Covid-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes

 , , and
, , and
Abstract
:1. Introduction
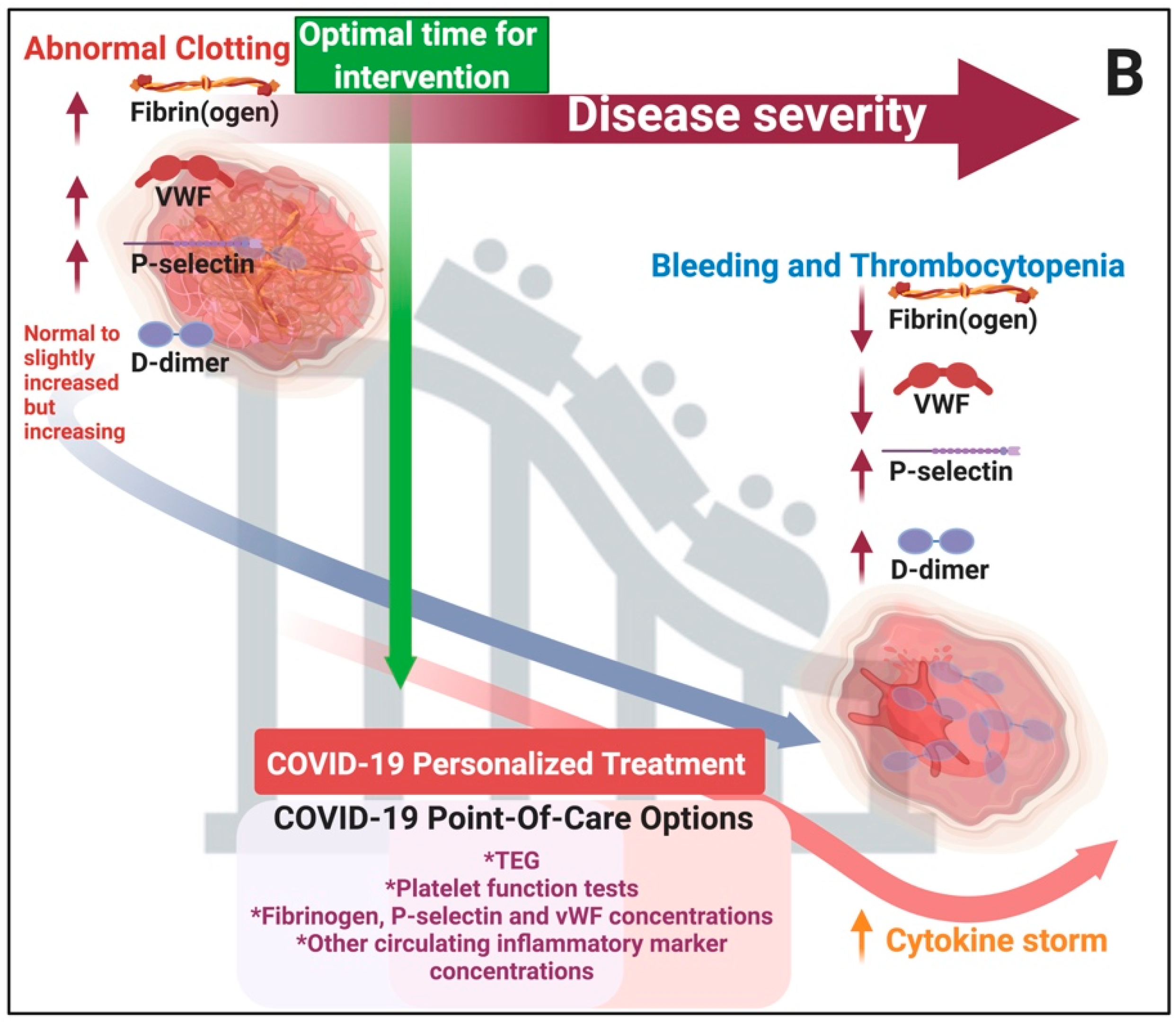
2. Discussion
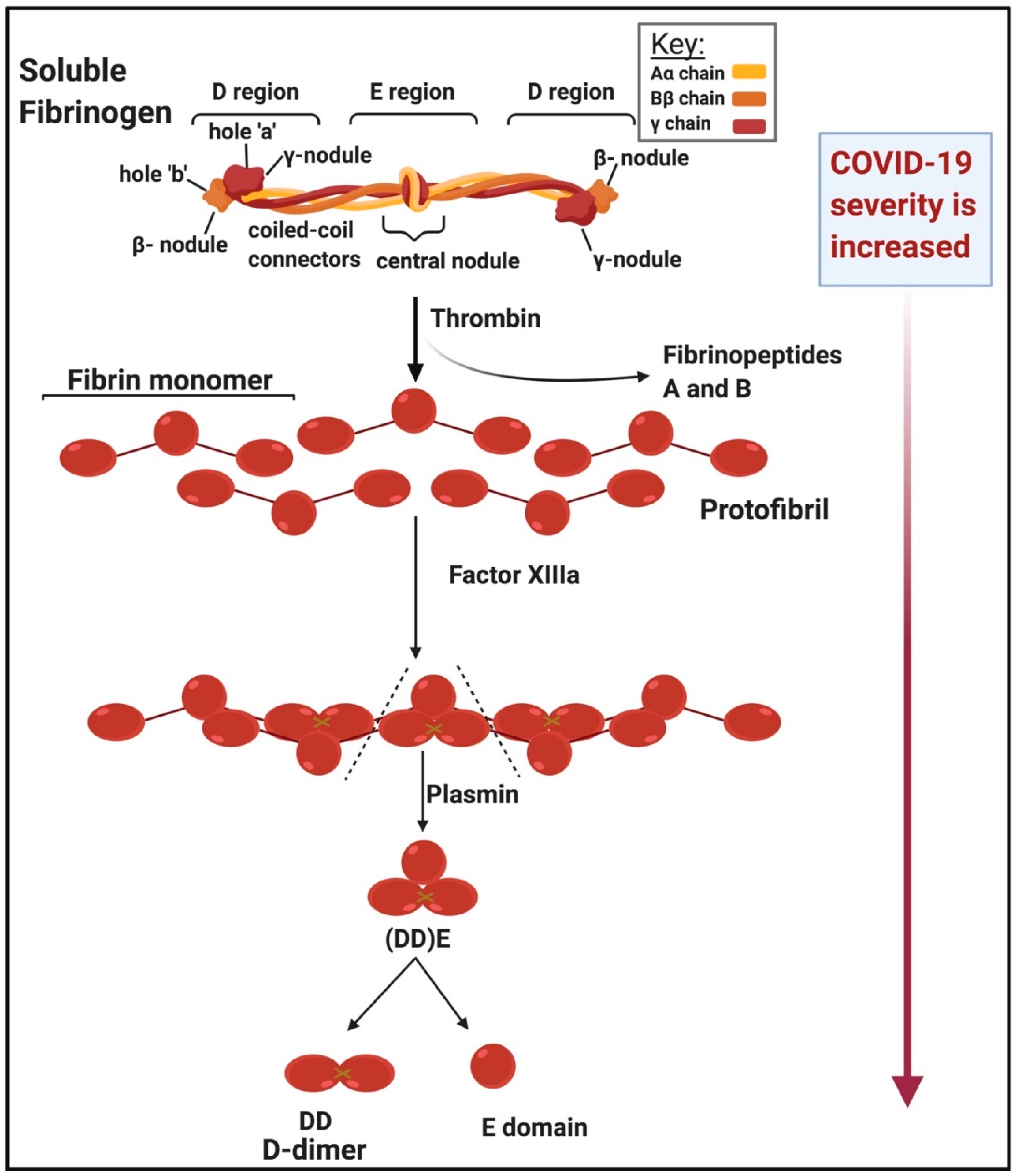
2.1. The Importance of Fibrin(Ogen) and Its Breakdown Product, D-Dimer, as Circulating Biomarkers
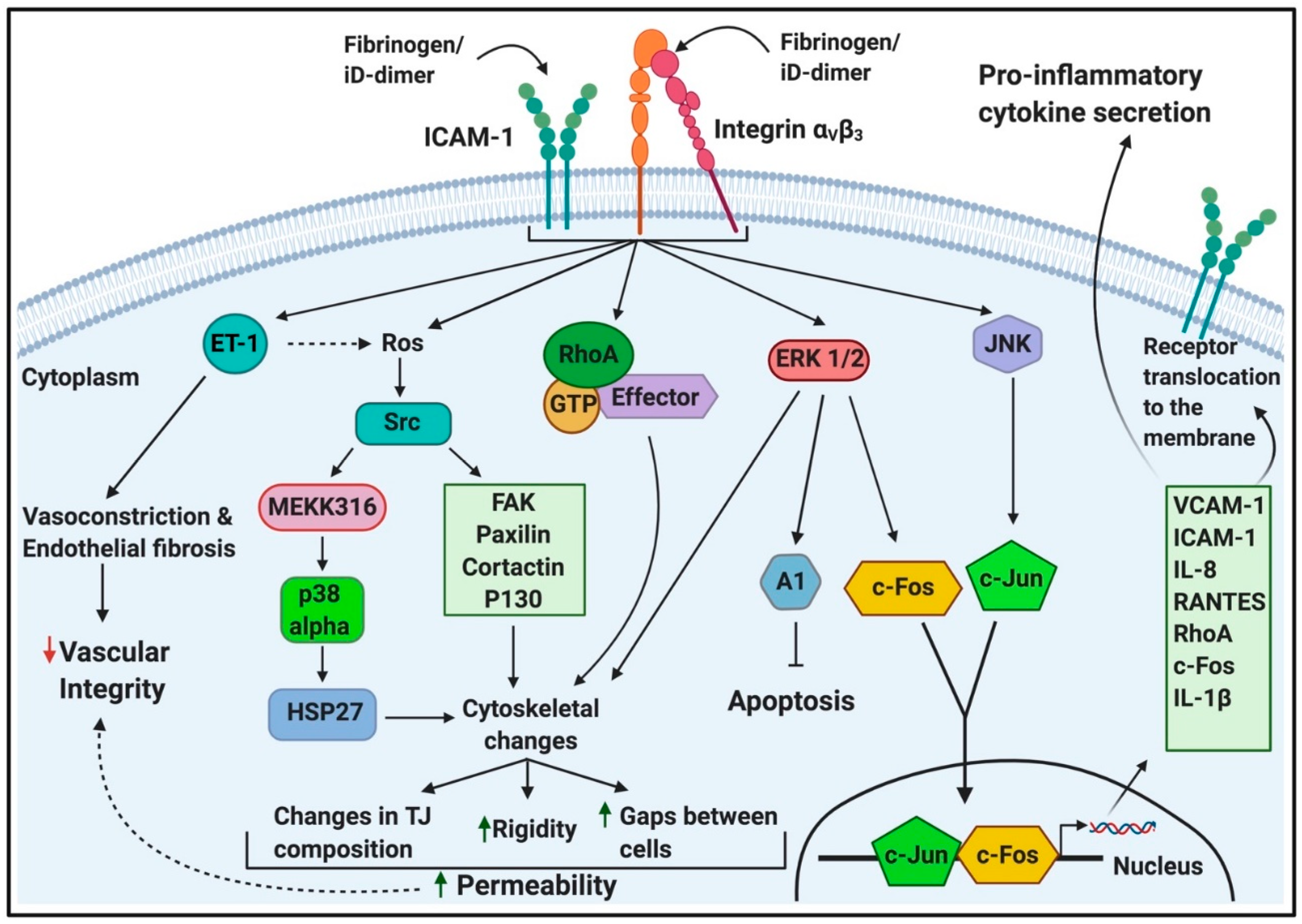
2.1.1. Interaction of Fibrin(Ogen) and D-Dimer with Cellular Receptors
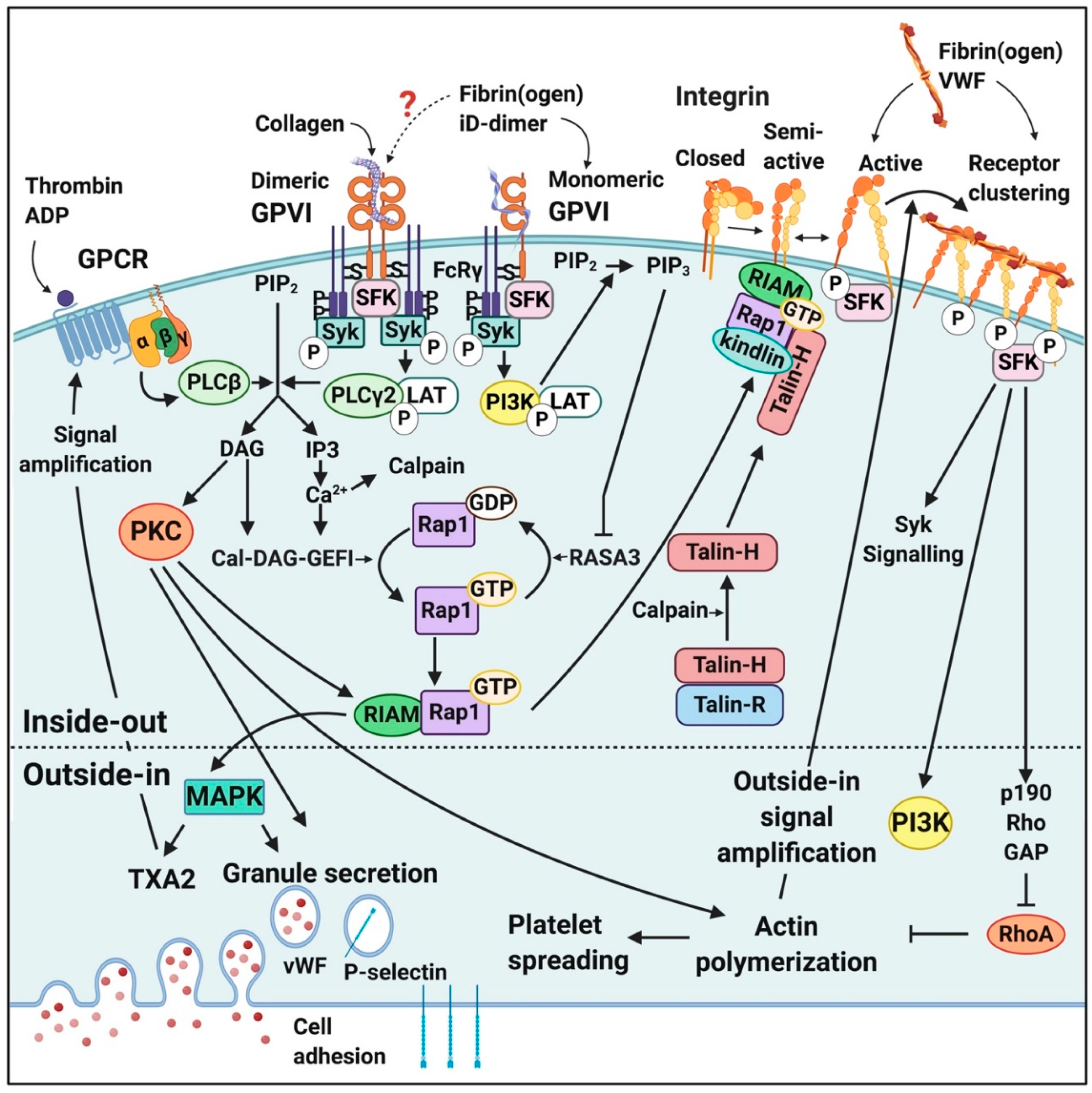
2.1.2. Fibrin(Ogen) and D-Dimer Receptors and Pathways in Platelets
2.1.3. Fibrin(Ogen) Binding to Endothelial Cells
2.1.4. Fibrin(Ogen) and D-Dimer Binding to Erythrocytes
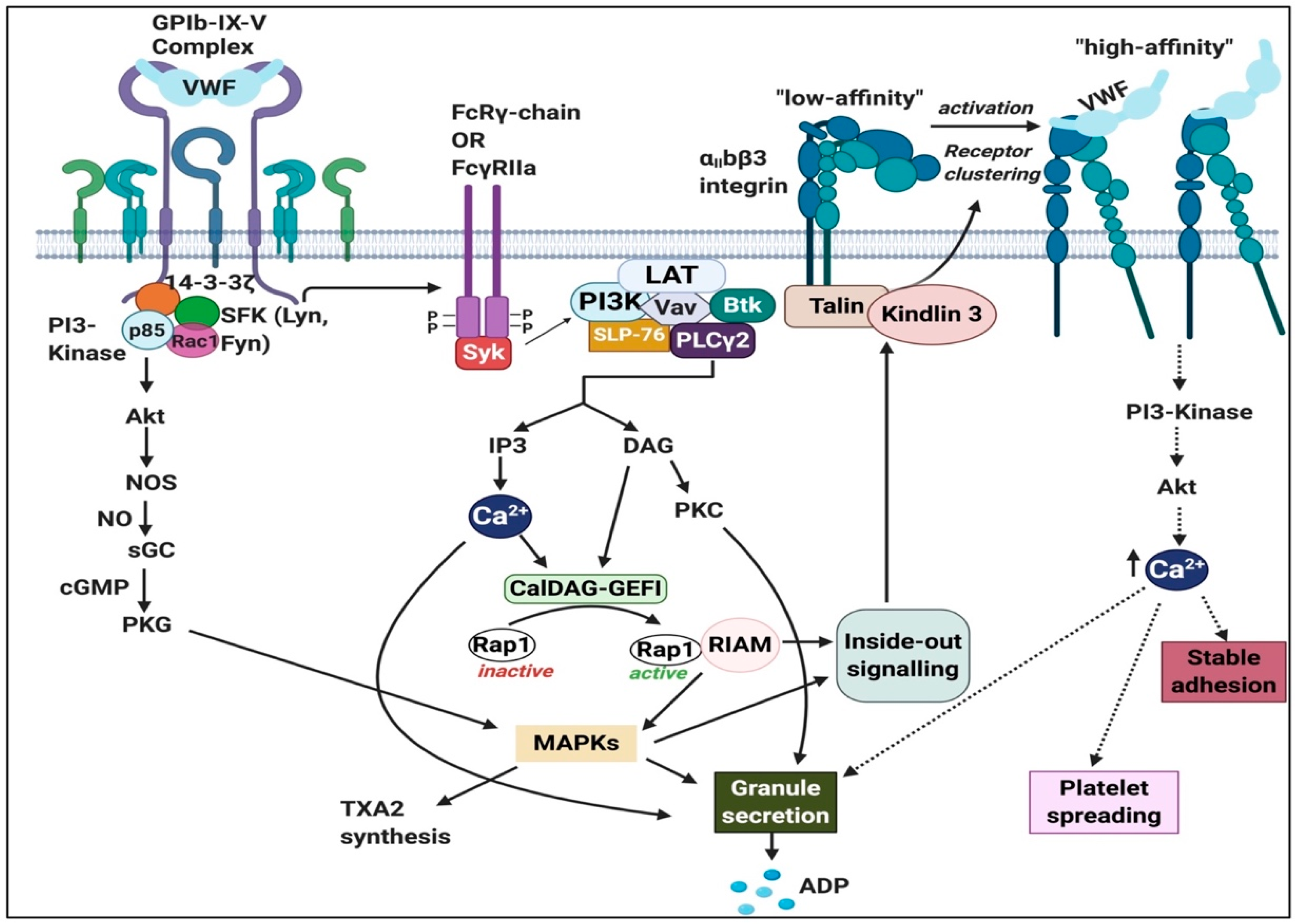
2.2. The Importance of Von Willebrand Factor (VWF) as a Circulating Biomarker
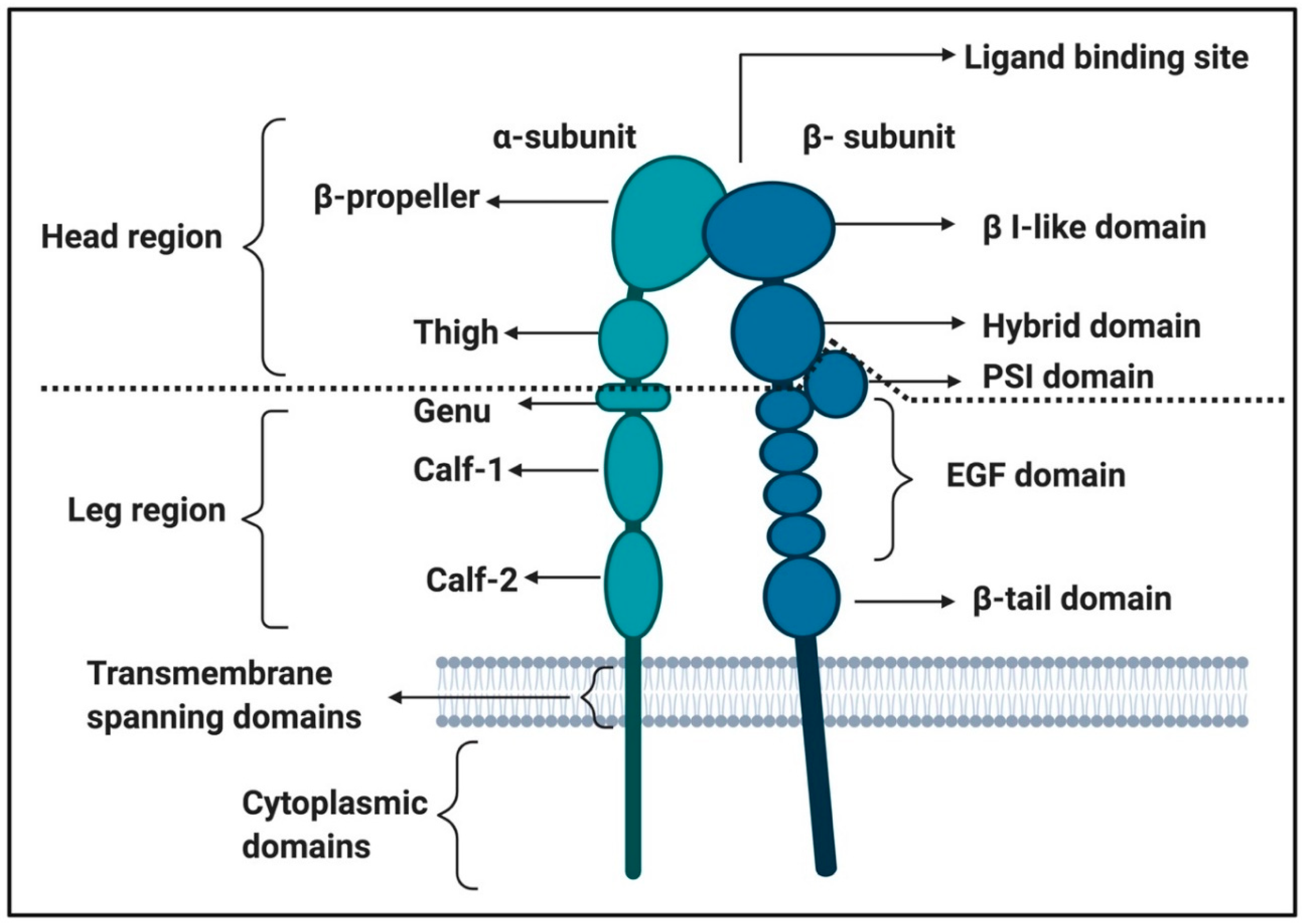
2.2.1. Von Willebrand Factor (VWF) Receptors and Pathways on Platelets
2.2.2. Von Willebrand Factor Receptors and Pathways on Endothelial Cells
2.2.3. Von Willebrand Factor (VWF) Signalling in Erythrocytes
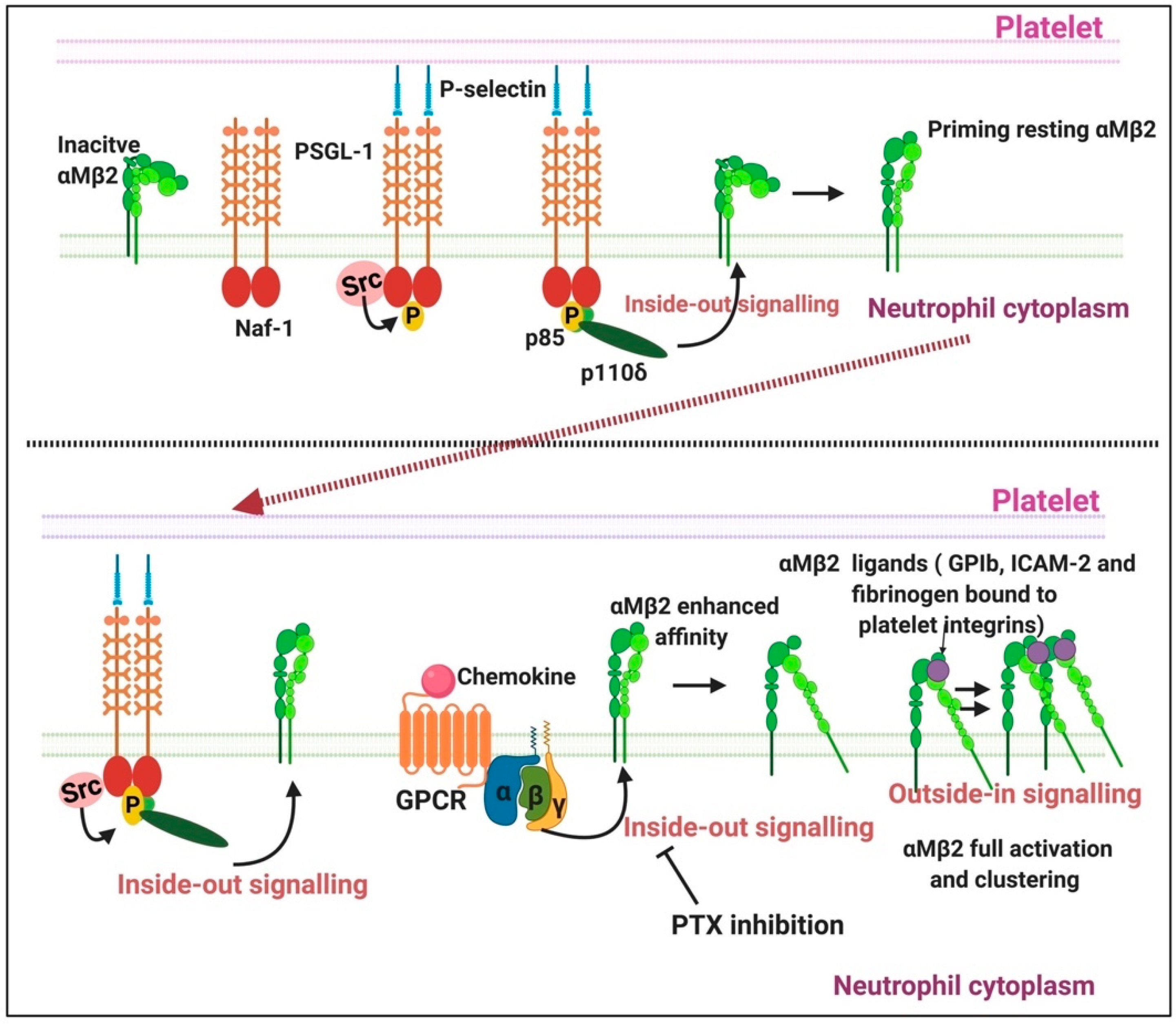
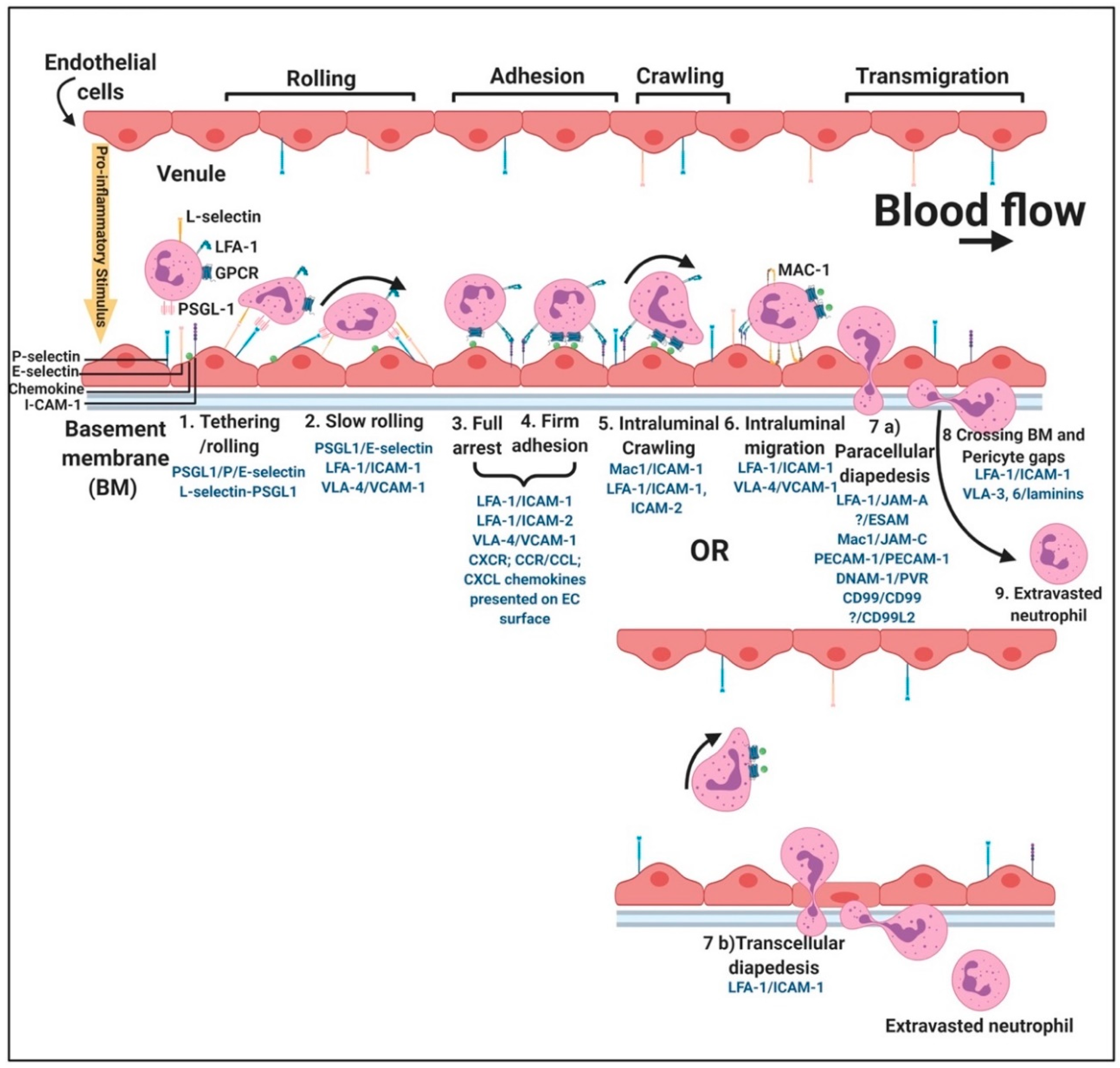
2.3. The Importance of P-Selectin as a Circulating Biomarker
2.3.1. P-Selectin Signalling in Platelets
2.3.2. P-Selectin Signalling in Endothelial Cells
2.3.3. P-Selectin Interaction with Erythrocytes
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| VWF | Von Willebrand Factor |
| EC | Endothelial cells |
| COVID-19 | Coronavirus disease 2019 |
| ICAM-1 | “Intracellular” adhesion molecule 1 |
| sP-selectin | Soluble P-selectin |
| GPCR | G-protein coupled receptor |
| GPVI | Glycoprotein VI |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| SFK | Scr family kinases |
| Syk | Spleen tyrosine kinase |
| PLC | Phospholipase C |
| LAT | Linker for activation of T-cells |
| DAG | Diacylglycerol |
| IP3 | Inositol triphosphate |
| PKC | Protein kinase C |
| Ca2+ | Calcium ions |
| Cal-DAG-GEFI | Diacylglycerol regulated guanine nucleotide exchange factor I |
| GDP | Guanine diphosphate |
| GTP | Guanine triphosphate |
| RASA3 | Ras GTPase-activating protein 3 |
| RIAM | Rap1-GTP interacting adapter molecule |
| MAPK | Mitogen-activated protein kinase |
| TXA2 | Thromboxane A2 |
| GAP | GTPase-activating protein |
| PI3K | Phosphatidylinositide 3-kinase |
References
- Marchandot, B.; Sattler, L.; Jesel, L.; Matsushita, K.; Schini-Kerth, V.; Grunebaum, L.; Morel, O. COVID-19 Related Coagulopathy: A Distinct Entity? J. Clin. Med. 2020, 9, 1651. [Google Scholar] [CrossRef] [PubMed]
- Mucha, S.R.; Dugar, S.; McCrae, K.; Joseph, D.E.; Bartholomew, J.; Sacha, G.; Militello, M. Coagulopathy in COVID-19. Cleve. Clin. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Boccia, M.; Aronne, L.; Celia, B.; Mazzeo, G.; Ceparano, M.; D’Agnano, V.; Parrella, R.; Valente, T.; Bianco, A.; Perrotta, F. COVID-19 and coagulative axis: Review of emerging aspects in a novel disease. Monaldi. Arch. Chest. Dis. 2020, 90, 271–276. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Pavan Bendapud, P.; Bornikova, L.; Gupta, S.; et al. COVID and Coagulation: Bleeding and Thrombotic Manifestations of SARS-CoV2 Infection. Blood 2020. [Google Scholar] [CrossRef]
- Zou, Y.; Guo, H.; Zhang, Y.; Zhang, Z.; Liu, Y.; Wang, J.; Lu, H.; Qian, Z. Analysis of coagulation parameters in patients with COVID-19 in Shanghai, China. Biosci. Trends 2020. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Kowalewski, M.; Fina, D.; Słomka, A.; Raffa, G.M.; Martucci, G.; Lo Coco, V.; De Piero, M.E.; Ranucci, M.; Suwalski, P.; Lorusso, R. COVID-19 and ECMO: The interplay between coagulation and inflammation—A narrative review. Crit. Care 2020, 24. [Google Scholar] [CrossRef]
