A Fish Leukocyte Immune-Type Receptor Uses a Novel Intracytoplasmic Tail Networking Mechanism to Cross-Inhibit the Phagocytic Response


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
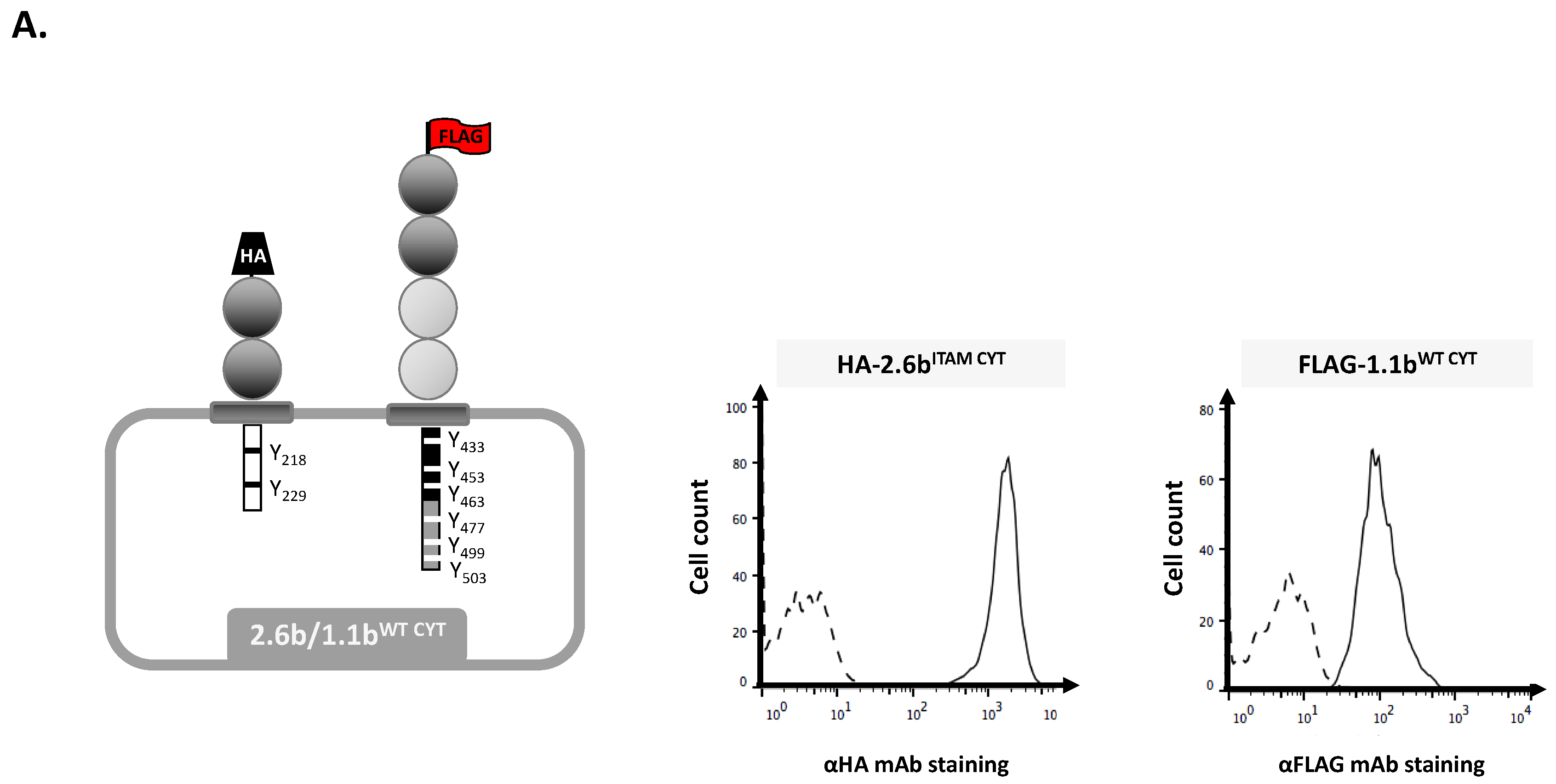
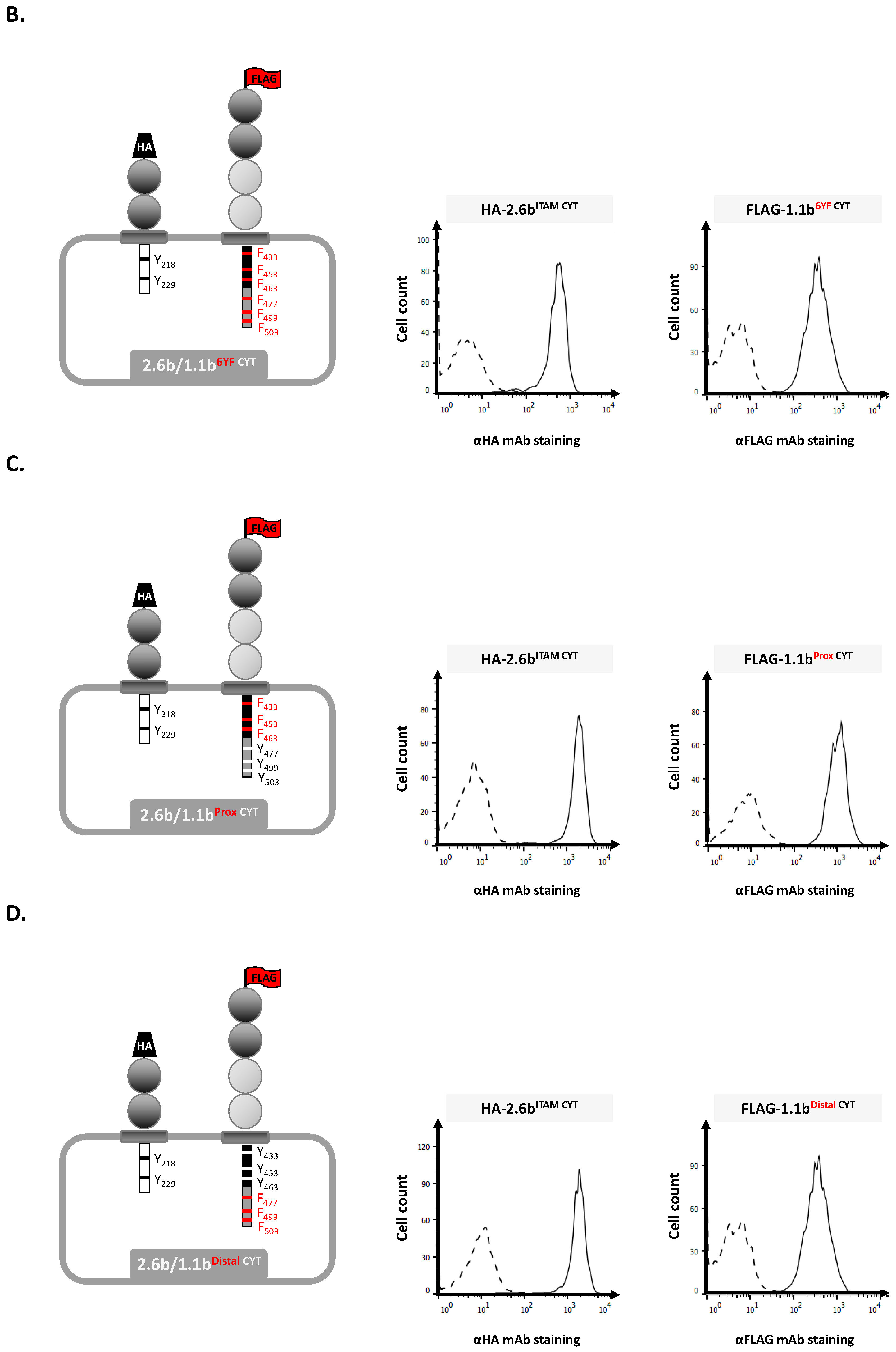
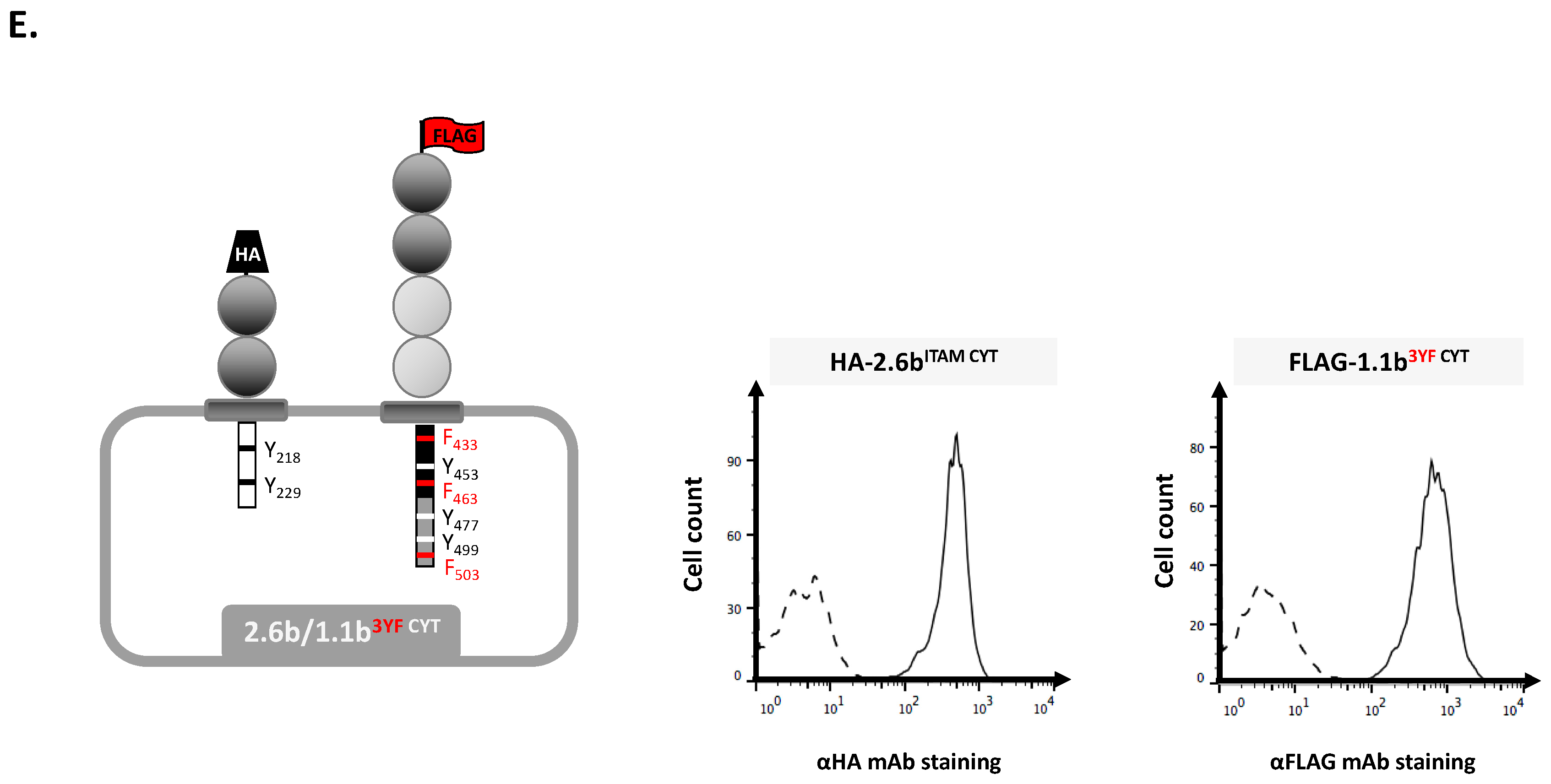
2.1. Examination of Independent Construct Activity in IpLITR Co-Expressing (2.6bITAM CYT/1.1bWT CYT) AD293 Cells Using Epitope Tag-Specific mAb-Opsonized Beads
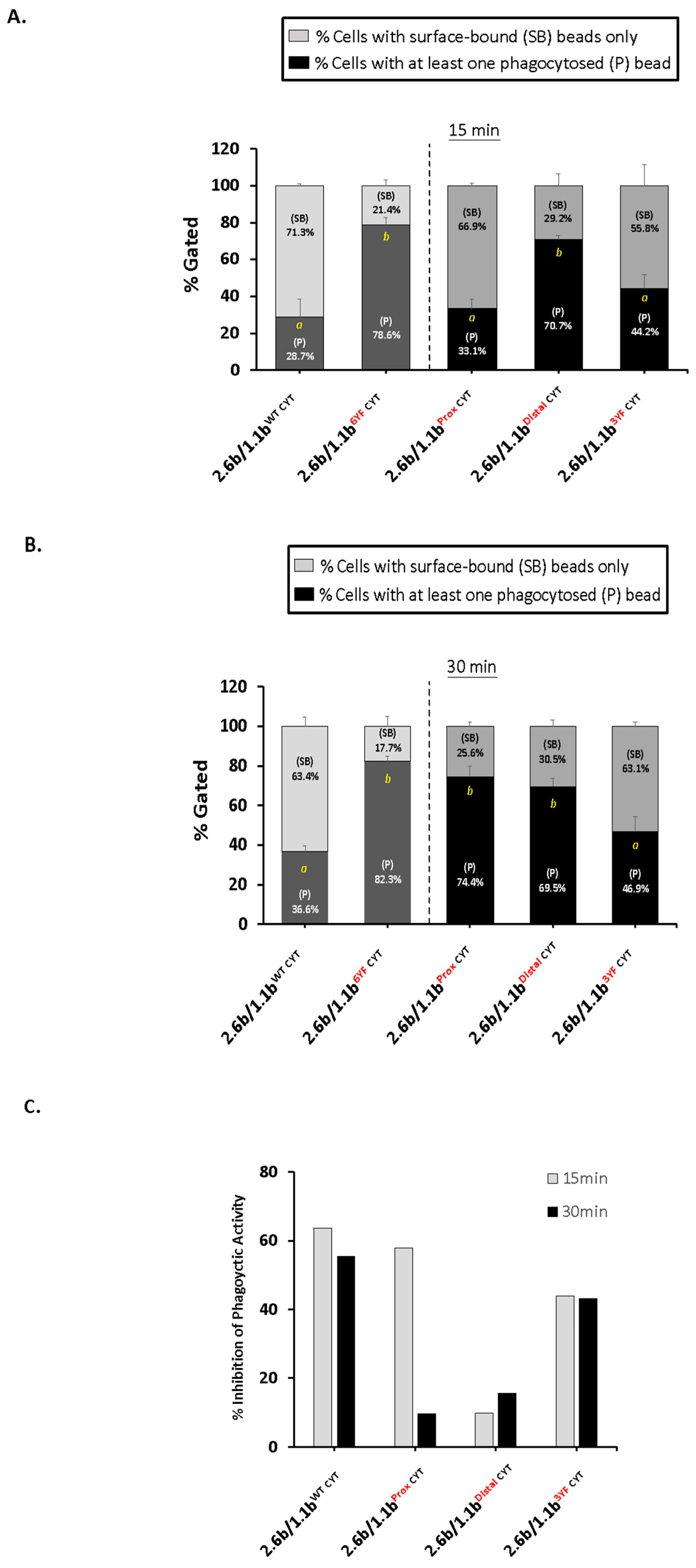
2.2. Co-Crosslinking 2.6bITAM CYT with 1.1bWT CYT Inhibits the ITAM-Mediated Phagocytic Response in IpLITR Co-Expressing AD293 Cells
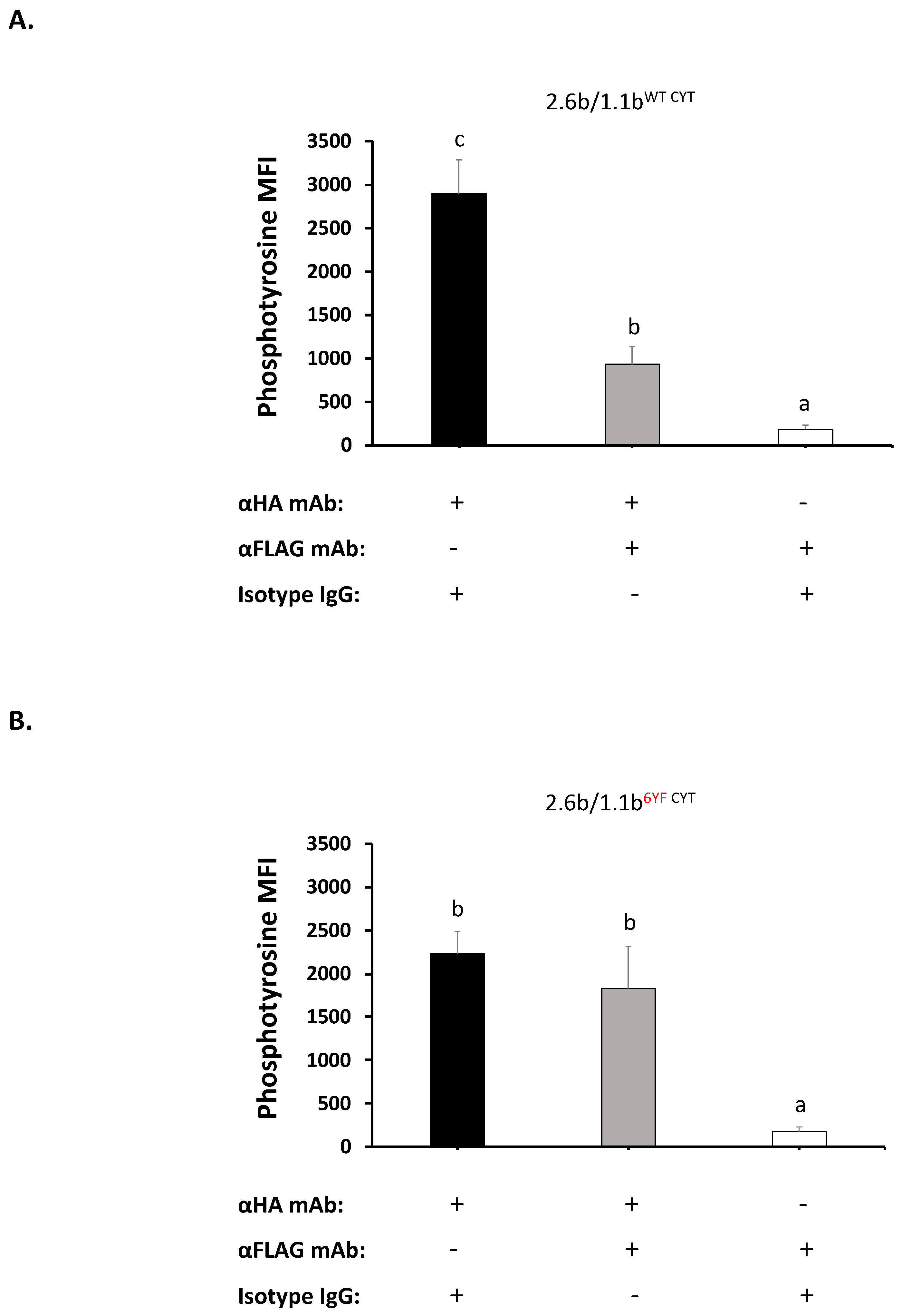
2.3. Co-Crosslinking 2.6bITAM CYT with 1.1bWT CYT Significantly Abrogates 2.6bITAM CYT-Induced Intracellular Phosphotyrosine Levels
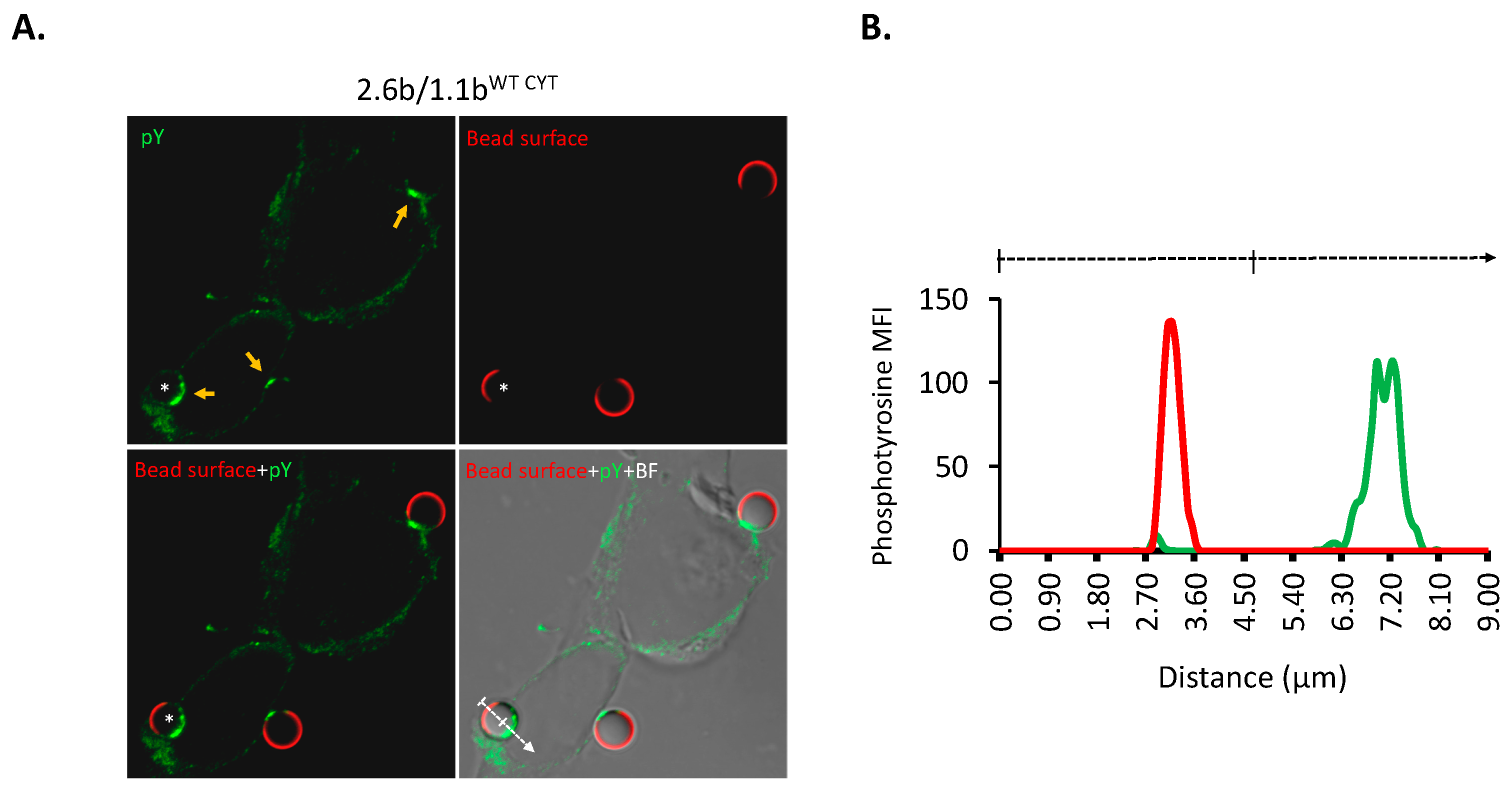
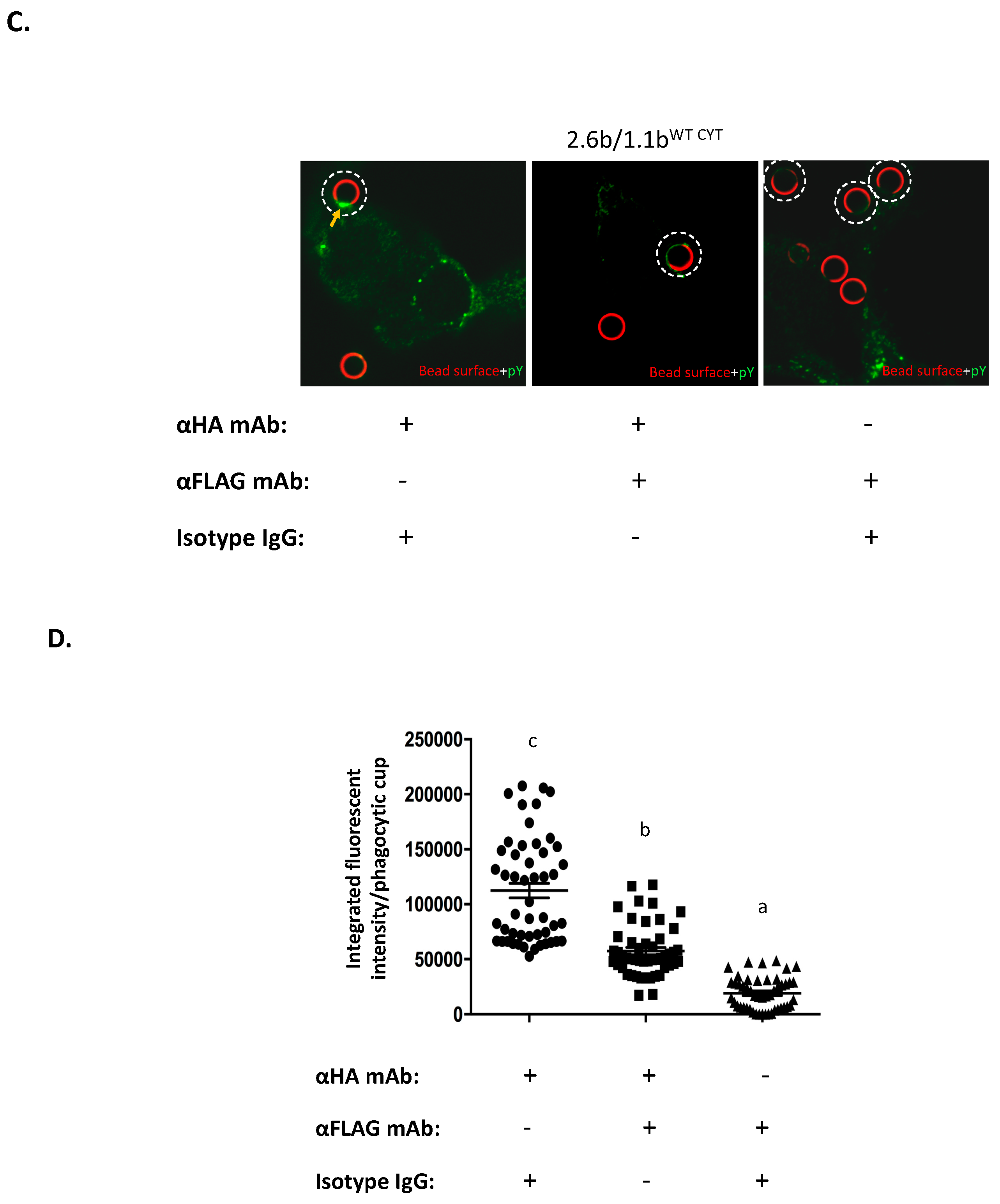
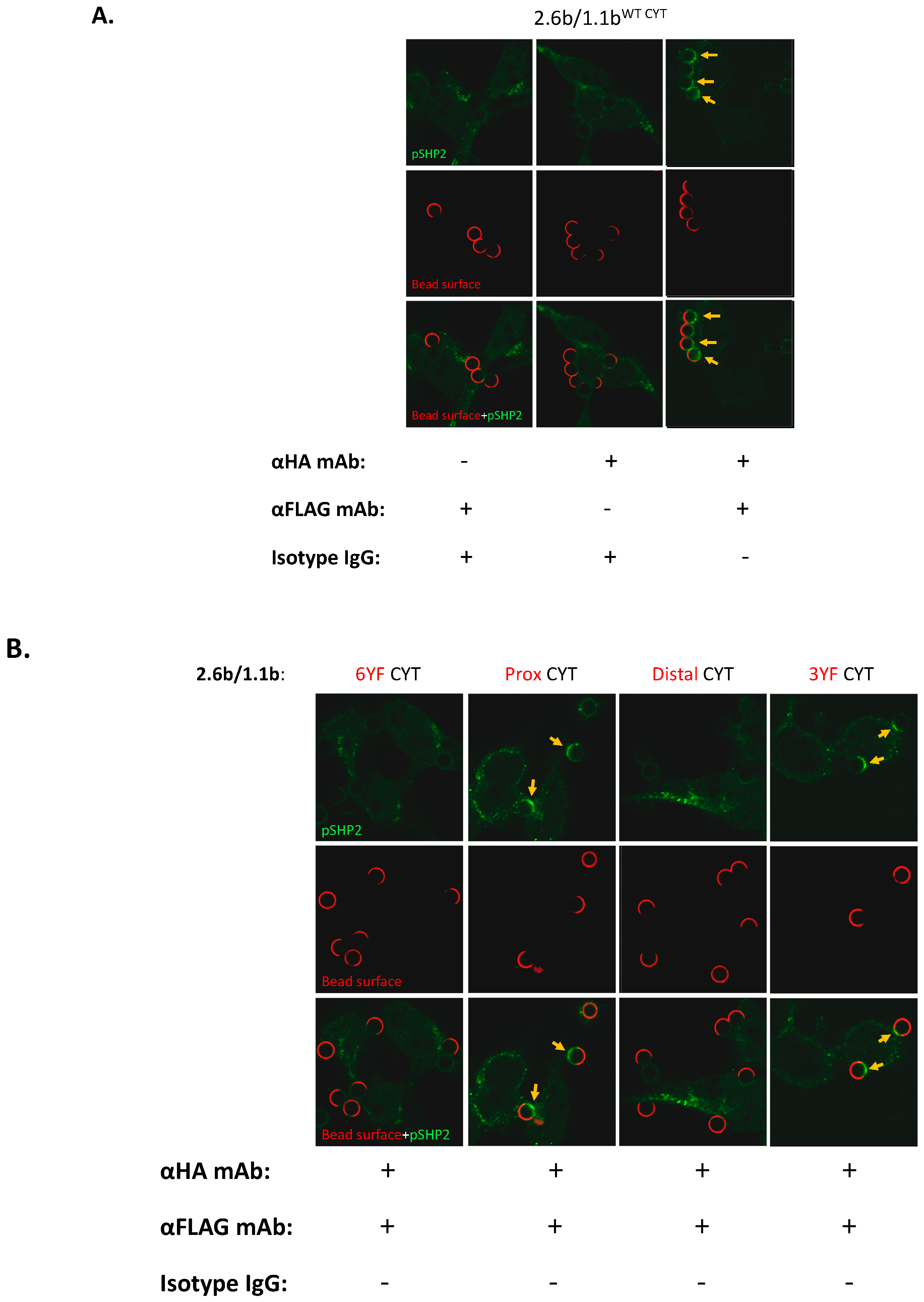
2.4. Examination of IpLITR-Mediated Tyrosine Phosphorylation Staining at Phagocytic Cups
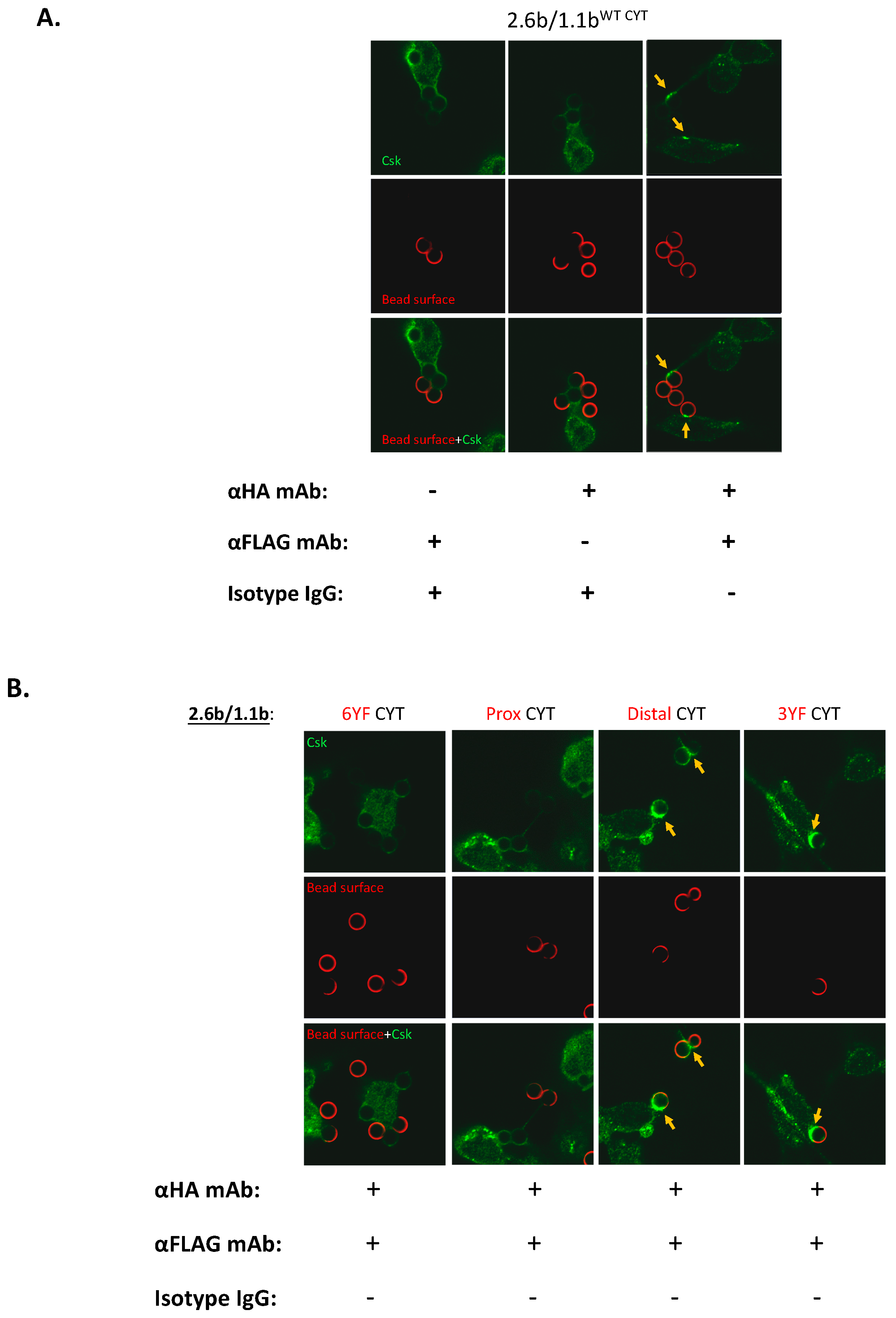
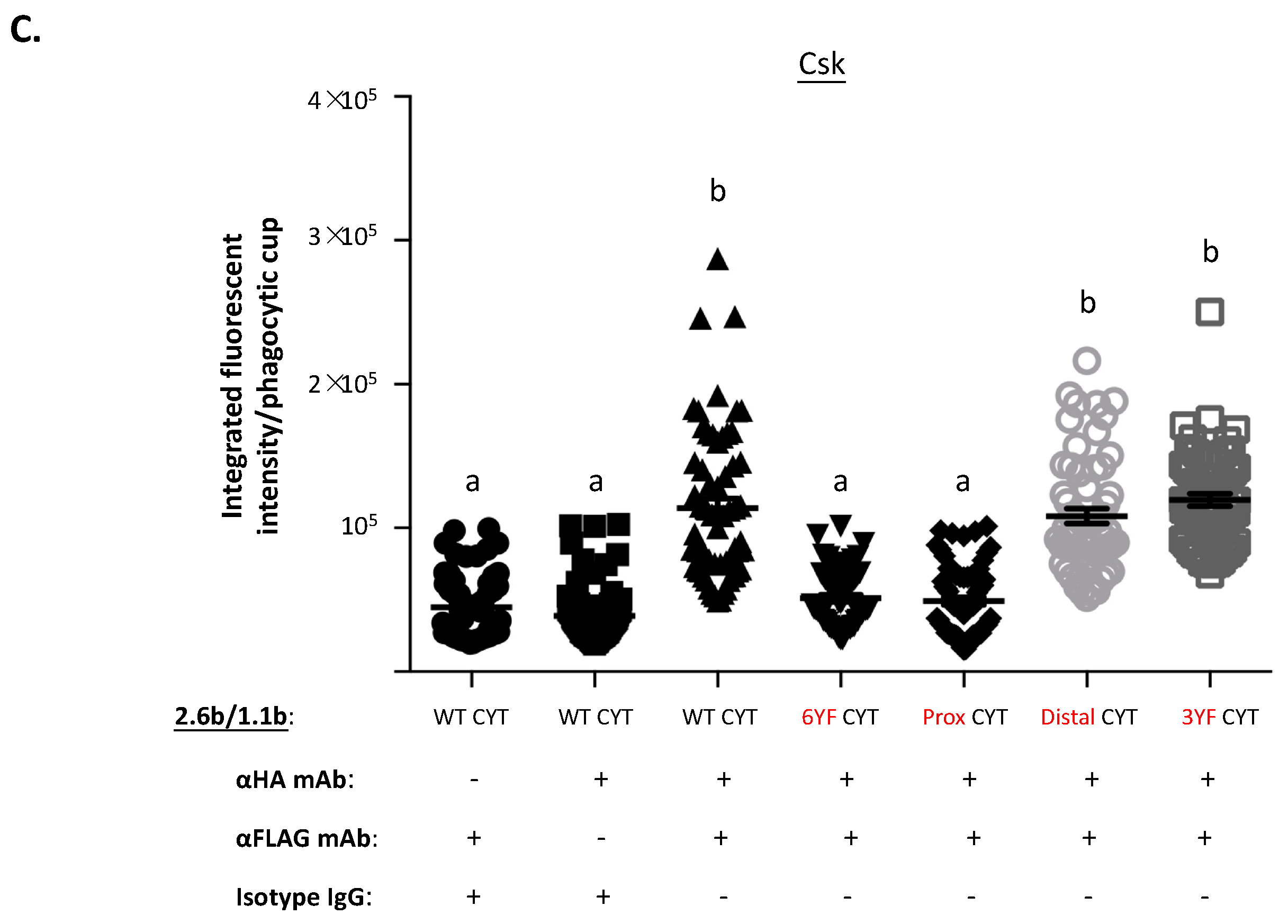
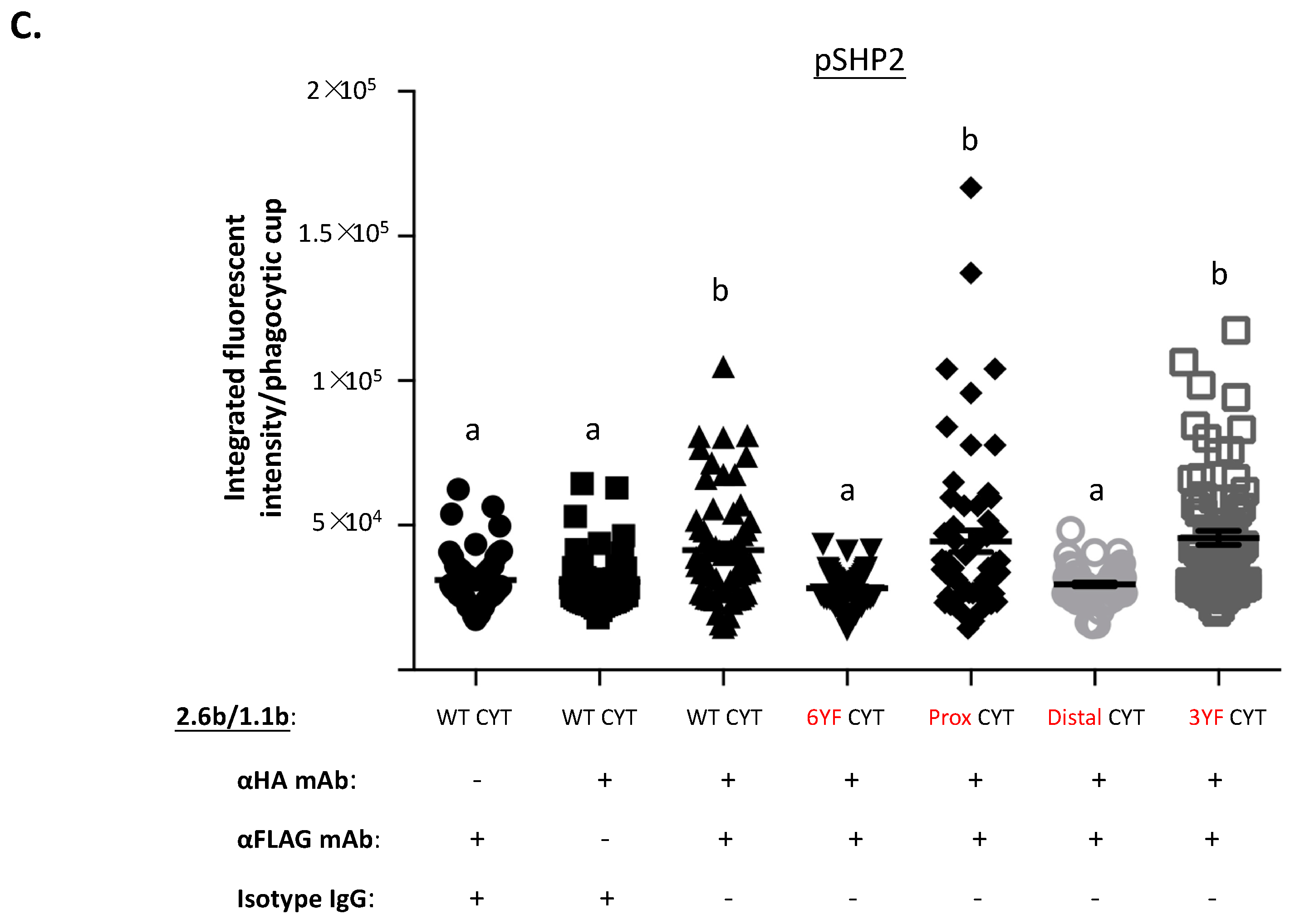
2.5. Examination of the Selective Recruitments of Csk and pSHP-2 Molecules to 1.1bCYT Constructs Using Confocal Microscopy
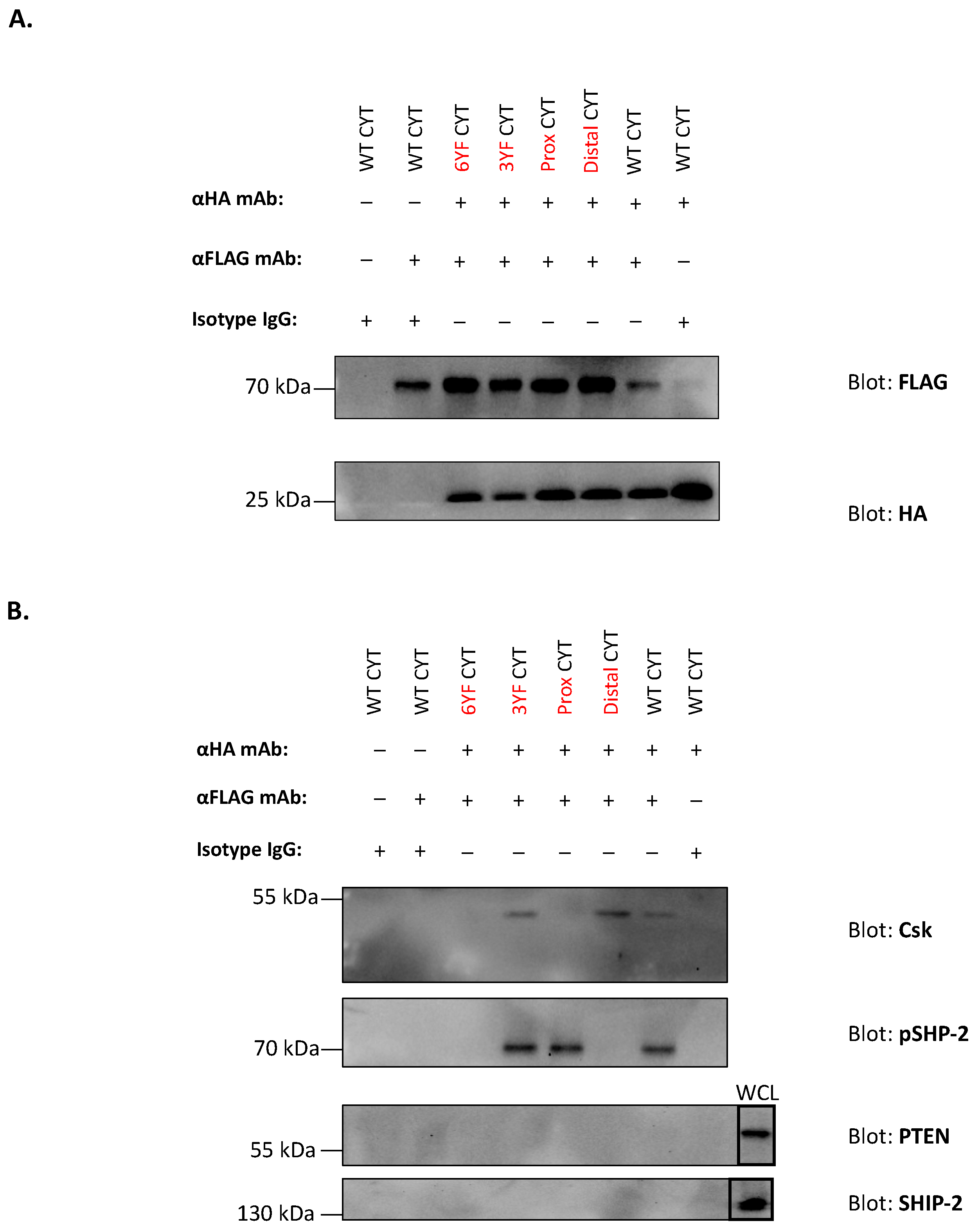
2.6. Co-Immunoprecipitation-Based Examination of the Selective Recruitments of Csk and pSHP-2 to 1.1bCYT Constructs during Co-Crosslinking with 2.6bITAM CYT
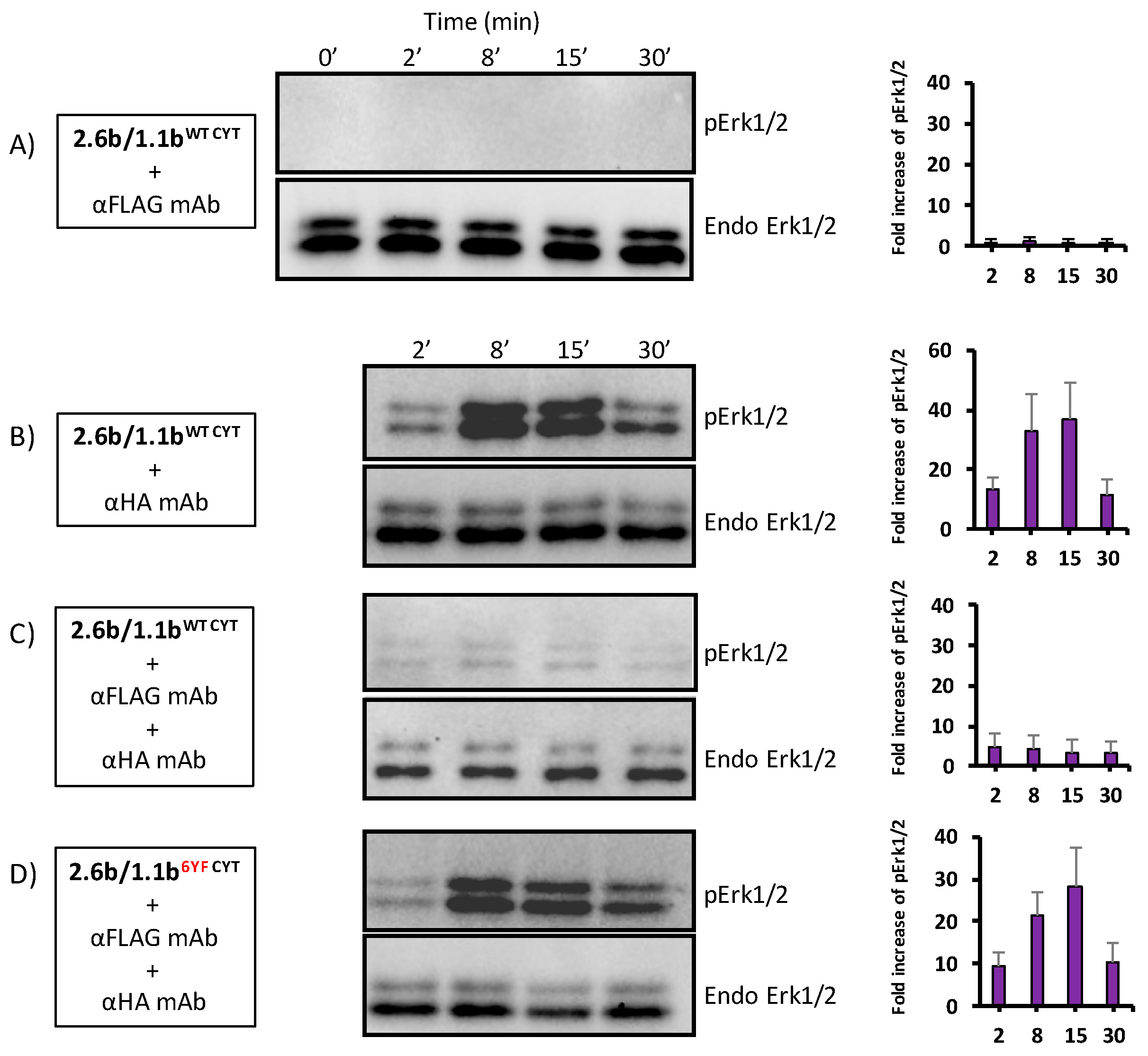
2.7. Inhibition of pErk1/2 Activation Occurs during IpLITR 1.1b-Mediated Cross-Talk Inhibition
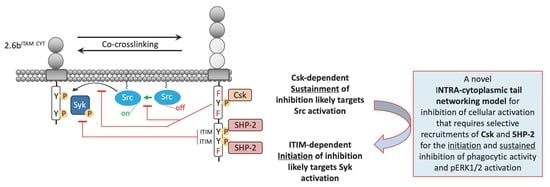
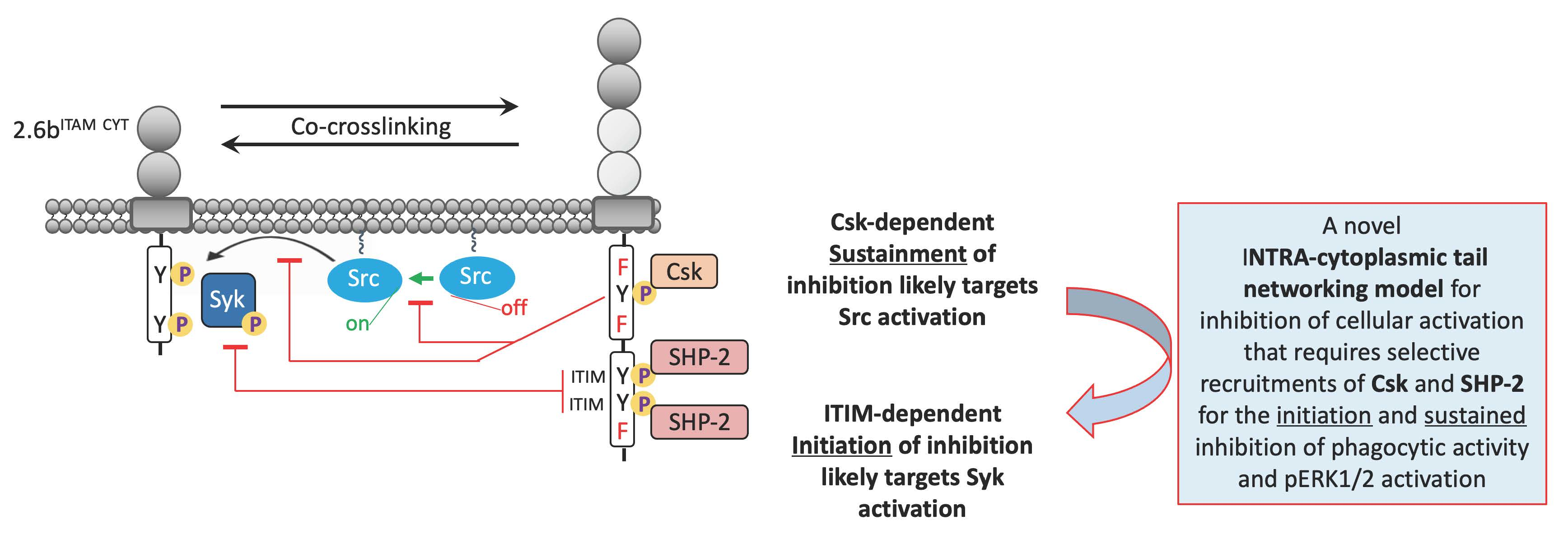
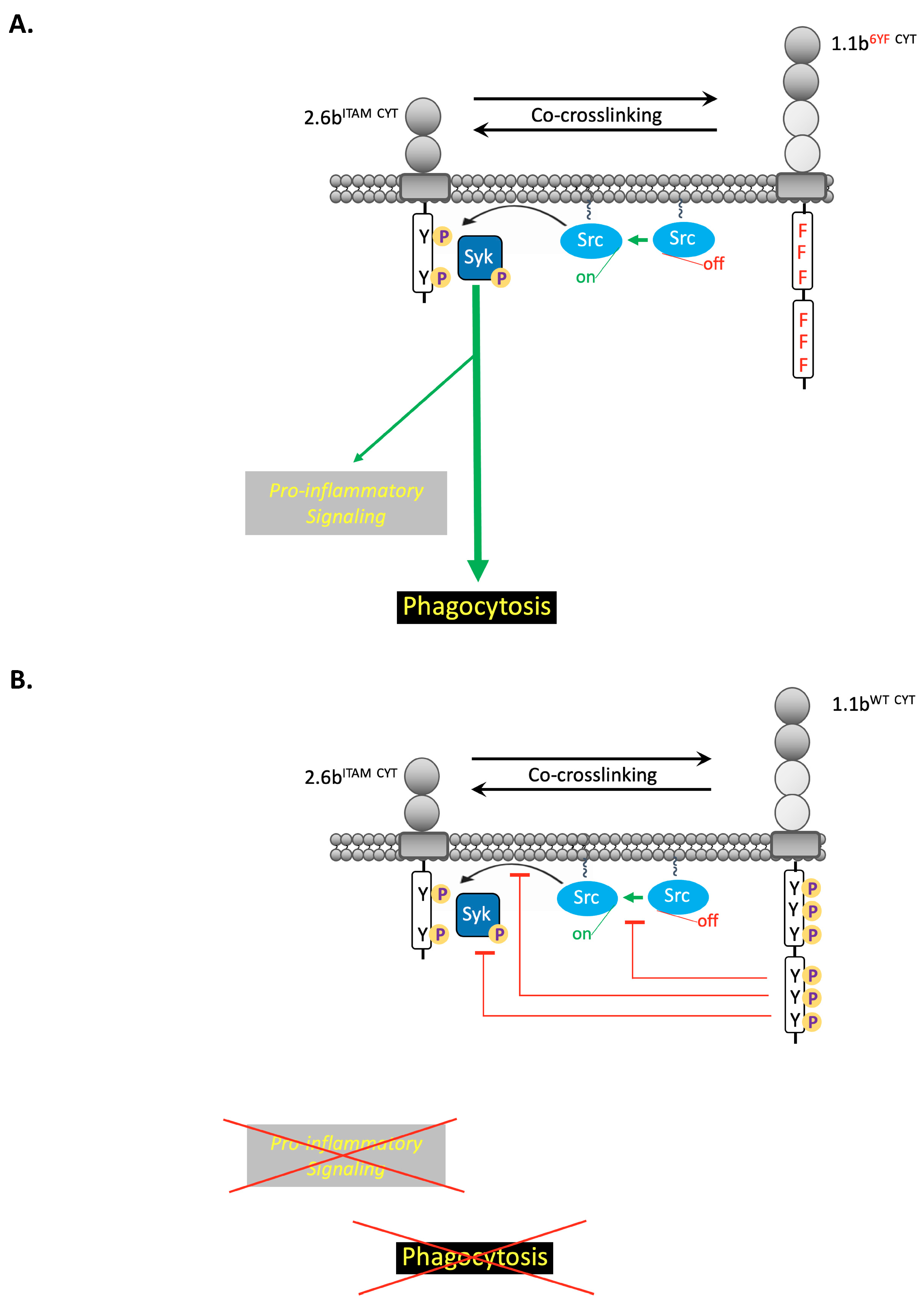
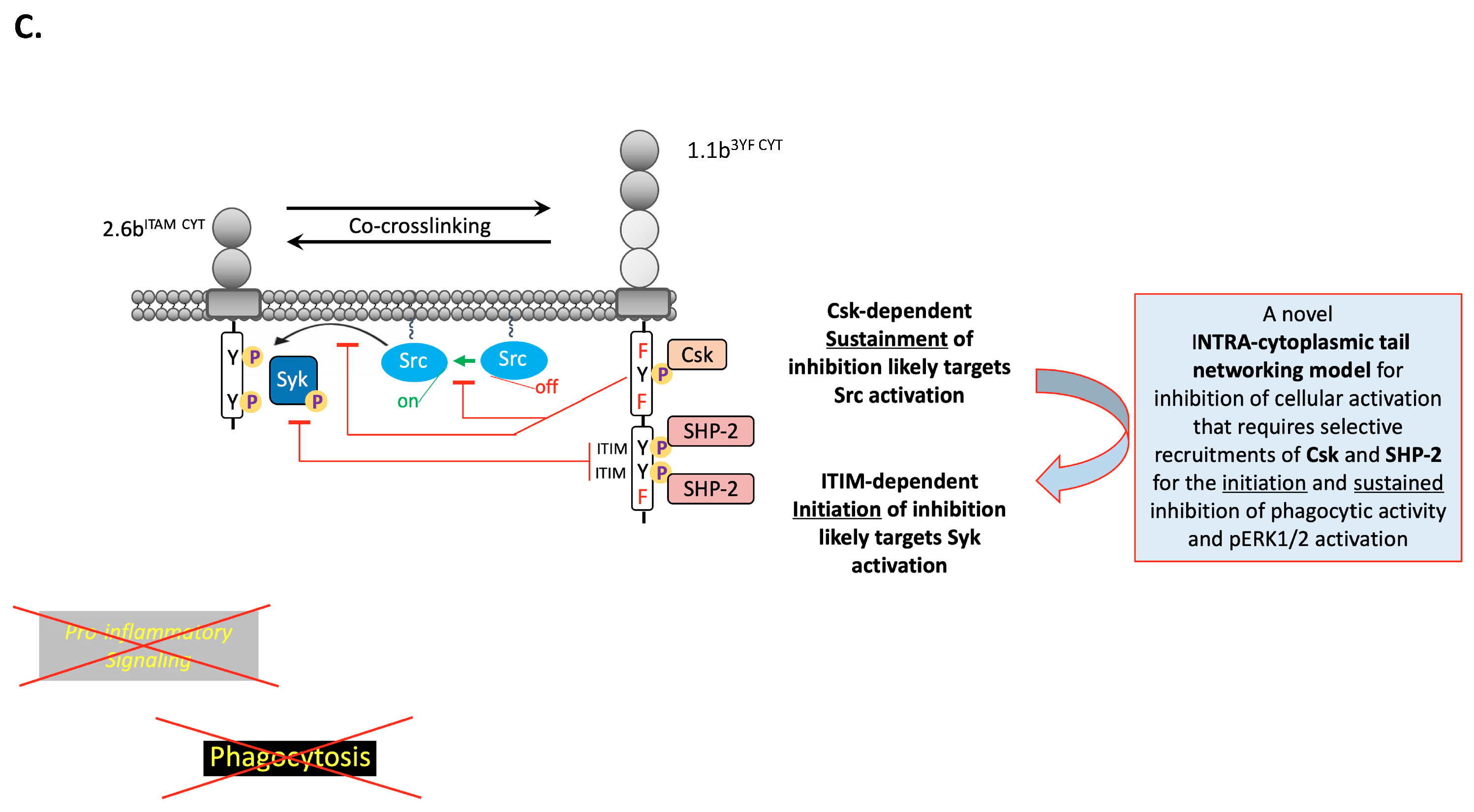
3. Discussion
4. Materials and Methods
4.1. Generation of AD293 Cell Lines Co-Expressing N-Terminal HA- and FLAG-Tagged IpLITR Constructs
4.2. Imaging Flow Cytometry-Based Bead-Binding and Phagocytosis Assays
4.3. Imaging Flow Cytometry-Based Intracellular Phospho-Tyrosine Staining
4.4. Confocal Microscopy-Based Examination of the Recruitment of Intracellular Signaling Molecules during IpLITR-Mediated Cross-Talk Regulation of Phagocytosis
4.5. Co-Immunoprecipitation of IpLITR Constructs with Intracellular Signaling Molecules
4.6. Examination of Erk1/2 Pathway Activation Following the Co-Crosslinking of 2.6bITAM CYT with Various 1.1bCYT Constructs
