].
Furthermore, ADAM17 might be additionally involved in the generation of inflammatory signalling, notably IL-6 trans-signalling through the generation of sIL-6R (Figure 2c). IL-6 trans-signalling was demonstrated to promote pancreatitis-associated lung injury [91] and the progression of pancreatic intraepithelial neoplasms [92]. However, direct experimental evidence for ADAM17 in an early inflammatory stage of pancreatic cancer is still lacking.
Poor prognosis of PDAC also correlates with high expression levels of ADAM8 and ADAM9 [93,94]. While enhanced expression of ADAM9 in PDAC cell lines facilitated anchorage-independent growth and was associated with increased vascularisation in a xenograft model, growth of tumour tissue was unaltered [94]. In contrast, a peptidomimetic inhibitor of ADAM8 reduced tumour growth of pancreatic tumour cells in a xenograft model but was also able to impair tumour growth in a KRAS-driven pancreas cancer model and significantly prolonged overall survival [95]. The pro-tumorigenic effect of ADAM8 was linked to its association with β1-integrin and the resulting increase in tumour cell motility, invasiveness and activation of the MAP kinases ERK1/2 [95]. Hence, ADAM8 represents a novel target for the treatment of pancreatic cancer [30].
3.4. ADAM Proteases in Hepatic Cancer
The liver is an organ with major functions in metabolism, innate immunity and detoxification. It serves as a first line defence against intestinal pathogens. It is therefore not surprising that the liver has a tremendous potential to regenerate. The liver consists of different resident cell types, including hepatocytes, cholangiocytes, Kupffer cells, hepatic stellate cells (HSCs) and liver sinusoidal endothelial cell. Hepatocytes, the hepatic epithelial cells make up the majority of cells in the liver. Upon acute hepatic damage, hepatocytes have the capacity to restore the lost liver tissue via proliferation and hypertrophy [96]. However, during chronic damage, hepatocytes undergo senescence and are unable to proliferate. Under these conditions, liver stem and progenitor cells (LPCs) proliferate and differentiate either into hepatocytes or cholangiocytes, the biliary epithelial cells [97]. ADAM10 was demonstrated to be essential for hepatocellular homeostasis via regulation of bile acid transporters [98]. Interestingly, in the liver ADAM10 is dispensable for Notch processing and ADAM10-deficient mice display normal formation of the biliary tree [98] which was linked to Notch2 activity [99].
Hepatocellular carcinoma (HCC) is one of the most frequently occurring tumour entities worldwide. Fibrotic livers can progress to liver cirrhosis which predisposes to HCC formation. The differentiation of HSCs into collagen-secreting myofibroblasts is key to fibrosis and liver cirrhosis development [100,101]. There are several indications that ADAM-induced signalling pathways are involved in both the induction and the prevention of liver fibrosis. This has been recently reviewed in depth elsewhere [102,103]. The release of EGFR ligands by HSCs can have pro- as well as anti-fibrotic activities, depending on the ligand, while the release of TNFα promotes liver fibrosis [103]. Consistent with a pro-fibrotic role of ADAM17, substrates of ADAM17 are elevated in the serum of patients suffering from liver cirrhosis [104]. In contrast, ADAM17 seems also to prevent the exacerbation of pro-fibrotic signalling (Figure 2d). HSCs lacking Rhbdf2 that is required for ADAM17 maturation, displayed reduced TNFR1 and 2 shedding and as a consequence of enhanced TNF signalling, Rhbdf2-/- mice displayed enhanced bile duct obstruction-induced liver fibrosis [104]. Increased expression of ADAM12 was detected in activated HSCs and in liver tissue sections from patients suffering from liver cirrhosis or HCC [105,106], suggesting that ADAM12 has a tumour-promoting role in the liver by actively remodelling the extracellular matrix.
ADAM10 was shown to be overexpressed in HCC and associated with a poor prognosis [107]. miRNAs targeting ADAM10 reduced the invasive potential of HCC cell lines [108,109]. While direct experimental evidence is still lacking, these findings suggest that ADAM10 has a tumour promoting role in the liver, potentially via induction of Notch signalling (Figure 2d), which was suggested to promote cholangiocarcinoma [110].
EGFR signalling has a dual role in the liver. EGFR signalling on hepatocytes was demonstrated to enhance intrinsic DNA damage repair and chronic inflammatory signalling in obese mice via IL-1β and TNFα abrogates this tumour suppressing effect of EGFR (Figure 2d) [111]. In contrast, similar to its role in intestinal tumour formation, autocrine activation of EGFR on Kupffer cells was demonstrated to promote IL-6 secretion (Figure 2d) [112]. The development of HCC in humans was associated with elevated serum levels of IL-6 [113,114], and murine models have demonstrated that HCC formation is blunted in the absence of IL-6 [115]. Interestingly, HCC formation seems to be completely dependent on IL-6 trans-signalling (Figure 2d) and beside IL-6, Kupffer cells also provide sIL-6R [116]. Although experimental evidence is still lacking, it is tempting to speculate that ADAM17 on Kupffer cells is essential to (i) release EGFR ligands to induce IL-6 release and (ii) generate sIL-6R in order to promote tumorigenesis via IL-6 trans-signalling.