- Neri, T.; Nieri, D.; Celi, A. P-selectin blockade in COVID-19-related ARDS. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L1237–L1238. [Google Scholar] [CrossRef]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 2020, 120, 998–1000. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in Intensive Care Unit. A Report of Thromboelastography Findings and other Parameters of Hemostasis. J. Thromb. Haemost. 2020, 8, 1738–1742. [Google Scholar] [CrossRef]
- Garcia-Olivé, I.; Sintes, H.; Radua, J.; Abad Capa, J.; Rosell, A. D-dimer in patients infected with COVID-19 and suspected pulmonary embolism. Acad. Emerg. Med. 2020, 169, 106023. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hu, B.; Zhang, Z.; Qin, W.; Zhu, Z.; Zhai, Z.; Davdson, B.L.; Wang, C. D-dimer triage for COVID-19. Acad. Emerg. Med. 2020, 27, 613. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, K.; Wei, H.; Chen, W.; Wang, W.; Jia, L.; Liu, Q.; Zhang, J.; Shan, T.; Peng, Z.; et al. Dynamic relationship between D-dimer and COVID-19 severity. Br. J. Haematol. 2020, 190, e24–e27. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Thachil, J. Reporting of D-dimer data in COVID-19: Some confusion and potential for misinformation. Clin. Chem. Lab. Med. 2020, 58, 1437–4331. [Google Scholar] [CrossRef]
- Lippi, G.; Favaloro, E.J. D-dimer is Associated with Severity of Coronavirus Disease 2019: A Pooled Analysis. Thromb. Haemost. 2020, 120, 876–878. [Google Scholar] [CrossRef] [Green Version]
- Escher, R.; Breakey, N.; Lämmle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef]
- Zachariah, U.; Nair, S.C.; Goel, A.; Balasubramanian, K.A.; Mackie, I.; Elias, E.; Eapen, C.E. Targeting raised von Willebrand factor levels and macrophage activation in severe COVID-19: Consider low volume plasma exchange and low dose steroid. Thromb. Res. 2020, 192. [Google Scholar] [CrossRef]
- Ramapanicker, R.; Sun, X.; Viljanen, J.; Baltzer, L. Powerful binders for the D-Dimer by conjugation of the GPRP peptide to polypeptides from a designed set—Illustrating a general route to new binders for proteins. Bioconjugate Chem. 2013, 24, 17–25. [Google Scholar] [CrossRef]
- Wada, H.; Sakuragawa, N. Are fibrin-related markers useful for the diagnosis of thrombosis? Semin. Thromb. Hemost. 2008, 34, 33–38. [Google Scholar] [CrossRef]
- Maier, C.L.; Truong, A.D.; Auld, S.C.; Polly, D.M.; Tanksley, C.L.; Duncan, A. COVID-19-associated hyperviscosity: A link between inflammation and thrombophilia? Lancet 2020, 395, 1758–1759. [Google Scholar] [CrossRef]
- Zeng, F.; Huang, Y.; Guo, Y.; Yin, M.; Chen, X.; Xiao, L.; Deng, G. Association of inflammatory markers with the severity of COVID-19: A meta-analysis. Int. J. Infect. Dis. 2020, 96, 467–474. [Google Scholar] [CrossRef]
- Beck, N. Diagnostic Hematology; Springer Science and Business Media LLC: London, UK, 2009. [Google Scholar]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dorgalaleh, A. Bleeding and Bleeding Risk in COVID-19. Semin. Thromb. Hemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, X.; Fan, Q.; Liu, H.; Liu, X.; Liu, Z.; Zhang, Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J. Thromb. Haemost. 2020, 18, 1324–1329. [Google Scholar] [CrossRef]
- Kollias, A.; Kyriakoulis, K.G.; Dimakakos, E.; Poulakou, G.; Stergiou, G.S.; Syrigos, K. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: Emerging evidence and call for action. Br. J. Haematol. 2020, 189, 846–847. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Q.; Wang, Y.; Wu, Y.; Xu, J.; Yu, Y.; Shang, Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1469–1472. [Google Scholar] [CrossRef]