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASB | antibody staining buffer |
| BSA | bovine serum albumin |
| Csk | C-terminal src kinase |
| CYT | cytoplasmic tail |
| Erk1/2 | extracellular signal-regulated kinase 1/2 |
| FcRs | Fc receptors |
| FcRLs | FcR-like proteins |
| FBS | fetal bovine serum |
| HA | hemagglutinin |
| HRP | horseradish peroxidase |
| IpLITRs | Ictalurus punctatus leukocyte immune-type receptors |
| ITAM | immunoreceptor tyrosine-based activation motif |
| ITIM | immunoreceptor tyrosine-based inhibitory motif |
| ITSM | immunoreceptor tyrosine-based switch motif |
| LILRs | leukocyte Ig-like receptors |
| LRC | leukocyte receptor complex |
| LY | light-yellow |
| MFIs | mean fluorescent intensities |
| NF | non-fluorescent |
| PFA | paraformaldehyde |
| YF | tyrosine to phenylalanine |
| PTEN | phosphatase and tensin homolog |
| PLCγ1 | phospholipase Cγ1 |
| ROI | region of interest |
| SHIP-2 | Src homology 2 domain containing inositol 5′-phosphatase |
| SHP-2 | Src homology 2-containing protein tyrosine phosphatase 2 |
| TLRs | toll-like receptors |
| TM | transmembrane |
| YG | yellow-green |
References
- Flannagan, R.S.; Jaumouille, V.; Grinstein, S. The Cell Biology of Phagocytosis. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Rajaram, M.V.S.; Curry, H.M.; Leander, R.; Schlesinger, L.S. Fine Tuning Inflammation at the Front Door: Macrophage Complement Receptor 3-mediates Phagocytosis and Immune Suppression for Francisella tularensis. PLoS Pathog. 2013, 9, e1003114. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.A.; Grinstein, S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef]
- Underhill, D.M.; Goodridge, H.S. Information processing during phagocytosis. Nat. Rev. Immunol. 2012, 12, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Doshi, N.; Mitragotri, S. Macrophages recognize size and shape of their targets. PLoS ONE 2010, 5, e10051. [Google Scholar] [CrossRef] [Green Version]
- Brewer, J.M.; Pollock, K.G.J.; Tetley, L.; Russell, D.G. Vesicle size influences the trafficking, processing, and presentation of antigens in lipid vesicles. J. Immunol. 2004, 173, 6143–6150. [Google Scholar] [CrossRef] [Green Version]