4. ADAM-Directed Therapeutic Approaches
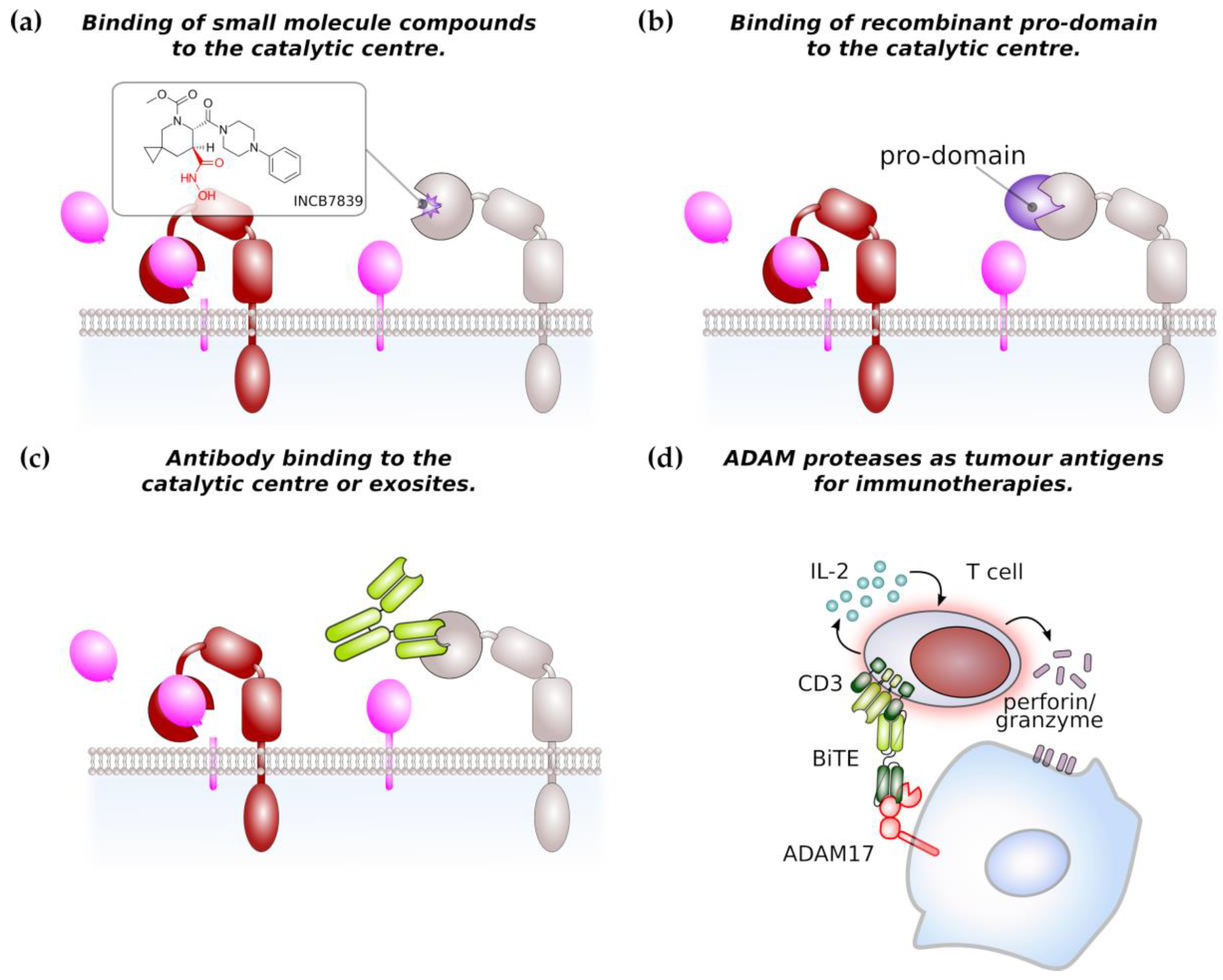
Due to its major role in TNF processing, ADAM17 has previously gained interest as therapeutic target for the treatment of chronic inflammatory diseases and has led to the development of a series of small molecular inhibitors. The majority of these inhibitors is hydroxamate-based (Figure 3a) and relatively specific for ADAM17 or dual inhibitors for ADAM10 and ADAM17. These inhibitors bind to the zinc ion in the catalytic centre and to the S1’ pocket of ADAM17 [117,118,119]. However, none of the earlier inhibitors reached beyond phase 3 in clinical trial due to musculoskeletal and liver toxicities [118]. This led to the development of more specific inhibitors such as INCB7839, a dual inhibitor of ADAM10 and ADAM17 which has been used in clinical trials for the treatment of HER2+ breast cancer, large B-cell non-Hodgkin lymphoma [118] and is currently in clinical testing for the treatment of pediatric glioma. These data will also show if severe side effects encountered by use of the earlier inhibitors is linked to their unspecificity or to the fact that ADAM10/17 have a broad substrate spectrum with proteins involved in different biological processes.
Another approach to target ADAM activity is the development of antibodies targeting the catalytic site and the use of recombinant pro-domains of ADAM proteases (Figure 3b,c). Two monoclonal antibodies D1 (A12) [90,120,121] and MEDI3622 [122] directed against ADAM17 were demonstrated to be efficient in pre-clinical cancer models. The monoclonal antibody 8C7 was directed against an active conformation of ADAM10 and was shown to inhibit lymphoma growth in a xenograft model and gastrointestinal tumour growth in a genetic mouse model [123].
The pro-domains of ADAM proteases share only a low level of similarity and are thought be highly specific for their cognate protease. The use of recombinant pro-domains has therefore been considered as an alternative approach to specifically target the catalytic site of ADAM proteases and the inhibitory selectivity was demonstrated for pro-domains of ADAM10, 12 and 17 [7,117,124,125]. ADAM17 recombinant pro-domain has been shown to specifically inhibit ADAM17 in different mouse models [8,126,127,128] and might enter into clinical development for the treatment of inflammatory bowel disease [118].
Peptidomimetics were shown to be useful to inhibit ADAM8 multimerisation and hence autoactivation in pancreatic cancer [95]. This approach might also be applicable to other ADAM family members, where the association with integrins enhances cancer cell motility.
Overexpression of ADAM family members on tumour cells make them an attractive target for immunotherapies. An ADAM17/CD3 bispecific T-cell enganger (BiTE) antibody was able to induce the T-cell mediated lysis of pancreatic cancer cells in vitro (Figure 3d) [129]. Furthermore, peptides derived from ADAM17 were found to be present on major histocompatibility complex (MHC) I molecules on prostate cancer cells and therefore represent an immunotherapeutic target [130]. However, ADAM protease-directed immunotherapy is only beginning to evolve and definitely warrants further investigation.
5. Conclusions
There is now a growing body of evidence that members of the ADAM protease family are over-expressed on tumour cells and are correlated with tumour formation and the progression of gastrointestinal tumours (summarized in Table 2). Some of the tumorigenic signalling pathways initiated by ADAM proteases have been deciphered. However, unbiased proteomics approaches are needed to identify ADAM substrates and ADAM protease-activated signalling pathways that might represent novel targets for tumour therapy.
Given the fact that ADAM proteases generate paracrine signals, the role of ADAM proteases in the tumour microenvironment needs more attention. Cell type-specific gene targeting in mouse models of cancer combined with unbiased proteomic substrate identification will improve our understanding of how ADAM proteases shape the tumour microenvironment and promote tumour metastasis. This will also help to design tissue-specific ADAM protease inhibitors or to target ADAM-mediated novel tumour cell vulnerabilities, with the aim to reduce the severe side effects encountered with current ADAM protease inhibitors.
Author Contributions
Conceptualization, D.S.-A.; writing—original draft preparation, D.S.-A. and N.S.; writing—review and editing, D.S.-A., S.R.-J. and N.S.; visualization, D.S.-A.; funding acquisition, D.S.-A. and S.R.-J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG), Bonn, (grant number SFB841, “Liver inflammation: Infection, immune regulation and consequences”, to D.S.-A., S.R.-J.; grant number SFB877 to S.R.-J.), and the Cluster of Excellence ‘Precision Medicine’ to S.R.-J.
Acknowledgments
In this section you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
S.R.-J. is an inventor of patents owned by the CONARIS Research Institute, which develops the sgp130Fc protein together with Ferring Pharmaceuticals, and he has stock ownership in CONARIS. No conflicts of interest, financial or otherwise, are declared by N.S. and D.S.-A.