- Xu, P.; Zhou, Q.; Xu, J. Mechanism of thrombocytopenia in COVID-19 patients. Ann. Hematol. 2020, 99, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amgalan, A.; Othman, M. Exploring possible mechanisms for COVID-19 induced thrombocytopenia: Unanswered questions. J. Thromb. Haemost. 2020, 18, 1514–1516. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; Pretorius, E. A Champion of Host Defense: A Generic Large-Scale Cause for Platelet Dysfunction and Depletion in Infection. Semin. Thromb. Hemost. 2020, in press. [Google Scholar] [CrossRef] [Green Version]
- Weisel, J.W.; Litvinov, R.I. Fibrin formation, structure and properties. Sub-Cell Biochem. 2017, 82, 405–456. [Google Scholar]
- Doolittle, R.F. Structural Basis of Signaling Events Involving Fibrinogen and Fibrin. In Handbook of Cell Signalling, 2nd ed.; Academic Press: San Diego, CA, USA, 2010; Chapter 17; pp. 111–114. [Google Scholar]
- Grover, S.P.; Mackman, N. Intrinsic Pathway of Coagulation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 331–338. [Google Scholar] [CrossRef]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef]
- Randeria, S.N.; Thomson, G.J.A.; Nell, T.A.; Roberts, T.; Pretorius, E. Inflammatory cytokines in type 2 diabetes mellitus as facilitators of hypercoagulation and abnormal clot formation. Cardiovasc. Diabetol. 2019, 18, 72. [Google Scholar] [CrossRef] [Green Version]
- Pieters, M.; Wolberg, A.S. Fibrinogen and fibrin: An illustrated review. Res. Pract. Thromb. Haemost. 2019, 3, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Weisel, J.W.; Litvinov, R.I. Red blood cells: The forgotten player in hemostasis and thrombosis. J. Thromb. Haemost. 2019, 17, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, E. Platelets as Potent Signaling Entities in Type 2 Diabetes Mellitus. Trends. Endocrinol. Metab. 2019, 30, 532–545. [Google Scholar] [CrossRef]
- Weitz, J.I.; Fredenburgh, J.C.; Eikelboom, J.W. A Test in Context: D-Dimer. J. Am. Coll. Cardiol. 2017, 70, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Linkins, L.A.; Takach Lapner, S. Review of D-dimer testing: Good, Bad, and Ugly. Int. J. Lab. Hematol. 2017, 39, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Favresse, J.; Lippi, G.; Roy, P.-M.; Chatelain, B.; Jacqmin, H.; Ten Cate, H.; Mullier, F. D-dimer: Preanalytical, analytical, postanalytical variables, and clinical applications. Crit. Rev. Clin. Lab. Sci. 2018, 55, 548–577. [Google Scholar] [CrossRef]
- Thachil, J.; Lippi, G.; Favaloro, E.J. D-Dimer testing: Laboratory aspects and current issues. Methods Mol. Biol. 2017, 1646, 91–104. [Google Scholar] [PubMed]
- Adam, S.S.; Key, N.S.; Greenberg, C.S. D-dimer antigen: Current concepts and future prospects. Blood 2009, 113, 2878–2887. [Google Scholar] [CrossRef] [Green Version]
- Tripodi, A. D-dimer testing in laboratory practice. Clin. Chem. 2011, 57, 1256–1262. [Google Scholar] [CrossRef] [Green Version]
- Lominadze, D.; Tsakadze, N.; Sen, U.; Falcone, J.C.; D’Souza, S.E. Fibrinogen and fragment D-induced vascular constriction. Am. J. Pysiol. Heart. Circ. Physiol. 2005, 288, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Onselaer, M.B.; Hardy, A.T.; Wilson, C.; Sanchez, X.; Babar, A.K.; Miller, J.L.C.; Watson, C.N.; Bonna, A.; Philippou, H.; Herr, A.B.; et al. Fibrin and D-dimer bind to monomeric GPVI. Blood Adv. 2017, 1, 1495–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bester, J.; Soma, P.; Kell, D.B.; Pretorius, E. Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget 2015, 6, 35284–35303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezuidenhout, J.A.; Venter, C.; Roberts, T.; Tarr, G.; Kell, D.B.; Pretorius, E. The Atypical Fibrin Fibre Network in Rheumatoid Arthritis and its Relation to Autoimmunity, Inflammation and Thrombosis. bioRxiv 2020. [Google Scholar] [CrossRef]