- Underhill, D.M.; Gantner, B. Integration of Toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. 2004, 6, 1368–1373. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lambris, J.D. More than complementing Tolls: Complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol. Rev. 2016, 274, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Tridandapani, S.; Anderson, C.L. Regulation of Phagocytosis by FcγRIIb and Phosphatases. In Molecular Mechanisms of Phagocytosis; Springer: Berlin/Heidelberg, Germany, 2013; pp. 85–96. [Google Scholar]
- Hunter, S.; Indik, Z.K.; Kim, M.-K.; Cauley, M.D.; Park, J.-G.; Schreiber, A.D. Inhibition of Fcγ Receptor-Mediated Phagocytosis by a Nonphagocytic Fcγ Receptor. Blood 1998, 91, 1762–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, J.S.; Mosser, D.M. Stimulatory and inhibitory signals originatingfrom the macrophage Fcγ receptors. Microbes Infect. 2001, 3, 131–139. [Google Scholar] [CrossRef]
- Hamerman, J.A.; Lanier, L.L. Inhibition of Immune Responses by ITAM-Bearing Receptors. Sci. Signal. 2006, 2006, re1. [Google Scholar] [CrossRef]
- Nimmerjahn, F. Activating and inhibitory FcγRs in autoimmune disorders. Springer Semin. Immunopathol. 2006, 28, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.; Maizes, J.S.; Guinamard, R.; Ono, M.; Takai, T.; Ravetch, J.V. Modulation of Immune Complex–induced Inflammation In Vivo by the Coordinate Expression of Activation and Inhibitory Fc Receptors. J. Exp. Med. 1999, 189, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stafford, J.L.; Bengten, E.; Du Pasquier, L.; McIntosh, R.D.; Quiniou, S.M.; Clem, L.W.; Miller, N.W.; Wilson, M. A novel family of diversified immunoregulatory receptors in teleosts is homologous to both mammalian Fc receptors and molecules encoded within the leukocyte receptor complex. Immunogenetics 2006, 58, 758–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stafford, J.L.; Bengten, E.; Du Pasquier, L.; Miller, N.W.; Wilson, M. Channel catfish leukocyte immune-type receptors contain a putative MHC class I binding site. Immunogenetics 2007, 59, 77–91. [Google Scholar] [CrossRef]
- Cortes, H.D.; Lillico, D.M.; Zwozdesky, M.A.; Pemberton, J.G.; O’Brien, A.; Montgomery, B.C.; Wiersma, L.; Chang, J.P.; Stafford, J.L. Induction of phagocytosis and intracellular signaling by an inhibitory channel catfish leukocyte immune-type receptor: Evidence for immunoregulatory receptor functional plasticity in teleosts. J. Innate Immun. 2014, 6, 435–455. [Google Scholar] [CrossRef]