References
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Huxley-Jones, J.; Clarke, T.-K.; Beck, C.; Toubaris, G.; Robertson, D.L.; Boot-Handford, R.P. The evolution of the vertebrate metzincins; insights from Ciona intestinalis and Danio rerio. BMC Evol. Biol. 2007, 7, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomis-Rüth, F.X. Catalytic domain architecture of metzincin metalloproteases. J. Biol. Chem. 2009, 284, 15353–15357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzen, I.; Lokau, J.; Düsterhöft, S.; Trad, A.; Garbers, C.; Scheller, J.; Rose-John, S.; Grötzinger, J. The membrane-proximal domain of A Disintegrin and Metalloprotease 17 (ADAM17) is responsible for recognition of the interleukin-6 receptor and interleukin-1 receptor II. FEBS Lett. 2012, 586, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Düsterhöft, S.; Michalek, M.; Kordowski, F.; Oldefest, M.; Sommer, A.; Röseler, J.; Reiss, K.; Grötzinger, J.; Lorenzen, I. Extracellular Juxtamembrane Segment of ADAM17 Interacts with Membranes and Is Essential for Its Shedding Activity. Biochemistry (Mosc.) 2015, 54, 5791–5801. [Google Scholar] [CrossRef]
- Endres, K.; Anders, A.; Kojro, E.; Gilbert, S.; Fahrenholz, F.; Postina, R. Tumor necrosis factor-alpha converting enzyme is processed by proprotein-convertases to its mature form which is degraded upon phorbol ester stimulation. Eur. J. Biochem. 2003, 270, 2386–2393. [Google Scholar] [CrossRef]
- Gonzales, P.E.; Solomon, A.; Miller, A.B.; Leesnitzer, M.A.; Sagi, I.; Milla, M.E. Inhibition of the tumor necrosis factor-alpha-converting enzyme by its pro domain. J. Biol. Chem. 2004, 279, 31638–31645. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.; Cohen, T.; Romi, E.; Levin, M.; Peleg, Y.; Arad, U.; Yaron, A.; Milla, M.E.; Sagi, I. Harnessing the natural inhibitory domain to control TNFα Converting Enzyme (TACE) activity in vivo. Sci. Rep. 2016, 6, 35598. [Google Scholar] [CrossRef] [Green Version]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 2012, 335, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Dornier, E.; Coumailleau, F.; Ottavi, J.-F.; Moretti, J.; Boucheix, C.; Mauduit, P.; Schweisguth, F.; Rubinstein, E. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J. Cell Biol. 2012, 199, 481–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prox, J.; Willenbrock, M.; Weber, S.; Lehmann, T.; Schmidt-Arras, D.; Schwanbeck, R.; Saftig, P.; Schwake, M. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell. Mol. Life Sci. Cmls 2012, 69, 2919–2932. [Google Scholar] [CrossRef] [PubMed]
- Christova, Y.; Adrain, C.; Bambrough, P.; Ibrahim, A.; Freeman, M. Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 2013, 14, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.A.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef] [Green Version]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef] [Green Version]
- Termini, C.M.; Gillette, J.M. Tetraspanins Function as Regulators of Cellular Signaling. Front. Cell Dev. Biol. 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Rodríguez, E.; Montero, J.C.; Esparís-Ogando, A.; Yuste, L.; Pandiella, A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735, a potential role in regulated shedding. Mol. Biol. Cell 2002, 13, 2031–2044. [Google Scholar] [CrossRef]
- Soond, S.M.; Everson, B.; Riches, D.W.H.; Murphy, G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 2005, 118, 2371–2380. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Derynck, R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 2010, 37, 551–566. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Ishiguro, J.; Kitamura, H.; Arima, N.; Okutani, M.; Shuto, A.; Higashiyama, S.; Ohwada, T.; Arai, H.; Makide, K.; et al. TGFα shedding assay: An accurate and versatile method for detecting GPCR activation. Nat. Methods 2012, 9, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Kordowski, F.; Büch, J.; Maretzky, T.; Evers, A.; Andrä, J.; Düsterhöft, S.; Michalek, M.; Lorenzen, I.; Somasundaram, P.; et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 2016, 7, 11523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleibaum, F.; Sommer, A.; Veit, M.; Rabe, B.; Andrä, J.; Kunzelmann, K.; Nehls, C.; Correa, W.; Gutsmann, T.; Grötzinger, J.; et al. ADAM10 sheddase activation is controlled by cell membrane asymmetry. J. Mol. Cell. Biol. 2019, 11, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalaris, A.; Rabe, B.; Paliga, K.; Lange, H.; Laskay, T.; Fielding, C.A.; Jones, S.A.; Rose-John, S.; Scheller, J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood 2007, 110, 1748–1755. [Google Scholar] [CrossRef] [PubMed]
- Tousseyn, T.; Thathiah, A.; Jorissen, E.; Raemaekers, T.; Konietzko, U.; Reiss, K.; Maes, E.; Snellinx, A.; Serneels, L.; Nyabi, O.; et al. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J. Biol. Chem. 2009, 284, 11738–11747. [Google Scholar] [CrossRef] [Green Version]
- Moss, M.L.; Powell, G.; Miller, M.A.; Edwards, L.; Qi, B.; Sang, Q.-X.A.; De Strooper, B.; Tesseur, I.; Lichtenthaler, S.F.; Taverna, M.; et al. ADAM9 inhibition increases membrane activity of ADAM10 and controls α-secretase processing of amyloid precursor protein. J. Biol. Chem. 2011, 286, 40443–40451. [Google Scholar] [CrossRef] [Green Version]
- Wichert, R.; Scharfenberg, F.; Colmorgen, C.; Koudelka, T.; Schwarz, J.; Wetzel, S.; Potempa, B.; Potempa, J.; Bartsch, J.W.; Sagi, I.; et al. Meprin β induces activities of A disintegrin and metalloproteinases 9, 10, and 17 by specific prodomain cleavage. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 11925–11940. [Google Scholar] [CrossRef] [Green Version]
- Scharfenberg, F.; Helbig, A.; Sammel, M.; Benzel, J.; Schlomann, U.; Peters, F.; Wichert, R.; Bettendorff, M.; Schmidt-Arras, D.; Rose-John, S.; et al. Degradome of soluble ADAM10 and ADAM17 metalloproteases. Cell. Mol. Life Sci. Cmls 2020, 77, 331–350. [Google Scholar] [CrossRef]
- Conrad, C.; Benzel, J.; Dorzweiler, K.; Cook, L.; Schlomann, U.; Zarbock, A.; Slater, E.P.; Nimsky, C.; Bartsch, J.W. ADAM8 in invasive cancers: Links to tumor progression, metastasis, and chemoresistance. Clin. Sci. (Lond. Engl. 1979) 2019, 133, 83–99. [Google Scholar] [CrossRef]