- De Waal, G.M.; Engelbrecht, L.; Davis, T.; De Villiers, W.J.S.; Kell, D.B.; Pretorius, E. Correlative Light-Electron Microscopy detects lipopolysaccharide and its association with fibrin fibres in Parkinson’s Disease, Alzheimer’s Disease and Type 2 Diabetes Mellitus. Sci. Rep. 2018, 8, 16798. [Google Scholar] [CrossRef]
- Kell, D.B.; Pretorius, E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 2014, 6, 748–773. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B.; Pretorius, E. The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr. Biol. 2015, 7, 24–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, D.B.; Pretorius, E. On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: The central roles of LPS and LPS-induced cell death. Integr. Biol. 2015, 7, 1339–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, D.B.; Pretorius, E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: Lessons from and for blood clotting. Prog. Biophys. Mol. Biol. 2017, 123, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Pretorius, E. No effects without causes. The Iron Dysregulation and Dormant Microbes hypothesis for chronic, inflammatory diseases: Evidence and consequences. Biol. Rev. 2018, 93, 1518–1557. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Pretorius, E. To What Extent Are the Terminal Stages of Sepsis, Septic Shock, Systemic Inflammatory Response Syndrome, and Multiple Organ Dysfunction Syndrome Actually Driven by a Prion/Amyloid Form of Fibrin? Semin. Thromb. Hemost. 2018, 44, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; Thomson, G.J.A.; Nunes, J.M.; Engelbrecht, A.M.; Nell, T.A.; de Villiers, W.J.S.; de Beer, M.C.; Engelbrecht, L.; Kell, D.B. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci. Rep. 2019, 9, 3102. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, E.; Bester, J.; Page, M.J.; Kell, D.B. The Potential of LPS-Binding Protein to Reverse Amyloid Formation in Plasma Fibrin of Individuals With Alzheimer-Type Dementia. Front. Aging Neurosci. 2018, 10, 257. [Google Scholar] [CrossRef]
- Pretorius, E.; Bester, J.; Vermeulen, N.; Alummoottil, S.; Soma, P.; Buys, A.V.; Kell, D.B. Poorly controlled type 2 diabetes is accompanied by significant morphological and ultrastructural changes in both erythrocytes and in thrombin-generated fibrin: Implications for diagnostics. Cardiovasc. Diabetol. 2015, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, E.; Bester, J.; Vermeulen, N.; Lipinski, B.; Gericke, G.S.; Kell, D.B. Profound morphological changes in the erythrocytes and fibrin networks of patients with hemochromatosis or with hyperferritinemia, and their normalization by iron chelators and other agents. PLoS ONE 2014, 9, e85271. [Google Scholar] [CrossRef]
- Pretorius, E.; Kell, D.B. Diagnostic morphology: Biophysical indicators for iron-driven inflammatory diseases. Integr. Biol. 2014, 6, 486–510. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, E.; Page, M.J.; Hendricks, L.; Nkosi, N.B.; Benson, S.R.; Kell, D.B. Both lipopolysaccharide and lipoteichoic acids potently induce anomalous fibrin amyloid formation: Assessment with novel Amytracker™ stains. J. R. Soc. Interface 2018, 15, 20170941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretorius, E.; Page, M.J.; Mbotwe, S.; Kell, D.B. Lipopolysaccharide-binding protein (LBP) can reverse the amyloid state of fibrin seen or induced in Parkinson’s disease: Implications. PLoS ONE 2018, 13, e0192121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020, 72, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suehiro, K.; Gailit, J.; Plow, E.F. Fibrinogen Is a Ligand for Integrin α5β1on Endothelial Cells. J. Biol. Chem. 1997, 272, 5360–5366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilder, R.L. Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann. Rheum. Dis. 2002, 61, ii96–ii99. [Google Scholar] [CrossRef]