- Montgomery, B.C.; Mewes, J.; Davidson, C.; Burshtyn, D.N.; Stafford, J.L. Cell surface expression of channel catfish leukocyte immune-type receptors (IpLITRs) and recruitment of both Src homology 2 domain-containing protein tyrosine phosphatase (SHP)-1 and SHP-2. Dev. Comp. Immunol. 2009, 33, 570–582. [Google Scholar] [CrossRef]
- Montgomery, B.C.; Cortes, H.D.; Burshtyn, D.N.; Stafford, J.L. Channel catfish leukocyte immune-type receptor mediated inhibition of cellular cytotoxicity is facilitated by SHP-1-dependent and -independent mechanisms. Dev. Comp. Immunol. 2012, 37, 151–163. [Google Scholar] [CrossRef]
- Lillico, D.M.E.; Pemberton, J.G.; Niemand, R.; Stafford, J.L. Selective recruitment of Nck and Syk contribute to distinct leukocyte immune-type receptor-initiated target interactions. Cell. Signal. 2020, 66, 109443. [Google Scholar] [CrossRef]
- Mewes, J.; Verheijen, K.; Montgomery, B.C.; Stafford, J.L. Stimulatory catfish leukocyte immune-type receptors (IpLITRs) demonstrate a unique ability to associate with adaptor signaling proteins and participate in the formation of homo- and heterodimers. Mol. Immunol. 2009, 47, 318–331. [Google Scholar] [CrossRef]
- Cortes, H.D.; Montgomery, B.C.; Verheijen, K.; Garcia-Garcia, E.; Stafford, J.L. Examination of the stimulatory signaling potential of a channel catfish leukocyte immune-type receptor and associated adaptor. Dev. Comp. Immunol. 2012, 36, 62–73. [Google Scholar] [CrossRef]
- Lillico, D.M.; Zwozdesky, M.A.; Pemberton, J.G.; Deutscher, J.M.; Jones, L.O.; Chang, J.P.; Stafford, J.L. Teleost leukocyte immune-type receptors activate distinct phagocytic modes for target acquisition and engulfment. J. Leukoc. Biol. 2015, 98, 235–248. [Google Scholar] [CrossRef]
- Zwozdesky, M.A.; Fei, C.; Lillico, D.M.E.; Stafford, J.L. Imaging flow cytometry and GST pulldown assays provide new insights into channel catfish leukocyte immune-type receptor-mediated phagocytic pathways. Dev. Comp. Immunol. 2017, 67, 126–138. [Google Scholar] [CrossRef]
- Lillico, D.M.E.; Pemberton, J.G.; Stafford, J.L. Selective Regulation of Cytoskeletal Dynamics and Filopodia Formation by Teleost Leukocyte Immune-Type Receptors Differentially Contributes to Target Capture During the Phagocytic Process. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Jones, L.O.; Stafford, J.L. Imaging flow cytometry and confocal microscopy-based examination of F-actin and phosphoinositide dynamics during leukocyte immune-type receptor-mediated phagocytic events. Dev. Comp. Immunol. 