- Sundberg, C.; Thodeti, C.K.; Kveiborg, M.; Larsson, C.; Parker, P.; Albrechtsen, R.; Wewer, U.M. Regulation of ADAM12 cell-surface expression by protein kinase C epsilon. J. Biol. Chem. 2004, 279, 51601–51611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17, Physiology and pathophysiology. Biochimica et biophysica acta. Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Müllberg, J.; Schooltink, H.; Stoyan, T.; Günther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, I.; Schwarz, J.; Lücke, K.; Schumacher, N.; Schumacher, V.; Schmidt, S.; Rabe, B.; Saftig, P.; Donners, M.; Rose-John, S.; et al. ADAM17 controls IL-6 signaling by cleavage of the murine IL-6Rα from the cell surface of leukocytes during inflammatory responses. J. Leukoc. Biol. 2016, 99, 749–760. [Google Scholar] [CrossRef]
- Borrell-Pagès, M.; Rojo, F.; Albanell, J.; Baselga, J.; Arribas, J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. Embo J. 2003, 22, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Gschwind, A.; Hart, S.; Fischer, O.M.; Ullrich, A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. Embo J. 2003, 22, 2411–2421. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.C.; Sunnarborg, S.W.; Hinkle, C.L.; Myers, T.J.; Stevenson, M.Y.; Russell, W.E.; Castner, B.J.; Gerhart, M.J.; Paxton, R.J.; Black, R.A.; et al. TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann. N.Y. Acad. Sci. 2003, 995, 22–38. [Google Scholar] [CrossRef]
- Egger, B.; Büchler, M.W.; Lakshmanan, J.; Moore, P.; Eysselein, V.E. Mice harboring a defective epidermal growth factor receptor (waved-2) have an increased susceptibility to acute dextran sulfate-induced colitis. Scand. J. Gastroenterol. 2000, 35, 1181–1187. [Google Scholar] [CrossRef]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Gijsen, M.; King, P.; Perera, T.; Parker, P.J.; Harris, A.L.; Larijani, B.; Kong, A. HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biol. 2010, 8, e1000563. [Google Scholar] [CrossRef] [Green Version]
- Kyula, J.N.; Van Schaeybroeck, S.; Doherty, J.; Fenning, C.S.; Longley, D.B.; Johnston, P.G. Chemotherapy-induced activation of ADAM-17, a novel mechanism of drug resistance in colorectal cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 3378–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.; Schumacher, N.; Schwarz, J.; Tangermann, S.; Kenner, L.; Schlederer, M.; Sibilia, M.; Linder, M.; Altendorf-Hofmann, A.; Knösel, T.; et al. ADAM17 is required for EGF-R-induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018, 215, 1205–1225. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a Notch. Development (Camb. Engl.) 2013, 140, 689–704. [Google Scholar] [CrossRef] [Green Version]
- Bigas, A.; Espinosa, L. The multiple usages of Notch signaling in development, cell differentiation and cancer. Curr. Opin. Cell Biol. 2018, 55, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, D.; de Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lübke, T.; Lena Illert, A.; von Figura, K.; et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.-H.; VanDussen, K.L.; Sawey, E.T.; Wade, A.W.; Kasper, C.; Rakshit, S.; Bhatt, R.G.; Stoeck, A.; Maillard, I.; Crawford, H.C.; et al. ADAM10 regulates Notch function in intestinal stem cells of mice. Gastroenterology 2014, 147, 822–834.e13. [Google Scholar] [CrossRef] [Green Version]
- Bozkulak, E.C.; Weinmaster, G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol. Cell. Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, A.; Seidl, S.; Vlachou, P.; Michel, L.; Mitova, N.; Schatz, N.; Specht, K.; Koch, I.; Schuster, T.; Grundler, R.; et al. ADAM17 regulates epidermal growth factor receptor expression through the activation of Notch1 in non-small cell lung cancer. Cancer Res. 2010, 70, 5368–5378. [Google Scholar] [CrossRef] [Green Version]
- Wallasch, C.; Crabtree, J.E.; Bevec, D.; Robinson, P.A.; Wagner, H.; Ullrich, A. Helicobacter pylori-stimulated EGF receptor transactivation requires metalloprotease cleavage of HB-EGF. Biochem. Biophys. Res. Commun. 2002, 295, 695–701. [Google Scholar] [CrossRef]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 2007, 117, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R.; Nomura, S. Differentiation of the gastric mucosa III. Animal models of oxyntic atrophy and metaplasia. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G999–G1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiem, S.; Eissmann, M.F.; Elzer, J.; Jonas, A.; Putoczki, T.L.; Poh, A.; Nguyen, P.; Preaudet, A.; Flanagan, D.; Vincan, E.; et al. Stomach-Specific Activation of Oncogenic KRAS and STAT3-Dependent Inflammation Cooperatively Promote Gastric Tumorigenesis in a Preclinical Model. Cancer Res. 2016, 76, 2277–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, T.; Tomita, T.; Dixon, M.F.; Axon, A.T.R.; Robinson, P.A.; Crabtree, J.E. ADAMs (a disintegrin and metalloproteinase) messenger RNA expression in Helicobacter pylori-infected, normal, and neoplastic gastric mucosa. J. Infect. Dis. 2002, 185, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Carl-McGrath, S.; Lendeckel, U.; Ebert, M.; Roessner, A.; Röcken, C. The disintegrin-metalloproteinases ADAM9, ADAM12, and ADAM15 are upregulated in gastric cancer. Int. J. Oncol. 2005, 26, 17–24. [Google Scholar] [CrossRef]
- Kim, S.; Takahashi, H.; Lin, W.-W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.-L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-Y.; Ye, Z.-Y.; Li, L.; Zhao, Z.-S.; Shao, Q.-S.; Tao, H.-Q. ADAM 10 is associated with gastric cancer progression and prognosis of patients. J. Surg. Oncol. 2011, 103, 116–123. [Google Scholar] [CrossRef]
- Shou, Z.-X.; Jin, X.; Zhao, Z.-S. Upregulated expression of ADAM17 is a prognostic marker for patients with gastric cancer. Ann. Surg. 2012, 256, 1014–1022. [Google Scholar] [CrossRef]
- Fang, W.; Qian, J.; Wu, Q.; Chen, Y.; Yu, G. ADAM-17 expression is enhanced by FoxM1 and is a poor prognostic sign in gastric carcinoma. J. Surg. Res. 2017, 220, 223–233. [Google Scholar] [CrossRef]
- Takada, H.; Imoto, I.; Tsuda, H.; Nakanishi, Y.; Ichikura, T.; Mochizuki, H.; Mitsufuji, S.; Hosoda, F.; Hirohashi, S.; Ohki, M.; et al. ADAM23, a possible tumor suppressor gene, is frequently silenced in gastric cancers by homozygous deletion or aberrant promoter hypermethylation. Oncogene 2005, 24, 8051–8060. [Google Scholar] [CrossRef] [Green Version]
- McClurg, U.L.; Danjo, K.; King, H.O.; Scott, G.B.; Robinson, P.A.; Crabtree, J.E. Epithelial cell ADAM17 activation by Helicobacter pylori: Role of ADAM17 C-terminus and Threonine-735 phosphorylation. Microbes Infect. 2015, 17, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimura, T.; Kataoka, H.; Ogasawara, N.; Kubota, E.; Sasaki, M.; Tanida, S.; Joh, T. Suppression of proHB-EGF carboxy-terminal fragment nuclear translocation: A new molecular target therapy for gastric cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 3956–3965. [Google Scholar] [CrossRef] [Green Version]