- Yokoyama, K.; Zhang, X.-P.; Medved, L.; Takada, Y. Specific Binding of Integrin αvβ3 to the Fibrinogen γ and αEChain C-Terminal Domains†. Biochemistry 1999, 38, 5872–5877. [Google Scholar] [CrossRef]
- Ponamarczuk, H.; Popielarski, M.; Stasiak, M.; Bednarek, R.; Studzian, M.; Pulaski, L.; Babinska, A.; Swiatkowska, M. Contribution of activated beta3 integrin in the PDI release from endothelial cells. Front. Biosci. 2018, 23, 1612–1627. [Google Scholar]
- Pluskota, E.; D’Souza, S.E. Fibrinogen interactions with ICAM-1 (CD54) regulate endothelial cell survival. JBIC J. Biol. Inorg. Chem. 2000, 267, 4693–4704. [Google Scholar] [CrossRef] [Green Version]
- Koenig, W. Fibrin(ogen) in cardiovascular disease: An update. Thromb Haemost. 2003, 89, 601–609. [Google Scholar] [CrossRef]
- Patibandla, P.K.; Tyagi, N.; Dean, W.L.; Tyagi, S.C.; Roberts, A.M.; Lominadze, D. Fibrinogen induces alterations of endothelial cell tight junction proteins. J. Cell. Physiol. 2009, 221, 195–203. [Google Scholar] [CrossRef]
- Altieri, D.C.; Duperray, A.; Plescia, J.; Thornton, G.; Languino, L.R. Structural Recognition of a Novel Fibrinogen Chain Sequence (117133) by Intercellular Adhesion Molecule-1 Mediates Leukocyte-Endothelium Interaction. J. Biol. Chem. 1995, 270, 696–699. [Google Scholar] [CrossRef] [Green Version]
- Gray, C.; Craig, A. Fibrinogen Binding to Intercellular Adhesion Molecule 1: Implications for Plasmodium falciparum Adhesion. Infect. Immun. 2002, 70, 3962–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowkes, R.C.; Wolf, S. ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 2009, 61, 22–32. [Google Scholar] [CrossRef]
- Mangin, P.H.; Onselaer, M.-B.; Receveur, N.; Le Lay, N.; Hardy, A.T.; Wilson, C.; Sánchez, X.; Loyau, S.; Dupuis, A.; Babar, A.K.; et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica 2018, 103, 898–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlShehri, O.M.; Hughes, C.; Montague, S.; Watson, S.K.; Frampton, J.; Bender, M.; Watson, S.P. Fibrin activates GPVI in human and mouse platelets. Blood 2015, 126, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Induruwa, I.; Moroi, M.; Bonna, A.; Malcor, J.-D.; Howes, J.-M.; Warburton, E.A.; Farndale, R.W.; Jung, S.M. Platelet collagen receptor Glycoprotein VI-dimer recognizes fibrinogen and fibrin through their D-domains, contributing to platelet adhesion and activation during thrombus formation. J. Thromb. Haemost. 2018, 16, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneva, V.N.; Martyanov, A.A.; Morozova, D.S.; Panteleev, M.A.; Sveshnikova, A.N. Platelet Integrin αIIbβ3: Mechanisms of Activation and Clustering; Involvement into the Formation of the Thrombus Heterogeneous Structure. Biochem. (Moscow) Suppl. Ser. A 2019, 13, 97–110. [Google Scholar] [CrossRef]
- Xu, X.R.; Carrim, N.; Neves, M.A.; McKeown, T.; Stratton, T.W.; Coelho, R.M.P.; Lei, X.; Chen, P.; Xu, J.; Dai, X.; et al. Platelets and platelet adhesion molecules: Novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb. J. 2016, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Springer, T.A.; Zhu, J.; Xiao, T. Structural basis for distinctive recognition of fibrinogen gammaC peptide by the platelet integrin alphaIIbbeta3. J. Cell Biol. 2008, 182, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, F.; De Oliveira, S.; Freitas, T.; Gonçalves, S.; Santos, N.C. Variations on Fibrinogen-Erythrocyte Interactions during Cell Aging. PLoS ONE 2011, 6, e18167. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, S.; De Almeida, V.V.; Calado, A.; Rosario, H.; Saldanha, C. Integrin-associated protein (CD47) is a putative mediator for soluble fibrinogen interaction with human red blood cells membrane. Biochim. Biophys. Acta 2012, 1818, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiewiora, M.; Piecuch, J.; Sędek, Ł.; Mazur, B.; Sosada, K. The effects of obesity on CD47 expression in erythrocytes. Cytom. Part B Clin. Cytom. 2015, 92, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Slater, A.; Perrella, G.; Onselaer, M.-B.; Martin, E.M.; Gauer, J.S.; Xu, R.-G.; Heemskerk, J.W.; Ariëns, R.A.S.; Watson, S.P. Does fibrin(ogen) bind to monomeric or dimeric GPVI, or not at all? Platelets 2018, 30, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling During Platelet Adhesion and Activation. Arter. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [Green Version]
- Estevez, B.; Shen, B.; Du, X. Targeting integrin and integrin signaling in treating thrombosis. Arter. Thromb. Vasc. Biol. 2014, 35, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Shi, P.; Chen, X.; Zhang, L.; Liu, J.; Fan, X.; Luo, X. Clathrin-mediated integrin alphaIIbbeta3 trafficking controls platelet spreading. Platelets 2018, 29, 610–621. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin Structure, Activation, and Interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [Green Version]
- Srichai, M.B.; Zent, R. Integrin structure and function. In Cell-Extracellular Matrix Interactions in Cancer; Springer: New York, NY, USA, 2010; pp. 19–41. [Google Scholar]
- Yu, Y.; Zhu, J.; Mi, L.-Z.; Walz, T.; Sun, H.; Chen, J.; Springer, T.A. Structural specializations of α4β7, an integrin that mediates rolling adhesion. J. Cell Biol. 2012, 196, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Boylan, B.; Gao, C.; Rathore, V.; Gill, J.C.; Newman, D.K.; Newman, P.J. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood 2008, 112, 2780–2786. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Xie, W.; He, A.D.; Da, X.W.; Liang, M.L.; Yao, G.Q.; Xiang, J.-Z.; Gao, C.-J.; Ming, Z.-Y. Antiplatelet activity of chrysin via inhibiting platelet alphaIIbbeta3-mediated signaling pathway. Mol. Nutr. Food. Res. 2016, 60, 1984–1993. [Google Scholar] [CrossRef]
- Loyau, S.; Dumont, B.; Ollivier, V.; Boulaftali, Y.; Feldman, L.; Ajzenberg, N.; Perrus, M.J. Platelet Glycoprotein VI Dimerization, an Active Process Inducing Receptor Competence, Is an Indicator of Platelet Reactivity. Arter. Thromb. Vasc. Biol. 2012, 32, 778–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dütting, S.; Bender, M.; Nieswandt, B. Platelet GPVI: A target for antithrombotic therapy?! Trends Pharmacol. Sci. 2012, 33, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Ollivier, V.; Loyau, S.; Schaff, M.; Dumont, B.; Favier, R.; Freyburger, G.; Latger-Cannard, V.; Nieswandt, B.; Gachet, C.; et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015, 126, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jooss, N.J.; De Simone, I.; Provenzale, I.; Fernández, D.I.; Brouns, S.L.; Farndale, R.W.; Henskens, Y.M.; Kuijpers, M.J.; Cate, H.T.; Van Der Meijden, P.E.; et al. Role of Platelet Glycoprotein VI and Tyrosine Kinase Syk in Thrombus Formation on Collagen-Like Surfaces. Int. J. Mol. Sci. 2019, 20, 2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga-Szabo, D.; Pleines, I.; Nieswandt, B. Cell Adhesion Mechanisms in Platelets. Arter. Thromb. Vasc. Biol. 2008, 28, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; A Middleton, E.; Portier, I.; Rowley, J.W.; Stubben, C.J.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.J.; et al. Platelet Gene Expression and Function in COVID-19 Patients. Blood 2020. [Google Scholar] [CrossRef]