2019, 92, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Fei, C.; Lillico, D.M.E.; Hall, B.; Rieger, A.M.; Stafford, J.L. Connected component masking accurately identifies the ratio of phagocytosed and cell-bound particles in individual cells by imaging flow cytometry. Cytom. Part A 2017, 91, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flannagan, R.S.; Canton, J.; Furuya, W.; Glogauer, M.; Grinstein, S. The phosphatidylserine receptor TIM4 utilizes integrins as coreceptors to effect phagocytosis. MBoC 2014, 25, 1511–1522. [Google Scholar] [CrossRef]
- Tridandapani, S.; Wang, Y.; Marsh, C.B.; Anderson, C.L. Src homology 2 domain-containing inositol polyphosphate phosphatase regulates NF-kappa B-mediated gene transcription by phagocytic Fc gamma Rs in human myeloid cells. J. Immunol. 2002, 169, 4370–4378. [Google Scholar] [CrossRef]
- Nakamura, K.; Malykhin, A.; Coggeshall, K.M. The Src homology 2 domain-containing inositol 5-phosphatase negatively regulates Fcgamma receptor-mediated phagocytosis through immunoreceptor tyrosine-based activation motif-bearing phagocytic receptors. Blood 2002, 100, 3374–3382. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Dale, B.M.; Kashiwada, M.; Helgason, C.D.; Greenberg, S. A Regulatory Role for Src Homology 2 Domain–Containing Inositol 5′-Phosphatase (Ship) in Phagocytosis Mediated by Fcγ Receptors and Complement Receptor 3 (αMβ2; Cd11b/Cd18). J. Exp. Med. 2001, 193, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.-Y.; Hunter, S.; Kim, M.-K.; Indik, Z.K.; Schreiber, A.D. The effect of phosphatases SHP-1 and SHIP-1 on signaling by the ITIM- and ITAM-containing Fcγ receptors FcγRIIB and FcγRIIA. J. Leukoc. Biol. 2003, 73, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, C.P.; Tridandapani, S.; Helgason, C.D.; Humphries, R.K.; Krystal, G.; Marsh, C.B. The Inositol 5′-Phosphatase SHIP-1 and the Src Kinase Lyn Negatively Regulate Macrophage Colony-stimulating Factor-induced Akt Activity. J. Biol. Chem. 2003, 278, 38628–38636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, A.W.; Jones, M.L. A SHPing tale: Perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell. Signal. 2005, 17, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.S.; Hui, C.C.; Pawson, T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science 1993, 259, 1607–1611. [Google Scholar] [CrossRef]