- Ebi, M.; Kataoka, H.; Shimura, T.; Kubota, E.; Hirata, Y.; Mizushima, T.; Mizoshita, T.; Tanaka, M.; Mabuchi, M.; Tsukamoto, H.; et al. TGFβ induces proHB-EGF shedding and EGFR transactivation through ADAM activation in gastric cancer cells. Biochem. Biophys. Res. Commun. 2010, 402, 449–454. [Google Scholar] [CrossRef]
- Tanida, S.; Joh, T.; Itoh, K.; Kataoka, H.; Sasaki, M.; Ohara, H.; Nakazawa, T.; Nomura, T.; Kinugasa, Y.; Ohmoto, H.; et al. The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology 2004, 127, 559–569. [Google Scholar] [CrossRef]
- Okumura, T.; Ericksen, R.E.; Takaishi, S.; Wang, S.S.W.; Dubeykovskiy, Z.; Shibata, W.; Betz, K.S.; Muthupalani, S.; Rogers, A.B.; Fox, J.G.; et al. K-ras mutation targeted to gastric tissue progenitor cells results in chronic inflammation, an altered microenvironment, and progression to intraepithelial neoplasia. Cancer Res. 2010, 70, 8435–8445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutto, M.; Chen, Z.; Bhat, A.A.; Wang, L.; Zhu, S.; Gomaa, A.; Bates, A.; Bhat, N.S.; Peng, D.; Belkhiri, A.; et al. Activation of STAT3 signaling is mediated by TFF1 silencing in gastric neoplasia. Nat. Commun. 2019, 10, 3039. [Google Scholar] [CrossRef] [PubMed]
- Balic, J.J.; Garbers, C.; Rose-John, S.; Yu, L.; Jenkins, B.J. Interleukin-11-driven gastric tumourigenesis is independent of trans-signalling. Cytokine 2017, 92, 118–123. [Google Scholar] [CrossRef]
- Nakagawa, M.; Nabeshima, K.; Asano, S.; Hamasaki, M.; Uesugi, N.; Tani, H.; Yamashita, Y.; Iwasaki, H. Up-regulated expression of ADAM17 in gastrointestinal stromal tumors: Coexpression with EGFR and EGFR ligands. Cancer Sci. 2009, 100, 654–662. [Google Scholar] [CrossRef]
- Mei, L.; Du, W.; Idowu, M.; von Mehren, M.; Boikos, S.A. Advances and Challenges on Management of Gastrointestinal Stromal Tumors. Front. Oncol. 2018, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, K.; Swendeman, S.; Saftig, P.; Blobel, C.P. Stimulation of platelet-derived growth factor receptor beta (PDGFRbeta) activates ADAM17 and promotes metalloproteinase-dependent cross-talk between the PDGFRbeta and epidermal growth factor receptor (EGFR) signaling pathways. J. Biol. Chem. 2010, 285, 25024–25032. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, D.; Sun, X.; Zhang, Y.; Wang, L.; Suo, J. ADAM17 promotes lymph node metastasis in gastric cancer via activation of the Notch and Wnt signaling pathways. Int. J. Mol. Med. 2019, 43, 914–926. [Google Scholar] [CrossRef]
- Ishimoto, T.; Miyake, K.; Nandi, T.; Yashiro, M.; Onishi, N.; Huang, K.K.; Lin, S.J.; Kalpana, R.; Tay, S.T.; Suzuki, Y.; et al. Activation of Transforming Growth Factor Beta 1 Signaling in Gastric Cancer-associated Fibroblasts Increases Their Motility, via Expression of Rhomboid 5 Homolog 2, and Ability to Induce Invasiveness of Gastric Cancer Cells. Gastroenterology 2017, 153, 191–204.e16. [Google Scholar] [CrossRef]
- Dempsey, P.J. Role of ADAM10 in intestinal crypt homeostasis and tumorigenesis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Brindley, C.D.; Elder, N.H.; Myers, M.G.; Rajala, M.W.; Dekaney, C.M.; McNamee, E.N.; Frey, M.R.; Shroyer, N.F.; Dempsey, P.J. Cellular Plasticity of Defa4Cre-Expressing Paneth Cells in Response to Notch Activation and Intestinal Injury. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 533–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanas, G.; Cortina, C.; Sevillano, M.; Batlle, E. Cleavage of E-cadherin by ADAM10 mediates epithelial cell sorting downstream of EphB signalling. Nat. Cell Biol. 2011, 13, 1100–1107. [Google Scholar] [CrossRef]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Horiuchi, K.; Sasaki, A.; Tsukamoto, T.; Okabayashi, K.; Hasegawa, H.; Kitagawa, Y.; Okada, Y. Epithelial Cell-Derived a Disintegrin and Metalloproteinase-17 Confers Resistance to Colonic Inflammation Through EGFR Activation. EBioMedicine 2016, 5, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Hofseth, L.J.; Hebert, J.R.; Chanda, A.; Chen, H.; Love, B.L.; Pena, M.M.; Murphy, E.A.; Sajish, M.; Sheth, A.; Buckhaults, P.J.; et al. Early-onset colorectal cancer: Initial clues and current views. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 352–364. [Google Scholar] [CrossRef]
- Merchant, N.B.; Voskresensky, I.; Rogers, C.M.; LaFleur, B.; Dempsey, P.J.; Graves-Deal, R.; Revetta, F.; Foutch, A.C.; Rothenberg, M.L.; Washington, M.K.; et al. TACE/ADAM-17, A Component of the Epidermal Growth Factor Receptor Axis and a Promising Therapeutic Target in Colorectal Cancer. Clin. Cancer Res. 2008, 14, 1182–1191. [Google Scholar] [CrossRef] [Green Version]
- Srivatsa, S.; Paul, M.C.; Cardone, C.; Holcmann, M.; Amberg, N.; Pathria, P.; Diamanti, M.A.; Linder, M.; Timelthaler, G.; Dienes, H.P.; et al. EGFR in Tumor-Associated Myeloid Cells Promotes Development of Colorectal Cancer in Mice and Associates With Outcomes of Patients. Gastroenterology 2017, 153, 178–190.e10. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Czarnek, M.; Bzowska, M.; Mężyk-Kopeć, R.; Stalińska, K.; Wyroba, B.; Sroka, J.; Jucha, J.; Deneka, D.; Stokłosa, P.; et al. ADAM17 silencing in mouse colon carcinoma cells: The effect on tumoricidal cytokines and angiogenesis. PLoS ONE 2012, 7, e50791. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Ardito, C.; Grüner, B.; Takeuchi, K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.; DelGiorno, K.; Carpenter, E.; Halbrook, C.; Hall, J.; et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.; Sibilia, M.; Guerra, C.; Barbacid, M. EGF Receptor Signaling Is Essential for K-Ras Oncogene-Driven Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Ringel, J.; Jesnowski, R.; Moniaux, N.; Lüttges, J.; Ringel, J.; Choudhury, A.; Batra, S.K.; Klöppel, G.; Löhr, M. Aberrant expression of a disintegrin and metalloproteinase 17/tumor necrosis factor-alpha converting enzyme increases the malignant potential in human pancreatic ductal adenocarcinoma. Cancer Res. 2006, 66, 9045–9053. [Google Scholar] [CrossRef] [Green Version]