- Tyagi, N.; Roberts, A.M.; Dean, W.L.; Tyagi, S.C.; Lominadze, D. Fibrinogen induces endothelial cell permeability. Mol. Cell. Biochem. 2007, 307, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shaw, S.K.; Ma, S.; Yang, L.; Luscinskas, F.W.; Parkos, C.A. Regulation of leukocyte transmigration: Cell surface interactions and signaling events. J. Immunol. 2004, 172, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Guedes, A.F.; Moreira, C.; Nogueira, J.B.; Santos, N.C.; Carvalho, F. Fibrinogen-erythrocyte binding and hemorheology measurements in the assessment of essential arterial hypertension patients. Nanoscale 2019, 11, 2757–2766. [Google Scholar] [CrossRef]
- Saldanha, C. Fibrinogen interaction with the red blood cell membrane. Clin. Hemorheol. Microcirc. 2013, 53, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Denis, P.A. COVID-19-related Complications and Decompression Illness Share Main Features. Could the SARS-CoV2-related complications rely on blood foaming? Med. Hypotheses 2020, 144, 109918. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E. Erythrocyte deformability and eryptosis during inflammation, and impaired blood rheology. Clin. Hemorheol. Microcirc. 2018, 69, 545–550. [Google Scholar] [CrossRef]
- Pretorius, E.; Bester, J.; Vermeulen, N.; Lipinski, B. Oxidation inhibits iron-induced blood coagulation. Curr. Drug. Targets 2013, 14, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Lenting, P.J.; Christophe, O.; Denis, C.V. Von Willebrand factor biosynthesis, secretion, and clearance: Connecting the far ends. Blood 2015, 125, 2019–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzari, M.A.; Sanchez-Luceros, A.; Woods, A.; Alberto, M.F.; Meschengieser, S.S. Lazzari Von Willebrand factor (VWF) as a risk factor for bleeding and thrombosis. Hematology 2012, 17, s150–s152. [Google Scholar] [CrossRef]
- Chen, J.; Ling, M.; Fu, X.; López, J.A.; Chung, D. Simultaneous Exposure of Sites in von Willebrand Factor for Glycoprotein Ib Binding and ADAMTS13 Cleavage. Arter. Thromb. Vasc. Biol. 2012, 32, 2625–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.L. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu. Rev. Med. 2015, 66, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Bryckaert, M.; Rosa, J.P.; Denis, C.V.; Lenting, P.J. Of von Willebrand factor and platelets. Cell Mol. Life Sci. 2015, 72, 307–326. [Google Scholar] [CrossRef] [Green Version]
- Gragnano, F.; Sperlongano, S.; Golia, E.; Natale, F.; Bianchi, R.; Crisci, M.; Fimiani, F.; Pariggiano, I.; Vincenzo, D.; Carbone, A.; et al. The Role of von Willebrand Factor in Vascular Inflammation: From Pathogenesis to Targeted Therapy. Mediat. Inflamm. 2017, 1–13. [Google Scholar] [CrossRef]
- Buchtele, N.; Schwameis, M.; Gilbert, J.C.; Schoergenhofer, C.; Jilma, B. Targeting von Willebrand Factor in Ischaemic Stroke: Focus on Clinical Evidence. Thromb. Haemost. 2018, 118, 959–978. [Google Scholar] [CrossRef] [Green Version]
- Kalagara, T.; Moutsis, T.; Yang, Y.; Pappelbaum, K.I.; Farken, A.; Cladder-Micus, L.; Vidal-Y-Sy, S.; John, A.; Bauer, A.T.; Moerschbacher, B.M.; et al. The endothelial glycocalyx anchors von Willebrand factor fibers to the vascular endothelium. Blood Adv. 2018, 2, 2347–2357. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Roth, R.; Heuser, J.E.; Sadler, J.E. Integrin alpha(v)beta(3) on human endothelial cells binds von Willebrand factor strings under fluid shear stress. Blood 2009, 113, 1589–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyvandi, F.; Garagiola, I.; Baronciani, L. Role of von Willebrand factor in the haemostasis. Blood Transfus. 2011, 9, s3–s8. [Google Scholar] [PubMed]