- Vogel, W.; Lammers, R.; Huang, J.; Ullrich, A. Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science 1993, 259, 1611–1614. [Google Scholar] [CrossRef]
- Lu, W.; Gong, D.; Bar-Sagi, D.; Cole, P.A. Site-Specific Incorporation of a Phosphotyrosine Mimetic Reveals a Role for Tyrosine Phosphorylation of SHP-2 in Cell Signaling. Mol. Cell 2001, 8, 759–769. [Google Scholar] [CrossRef]
- Aman, M.J.; Walk, S.F.; March, M.E.; Su, H.-P.; Carver, D.J.; Ravichandran, K.S. Essential Role for the C-Terminal Noncatalytic Region of SHIP in FcγRIIB1-Mediated Inhibitory Signaling. Mol. Cell. Biol. 2000, 20, 3576–3589. [Google Scholar] [CrossRef] [PubMed]
- Damen, J.E.; Ware, M.D.; Kalesnikoff, J.; Hughes, M.R.; Krystal, G. SHIP’s C-terminus is essential for its hydrolysis of PIP3 and inhibition of mast cell degranulation. Blood 2001, 97, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Malbec, O.; Fong, D.C.; Turner, M.; Tybulewicz, V.L.J.; Cambier, J.C.; Fridman, W.H.; Daëron, M. Fcε Receptor I-Associated lyn-Dependent Phosphorylation of Fcγ Receptor IIB During Negative Regulation of Mast Cell Activation. J. Immunol. 1998, 160, 1647–1658. [Google Scholar]
- Malbec, O.; Cassard, L.; Albanesi, M.; Jönsson, F.; Mancardi, D.; Chicanne, G.; Payrastre, B.; Dubreuil, P.; Vivier, E.; Daëron, M. Trans-inhibition of activation and proliferation signals by Fc receptors in mast cells and basophils. Sci. Signal. 2016, 9, ra126. [Google Scholar] [CrossRef]
- Dréan, E.L.; Vély, F.; Olcese, L.; Cambiaggi, A.; Guia, S.; Krystal, G.; Gervois, N.; Moretta, A.; Jotereau, F.; Vivier, E. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: Association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. Eur. J. Immunol. 1998, 28, 264–276. [Google Scholar] [CrossRef]
- Marengère, L.E.M.; Waterhouse, P.; Duncan, G.S.; Mittrücker, H.-W.; Feng, G.-S.; Mak, T.W. Regulation of T Cell Receptor Signaling by Tyrosine Phosphatase SYP Association with CTLA-4. Science 1996, 272, 1170–1173. [Google Scholar] [CrossRef]
- Lee, K.-M.; Chuang, E.; Griffin, M.; Khattri, R.; Hong, D.K.; Zhang, W.; Straus, D.; Samelson, L.E.; Thompson, C.B.; Bluestone, J.A. Molecular Basis of T Cell Inactivation by CTLA-4. Science 1998, 282, 2263–2266. [Google Scholar] [CrossRef] [Green Version]
- Mócsai, A.; Ruland, J.; Tybulewicz, V.L.J. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef]