- Nagathihalli, N.S.; Beesetty, Y.; Lee, W.; Washington, M.K.; Chen, X.; Lockhart, A.C.; Merchant, N.B. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-α: Impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res. 2014, 74, 2062–2072. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Yuen, S.M.; Murphy, G.; Xie, R.; Kwok, H.F. Anti-tumor effects of a ‘human & mouse cross-reactive’ anti-ADAM17 antibody in a pancreatic cancer model in vivo. Eur. J. Pharm. Sci. 2017, 110, 62–69. [Google Scholar] [CrossRef]
- Zhang, H.; Neuhöfer, P.; Song, L.; Rabe, B.; Lesina, M.; Kurkowski, M.U.; Treiber, M.; Wartmann, T.; Regnér, S.; Thorlacius, H.; et al. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J. Clin. Investig. 2013, 123, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Lesina, M.; Kurkowski, M.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Valkovskaya, N.; Kayed, H.; Felix, K.; Hartmann, D.; Giese, N.A.; Osinsky, S.P.; Friess, H.; Kleeff, J. ADAM8 expression is associated with increased invasiveness and reduced patient survival in pancreatic cancer. J. Cell. Mol. Med. 2007, 11, 1162–1174. [Google Scholar] [CrossRef]
- Oria, V.O.; Lopatta, P.; Schmitz, T.; Preca, B.-T.; Nyström, A.; Conrad, C.; Bartsch, J.W.; Kulemann, B.; Hoeppner, J.; Maurer, J.; et al. ADAM9 contributes to vascular invasion in pancreatic ductal adenocarcinoma. Mol. Oncol. 2019, 13, 456–479. [Google Scholar] [CrossRef]
- Schlomann, U.; Koller, G.; Conrad, C.; Ferdous, T.; Golfi, P.; Garcia, A.M.; Höfling, S.; Parsons, M.; Costa, P.; Soper, R.; et al. ADAM8 as a drug target in pancreatic cancer. Nat. Commun. 2015, 6, 6175. [Google Scholar] [CrossRef] [Green Version]
- Gilgenkrantz, H.; Collin de l’Hortet, A. Understanding Liver Regeneration: From Mechanisms to Regenerative Medicine. Am. J. Pathol. 2018, 188, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Boulter, L.; Lu, W.-Y.; Forbes, S.J. Differentiation of progenitors in the liver: A matter of local choice. J. Clin. Investig. 2013, 123, 1867–1873. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Wetzel, S.; Köhn-Gaone, J.; Chalupsky, K.; Lüllmann-Rauch, R.; Barikbin, R.; Bergmann, J.; Wöhner, B.; Zbodakova, O.; Leuschner, I.; et al. A disintegrin and metalloprotease 10 (ADAM10) is a central regulator of murine liver tissue homeostasis. Oncotarget 2016, 7, 17431–17441. [Google Scholar] [CrossRef] [PubMed]
- Geisler, F.; Nagl, F.; Mazur, P.K.; Lee, M.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Schmid, R.M.; Siveke, J.T. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology 2008, 48, 607–616. [Google Scholar] [CrossRef]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Bolik, J.; Tirnitz-Parker, J.E.E.; Schmidt-Arras, D. ADAM and ADAMTS Proteases in Hepatic Disorders. J. Ren. Hepatic Disord. 2019, 3, 23–32. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. Regulation of Fibrotic Processes in the Liver by ADAM Proteases. Cells 2019, 8, 1226. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, B.; Behnke, K.; Belancic, A.; Al-Salihi, M.A.; Thabet, Y.; Polz, R.; Pellegrino, R.; Zhuang, Y.; Shinde, P.V.; Xu, H.C.; et al. iRhom2 inhibits bile duct obstruction-induced liver fibrosis. Sci. Signal. 2019, 12, eaax1194. [Google Scholar] [CrossRef]
- Le Pabic, H.; Bonnier, D.; Wewer, U.M.; Coutand, A.; Musso, O.; Baffet, G.; Clément, B.; Théret, N. ADAM12 in human liver cancers: TGF-beta-regulated expression in stellate cells is associated with matrix remodeling. Hepatology 2003, 37, 1056–1066. [Google Scholar] [CrossRef]
- Daduang, J.; Limpaiboon, T.; Daduang, S. Biomarker to distinguish hepatocellular carcinoma from cholangiocarcinoma by serum a disintegrin and metalloprotease 12. Arch. Med Sci. AMS 2011, 7, 1013–1016. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, S.; Liu, K.; Wang, Y.; Ji, B.; Zhang, X.; Liu, Y. A disintegrin and metalloprotease (ADAM)10 is highly expressed in hepatocellular carcinoma and is associated with tumour progression. J. Int. Med Res. 2014, 42, 611–618. [Google Scholar] [CrossRef]
- Liu, R.; Lauridsen, H.M.; Amezquita, R.A.; Pierce, R.W.; Jane-Wit, D.; Fang, C.; Pellowe, A.S.; Kirkiles-Smith, N.C.; Gonzalez, A.L.; Pober, J.S. IL-17 Promotes Neutrophil-Mediated Immunity by Activating Microvascular Pericytes and Not Endothelium. J. Immunol. 2016, 197, 2400–2408. [Google Scholar] [CrossRef]
- Liu, C.; Ren, Y.-F.; Dong, J.; Ke, M.-Y.; Ma, F.; Monga, S.P.S.; Wu, R.; Lv, Y.; Zhang, X.-F. Activation of SRY accounts for male-specific hepatocarcinogenesis: Implication in gender disparity of hepatocellular carcinoma. Cancer Lett. 2017, 410, 20–31. [Google Scholar] [CrossRef]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sörensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Guedj, A.; Volman, Y.; Geiger-Maor, A.; Bolik, J.; Schumacher, N.; Künzel, S.; Baines, J.F.; Nevo, Y.; Elgavish, S.; Galun, E.; et al. Gut microbiota shape ‘inflamm-ageing’ cytokines and account for age-dependent decline in DNA damage repair. Gut 2020, 69, 1064–1075. [Google Scholar] [CrossRef]
- Lanaya, H.; Natarajan, A.; Komposch, K.; Li, L.; Amberg, N.; Chen, L.; Wculek, S.K.; Hammer, M.; Zenz, R.; Peck-Radosavljevic, M.; et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat. Cell Biol. 2014, 16, 972–977. [Google Scholar] [CrossRef]
- Soresi, M.; Giannitrapani, L.; D’Antona, F.; Florena, A.-M.; La Spada, E.; Terranova, A.; Cervello, M.; D’Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 2563–2568. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Boeing, H.; Nöthlings, U.; Jenab, M.; Fedirko, V.; Kaaks, R.; Lukanova, A.; Trichopoulou, A.; Trichopoulos, D.; Boffetta, P.; et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology 2014, 60, 858–871. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, J.; Müller, M.; Baumann, N.; Reichert, M.; Heneweer, C.; Bolik, J.; Lücke, K.; Gruber, S.; Carambia, A.; Boretius, S.; et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017, 65, 89–103. [Google Scholar] [CrossRef]