- Kiefer, F.; Brumell, J.; Al-Alawi, N.; Latour, S.; Cheng, A.; Veillette, A.; Grinstein, S.; Pawson, T. The Syk Protein Tyrosine Kinase Is Essential for Fcγ Receptor Signaling in Macrophages and Neutrophils. Mol. Cell. Biol. 1998, 18, 4209–4220. [Google Scholar] [CrossRef] [Green Version]
- Berton, G.; Mócsai, A.; Lowell, C.A. Src and Syk kinases: Key regulators of phagocytic cell activation. Trends Immunol. 2005, 26, 208–214. [Google Scholar] [CrossRef]
- Simon, M.; Vanes, L.; Geahlen, R.L.; Tybulewicz, V.L.J. Distinct roles for the linker region tyrosines of Syk in FcepsilonRI signaling in primary mast cells. J. Biol. Chem. 2005, 280, 4510–4517. [Google Scholar] [CrossRef] [Green Version]
- Moon, K.D.; Post, C.B.; Durden, D.L.; Zhou, Q.; De, P.; Harrison, M.L.; Geahlen, R.L. Molecular Basis for a Direct Interaction between the Syk Protein-tyrosine Kinase and Phosphoinositide 3-Kinase. J. Biol. Chem. 2005, 280, 1543–1551. [Google Scholar] [CrossRef] [Green Version]
- Greuber, E.K.; Pendergast, A.M. Abl Family Kinases Regulate FcγR-Mediated Phagocytosis in Murine Macrophages. J. Immunol. 2012, 189, 5382–5392. [Google Scholar] [CrossRef]
- Ren, L.; Chen, X.; Luechapanichkul, R.; Selner, N.G.; Meyer, T.M.; Wavreille, A.-S.; Chan, R.; Iorio, C.; Zhou, X.; Neel, B.G.; et al. Substrate Specificity of Protein Tyrosine Phosphatases 1B, RPTPα, SHP-1, and SHP-2. Biochemistry 2011, 50, 2339–2356. [Google Scholar] [CrossRef] [Green Version]
- Law, C.L.; Chandran, K.A.; Sidorenko, S.P.; Clark, E.A. Phospholipase C-gamma1 interacts with conserved phosphotyrosyl residues in the linker region of Syk and is a substrate for Syk. Mol. Cell. Biol. 1996, 16, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Pérez de Castro, I.; Ahearn, I.M.; Grana, T.M.; Chiu, V.K.; Lockyer, P.J.; Cullen, P.J.; Pellicer, A.; Cox, A.D.; Philips, M.R. Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nature 2003, 424, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Yang, W.; Kontaridis, M.I.; Bivona, T.G.; Wen, G.; Araki, T.; Luo, J.; Thompson, J.A.; Schraven, B.L.; Philips, M.R.; et al. Shp2 Regulates Src Family Kinase Activity and Ras/Erk Activation by Controlling Csk Recruitment. Mol. Cell 2004, 13, 341–355. [Google Scholar] [CrossRef]
- Chong, Y.-P.; Mulhern, T.D.; Cheng, H.-C. C-terminal Src kinase (CSK) and CSK-homologous kinase (CHK)—Endogenous negative regulators of Src-family protein kinases. Growth Factors 2005, 23, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Okada, M. Regulation of the Src Family Kinases by Csk. Int. J. Biol. Sci. 2012, 8, 1385–1397. [Google Scholar] [CrossRef] [Green Version]