- Moss, M.L.; Sklair-Tavron, L.; Nudelman, R. Drug insight: Tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 2008, 4, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, N.; Robev, D.; Himanen, J.P.; Nikolov, D.B. ADAM proteases: Emerging role and targeting of the non-catalytic domains. Cancer Lett. 2019, 467, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M.; Tape, C.J.; Jodrell, D.I.; Murphy, G. Anti-Tumour Effects of a Specific Anti-ADAM17 Antibody in an Ovarian Cancer Model In Vivo. PLoS ONE 2012, 7, e40597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Benaich, N.; Tape, C.; Kwok, H.F.; Murphy, G. Targeting the sheddase activity of ADAM17 by an anti-ADAM17 antibody D1(A12) inhibits head and neck squamous cell carcinoma cell proliferation and motility via blockage of bradykinin induced HERs transactivation. Int. J. Biol. Sci. 2014, 10, 702–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios-Doria, J.; Sabol, D.; Chesebrough, J.; Stewart, D.; Xu, L.; Tammali, R.; Cheng, L.; Du, Q.; Schifferli, K.; Rothstein, R.; et al. A Monoclonal Antibody to ADAM17 Inhibits Tumor Growth by Inhibiting EGFR and Non–EGFR-Mediated Pathways. Mol. Cancer 2015, 14, 1637–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atapattu, L.; Saha, N.; Chheang, C.; Eissman, M.F.; Xu, K.; Vail, M.E.; Hii, L.; Llerena, C.; Liu, Z.; Horvay, K.; et al. An activated form of ADAM10 is tumor selective and regulates cancer stem-like cells and tumor growth. J. Exp. Med. 2016, 213, 1741–1757. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Bomar, M.; Liu, Q.; Sage, H.; Dempsey, P.; Lenhart, P.M.; Gillispie, P.A.; Stoeck, A.; Wildeboer, D.; Bartsch, J.W.; et al. The ADAM10 prodomain is a specific inhibitor of ADAM10 proteolytic activity and inhibits cellular shedding events. J. Biol. Chem. 2007, 282, 35712–35721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Moss, M.L.; Powell, G.; Petrovich, R.; Edwards, L.; Meyer, A.S.; Griffith, L.G.; Lauffenburger, D.A. Targeting autocrine HB-EGF signaling with specific ADAM12 inhibition using recombinant ADAM12 prodomain. Sci. Rep. 2015, 5, 15150. [Google Scholar] [CrossRef] [Green Version]
- Kefaloyianni, E.; Muthu, M.L.; Kaeppler, J.; Sun, X.; Sabbisetti, V.; Chalaris, A.; Rose-John, S.; Wong, E.; Sagi, I.; Waikar, S.S.; et al. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.I.; Alhayyani, S.; McLeod, L.; Yu, L.; Alanazi, M.; Deswaerte, V.; Tang, K.; Jarde, T.; Smith, J.A.; Prodanovic, Z.; et al. ADAM17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol. Med. 2019, 11, e9976. [Google Scholar] [CrossRef]
- Saad, M.I.; McLeod, L.; Yu, L.; Ebi, H.; Ruwanpura, S.; Sagi, I.; Rose-John, S.; Jenkins, B.J. The ADAM17 Protease Promotes Tobacco Smoke Carcinogen-induced Lung Tumourigenesis. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Yamamoto, K.; Trad, A.; Baumgart, A.; Hüske, L.; Lorenzen, I.; Chalaris, A.; Grötzinger, J.; Dechow, T.; Scheller, J.; Rose-John, S. A novel bispecific single-chain antibody for ADAM17 and CD3 induces T-cell-mediated lysis of prostate cancer cells. Biochem. J. 2012, 445, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Sinnathamby, G.; Zerfass, J.; Hafner, J.; Block, P.; Nickens, Z.; Hobeika, A.; Secord, A.A.; Lyerly, H.K.; Morse, M.A.; Philip, R. ADAM metallopeptidase domain 17 (ADAM17) is naturally processed through major histocompatibility complex (MHC) class I molecules and is a potential immunotherapeutic target in breast, ovarian and prostate cancers. Clin. Exp. Immunol. 2011, 163, 324–332. [Google Scholar] [CrossRef]
Figure 1.
ADAMs and ADAM-mediated signalling pathways. (a) General domain structure of ADAM proteases. Pro, pro-domain; Cat;catalytic domain; Dis, disintegrin domain; CR, cysteine-rich domain; EGF, EGF-like domain; TM, transmembrane domain; CD, cytoplasmic domain (b) Maturation of ADAM protease, notably ADAM10 and ADAM17 is facilitated by tetraspanins (Tspans) and inactive rhomboid proteases (iRhom), respectively. Activation of ADAM proteases, notably ADAM17, via C-terminal phosphorylation is mediated by protein kinase C (PKC) or the mitogen-activated kinase p38. (c) ADAM proteases have diverse substrates and are involved in cytokine/growth factor release, shedding of receptor molecules and the induction of intracellular signal transduction via limited proteolysis, followed by regulated intramembrane proteolysis (RIP).
Figure 1.
ADAMs and ADAM-mediated signalling pathways. (a) General domain structure of ADAM proteases. Pro, pro-domain; Cat;catalytic domain; Dis, disintegrin domain; CR, cysteine-rich domain; EGF, EGF-like domain; TM, transmembrane domain; CD, cytoplasmic domain (b) Maturation of ADAM protease, notably ADAM10 and ADAM17 is facilitated by tetraspanins (Tspans) and inactive rhomboid proteases (iRhom), respectively. Activation of ADAM proteases, notably ADAM17, via C-terminal phosphorylation is mediated by protein kinase C (PKC) or the mitogen-activated kinase p38. (c) ADAM proteases have diverse substrates and are involved in cytokine/growth factor release, shedding of receptor molecules and the induction of intracellular signal transduction via limited proteolysis, followed by regulated intramembrane proteolysis (RIP).

Figure 2.
Major ADAM-mediated signalling pathways in gastrointestinal cancer formation. (a) ADAM proteases in particular ADAM10 and ADAM17 promote gastric tumorigenesis mainly through activation of epidermal growth factor (EGF) receptor (EGFR) signalling on cancer cells and through inhibition of transforming growth factor (TGF) β receptor (TGFβR), thereby enhancing motility of cancer-associated fibroblasts (in purple). (b) Colonic tumorigenesis depends on Notch signalling promoted by ADAM10. In addition, myeloid ADAM17 is supposed to promote inflammatory signalling via proteolytic release of EGFR ligands, such as heparin-bound EGF (HB-EGF) and concomitant induction of interleukin (IL) 6 (green circles). In concert with the soluble IL-6 receptor (sIL-6R), IL-6 induces trans-signalling on colon epithelial cells. (c) EGFR signalling and IL-6 trans-signalling promotes progression of pancreatic intraepithelial neoplasia (PanIN) to pancreatic ductal adenocarcinoma (PDAC), presumably via ADAM17 activity. (d) DNA damage response (DDR) in hepatocytes is enhanced via EGFR signalling. Intestinal dysbiosis induces tumour necrosis factor (TNF) secretion from Kupffer cells (KCs), the liver-resident macrophages that impairs the beneficial effects of EGFR signalling on DDR. Both, EGFR ligands and TNF are presumably released by ADAM17 from KCs. Liver fibrosis is reduced by ADAM17 via impaired TNF receptor signalling in hepatic stellate cells. Hepatocarcinogenesis is promoted by IL-6 trans-signalling that is presumably induced by ADAM17 on KCs. Notch signalling mediated by ADAM10 in hepatocytes promotes hepatic tumorigenesis. Abbreviations: CAF: cancer associated fibroblast; EC: epithelial cell; GC: goblet cell; HSC: hepatic stellate cell; IEC: intestinal epithelial cell; iMB: intestinal microbiome ISC: intestinal stem cell; KC: Kupffer cell; PC: Paneth cell; TAM: tumour associated macrophage; TC: tumour cell.
Figure 2.
Major ADAM-mediated signalling pathways in gastrointestinal cancer formation. (a) ADAM proteases in particular ADAM10 and ADAM17 promote gastric tumorigenesis mainly through activation of epidermal growth factor (EGF) receptor (EGFR) signalling on cancer cells and through inhibition of transforming growth factor (TGF) β receptor (TGFβR), thereby enhancing motility of cancer-associated fibroblasts (in purple). (b) Colonic tumorigenesis depends on Notch signalling promoted by ADAM10. In addition, myeloid ADAM17 is supposed to promote inflammatory signalling via proteolytic release of EGFR ligands, such as heparin-bound EGF (HB-EGF) and concomitant induction of interleukin (IL) 6 (green circles). In concert with the soluble IL-6 receptor (sIL-6R), IL-6 induces trans-signalling on colon epithelial cells. (c) EGFR signalling and IL-6 trans-signalling promotes progression of pancreatic intraepithelial neoplasia (PanIN) to pancreatic ductal adenocarcinoma (PDAC), presumably via ADAM17 activity. (d) DNA damage response (DDR) in hepatocytes is enhanced via EGFR signalling. Intestinal dysbiosis induces tumour necrosis factor (TNF) secretion from Kupffer cells (KCs), the liver-resident macrophages that impairs the beneficial effects of EGFR signalling on DDR. Both, EGFR ligands and TNF are presumably released by ADAM17 from KCs. Liver fibrosis is reduced by ADAM17 via impaired TNF receptor signalling in hepatic stellate cells. Hepatocarcinogenesis is promoted by IL-6 trans-signalling that is presumably induced by ADAM17 on KCs. Notch signalling mediated by ADAM10 in hepatocytes promotes hepatic tumorigenesis. Abbreviations: CAF: cancer associated fibroblast; EC: epithelial cell; GC: goblet cell; HSC: hepatic stellate cell; IEC: intestinal epithelial cell; iMB: intestinal microbiome ISC: intestinal stem cell; KC: Kupffer cell; PC: Paneth cell; TAM: tumour associated macrophage; TC: tumour cell.

Figure 3.
ADAM proteases as potential therapeutic target in cancer therapy. (a) Most of small molecule compounds targeting ADAM protease catalytic domain are based on a hydroxamate structure (in red). The structure of the ADM17-specific inhibitor INCB7839 is shown as an example. (b) Recombinant pro-domain binds to the catalytic centre of its cognate ADAM protease thereby limiting access to the catalytic domain. Inhibition by pro-domains is considered to be highly specific. (c) Monoclonal antibodies targeting the catalytic site or regulatory exosites of ADAM proteases impair their catalytic activity. (d) ADAM protease can be target of immunotherapies. As an example, a bispecific T-cell engager (BiTE) single-chain antibody against CD3 and ADAM17 induces the T-cell-mediated killing of pancreatic cancer cells in vitro. Arrows indicate secretion of IL-2 or perforin/granzyme.
Figure 3.
ADAM proteases as potential therapeutic target in cancer therapy. (a) Most of small molecule compounds targeting ADAM protease catalytic domain are based on a hydroxamate structure (in red). The structure of the ADM17-specific inhibitor INCB7839 is shown as an example. (b) Recombinant pro-domain binds to the catalytic centre of its cognate ADAM protease thereby limiting access to the catalytic domain. Inhibition by pro-domains is considered to be highly specific. (c) Monoclonal antibodies targeting the catalytic site or regulatory exosites of ADAM proteases impair their catalytic activity. (d) ADAM protease can be target of immunotherapies. As an example, a bispecific T-cell engager (BiTE) single-chain antibody against CD3 and ADAM17 induces the T-cell-mediated killing of pancreatic cancer cells in vitro. Arrows indicate secretion of IL-2 or perforin/granzyme.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Classification of the human ADAM protease family.
| Proteolytically Active (HEXXHXXGXXH) | testis specific | ADAM 20,21,30 | |
| not testis specific | haematopoietic | ADAM 8,28 | |
| non haematopoietic | ADAM 9,10,12,15,17,19,33,DEC1 | ||
| Proteolytically Inactive | testis specific | ADAM 2,7, 18,29 | |
| not testis specific | ADAM 11,22,23,32 |
Table 2.
Overview of ADAM protease activity in gastroenterological tumours.
| ADAM ProTeases | Signals UpStream of ADAMs | Pathways Activated by ADAMs | Biological Effect | ||
|---|---|---|---|---|---|
| Gastric Cancer | ADAM9, 10,12,15,17,33↑ | IL-1β, IL-8, TGFβ | TC | AR, HB-EGF/EGFR↑ | tumour growth |
| ADAM17 | PDGFR/c-Kit | ||||
| ADAM17 | IL-1β, TNF/iRhom2 | CAF | TGFβR↓ | CAF, TC motility | |
| Colorectal Cancer | ADAM10 | n.i. | TC | Notch signalling↑ | block in differentiation, adenoma formation |
| ADAM17 | n.i. | TC | IL-6/gp130 trans-signalling↑ | tumour growth | |
| n.i. | EC | VEGF/VEGFR↑ | vascularisation | ||
| Pancreatic Cancer | ADAM8 | n.i. | TC | β1 integrin/ERK1/2 | tumour growth, TC motility |
| ADAM9 | n.i. | TC | integrins, HB-EGF/EGFR | vascularisation | |
| ADAM17 | deoxycholic acid | TC, SC | AR, TGFα/EGFR↑ | PDAC formation | |
| n.i. | TAM/TC | IL-6/gp130 trans-signalling↑ | PaNIN progression | ||
| Hepatic Cancer | ADAM10 | n.i. | TC | Notch signalling | tumour progression |
| ADAM17 | dysbiotic iMB | KC/HC | TNF/TNFR1/2↑ | reduced DNA damage repair | |
| ADAM17 | n.i. | KC/TC | IL-6/gp130 trans-signalling↑ | HCC formation | |
| ADAM17 | iRhom2 | HSC | TNF/TNFR1/2↓ | limiting fibrosis | |
Abbreviations used: CAF: cancer associated fibroblast, EC: endothelial cells, HC: hepatocyte, HSC: hepatic stellate cell, KC: Kupffer cell, TC: tumour cell, n.i.: not identified, all other abbreviations are spelled out in the main text. ↑ indicates up-regulated; ↓ indicates down-regulated.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schumacher, N.; Rose-John, S.; Schmidt-Arras, D. ADAM-Mediated Signalling Pathways in Gastrointestinal Cancer Formation. Int. J. Mol. Sci. 2020, 21, 5133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145133
AMA Style
Schumacher N, Rose-John S, Schmidt-Arras D. ADAM-Mediated Signalling Pathways in Gastrointestinal Cancer Formation. International Journal of Molecular Sciences. 2020; 21(14):5133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145133
Chicago/Turabian StyleSchumacher, Neele, Stefan Rose-John, and Dirk Schmidt-Arras. 2020. "ADAM-Mediated Signalling Pathways in Gastrointestinal Cancer Formation" International Journal of Molecular Sciences 21, no. 14: 5133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145133
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.