- Ai, J.; Maturu, A.; Johnson, W.; Wang, Y.; Marsh, C.B.; Tridandapani, S. The inositol phosphatase SHIP-2 down-regulates FcγR-mediated phagocytosis in murine macrophages independently of SHIP-1. Blood 2006, 107, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Wei, G.; Fang, H.; Guo, J.; Weinstein, M.; Marsh, C.B.; Ostrowski, M.C.; Tridandapani, S. The Inositol 3-Phosphatase PTEN Negatively Regulates Fcγ Receptor Signaling, but Supports Toll-Like Receptor 4 Signaling in Murine Peritoneal Macrophages. J. Immunol. 2004, 172, 4851–4857. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Peng, X.; De, P.K.; Geahlen, R.L.; Durden, D.L. PTEN controls immunoreceptor (immunoreceptor tyrosine-based activation motif) signaling and the activation of Rac. Blood 2002, 99, 694–697. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; Wavreille, A.-S.; Justiniano, S.E.; Marsh, R.L.; Yu, J.; Burry, R.W.; Jarjoura, D.; Eubank, T.; Caligiuri, M.A.; Butchar, J.P.; et al. LyGDI, a Novel SHIP-Interacting Protein, Is a Negative Regulator of FcγR-Mediated Phagocytosis. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Strzelecka-Kiliszek, A.; Kwiatkowska, K.; Sobota, A. Lyn and Syk Kinases Are Sequentially Engaged in Phagocytosis Mediated by FcγR. J. Immunol. 2002, 169, 6787–6794. [Google Scholar] [CrossRef] [Green Version]
















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fei, C.; Zwozdesky, M.A.; Stafford, J.L. A Fish Leukocyte Immune-Type Receptor Uses a Novel Intracytoplasmic Tail Networking Mechanism to Cross-Inhibit the Phagocytic Response. Int. J. Mol. Sci. 2020, 21, 5146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145146
Fei C, Zwozdesky MA, Stafford JL. A Fish Leukocyte Immune-Type Receptor Uses a Novel Intracytoplasmic Tail Networking Mechanism to Cross-Inhibit the Phagocytic Response. International Journal of Molecular Sciences. 2020; 21(14):5146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145146
Chicago/Turabian StyleFei, Chenjie, Myron A. Zwozdesky, and James L. Stafford. 2020. "A Fish Leukocyte Immune-Type Receptor Uses a Novel Intracytoplasmic Tail Networking Mechanism to Cross-Inhibit the Phagocytic Response" International Journal of Molecular Sciences 21, no. 14: 5146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145146