- Estevez, B.; Du, X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, Y.; Inoue, O.; Suzuki-Inoue, K. Platelet receptors activated via mulitmerization: Glycoprotein VI, GPIb-IX-V, and CLEC-2. J. Thromb. Haemost. 2013, 11, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Gibbins, J.M. Platelet adhesion signalling and the regulation of thrombus formation. J. Cell Sci. 2004, 117, 3415–3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, C.L.; Anderson, K.E.; Hughan, S.C.; Dopheide, S.M.; Salem, H.H.; Jackson, S.P. Essential role for phosphoinositide 3-kinase in shear-dependent signaling between platelet glycoprotein Ib/V/IX and integrin alpha(IIb)beta(3). Blood 2002, 99, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, R.D.; Ferraro, F.; Paschalaki, K.; Dryden, N.H.; McKinnon, T.A.J.; Sutton, R.E.; Payne, E.M.; Haskard, D.O.; Hughes, A.; Cutler, D.F.; et al. Endothelial von Willebrand factor regulates angiogenesis. Blood 2011, 117, 1071–1080. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ying, X.; Fu, C.; Patel, R.; Kuebler, W.; Greenberg, S.; Bhattacharya, J. Alpha(v)beta(3) integrin induces tyrosine phosphorylation-dependent Ca(2+) influx in pulmonary endothelial cells. Circ. Res. 2000, 86, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Randi, A.; Smith, K.E.; Castaman, G. Von Willebrand factor regulation of blood vessel formation. Blood 2018, 132, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeets, M.W.; Mourik, M.J.; Niessen, H.W.; Hordijk, P.L. Stasis Promotes Erythrocyte Adhesion to von Willebrand Factor. Arter. Thromb. Vasc. Biol. 2017, 37, 1618–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, S.L. When Flow Goes Slow, von Willebrand Factor Can Bind Red Blood Cells. Arter. Thromb. Vasc. Biol. 2017, 37, 1595. [Google Scholar] [CrossRef] [Green Version]
- Oggianu, L.; Lancellotti, S.; Pitocco, D.; Zaccardi, F.; Rizzo, P.; Martini, F.; Ghirlanda, G.; De Cristofaro, R. The Oxidative Modification of Von Willebrand Factor Is Associated with Thrombotic Angiopathies in Diabetes Mellitus. PLoS ONE 2013, 8, e55396. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chung, D. Inflammation, von Willebrand factor, and ADAMTS13. Blood 2018, 132, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Lang, K.S.; Lang, P.A.; Huber, S.M.; Wieder, T. Mechanisms and Significance of Eryptosis. Antioxid. Redox Signal. 2006, 8, 1183–1192. [Google Scholar] [CrossRef]
- Lang, E.; Lang, F. Triggers, Inhibitors, Mechanisms, and Significance of Eryptosis: The Suicidal Erythrocyte Death. BioMed Res. Int. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolay, J.P.; Thorn, V.; Daniel, C.; Amann, K.; Siraskar, B.; Lang, F.; Hillgruber, C.; Goerge, T.; Hoffmann, S.; Gorzelanny, C.; et al. Cellular stress induces erythrocyte assembly on intravascular von Willebrand factor strings and promotes microangiopathy. Sci. Rep. 2018, 8, 10945. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Nadar, S.K.; Lip, G.Y. The adhesion molecule P-selectin and cardiovascular disease. Eur. Hear. J. 2003, 24, 2166–2179. [Google Scholar] [CrossRef] [Green Version]
- Lorant, D.E.; Topham, M.K.; Whatley, R.E.; McEver, R.P.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. Inflammatory roles of P-selectin. J. Clin. Investig. 1993, 92, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Cambien, B.; Wagner, D.D. A new role in hemostasis for the adhesion receptor P-selectin. Trends Mol. Med. 2004, 10, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ghasemzadeh, M.; Hosseini, E. Platelet-leukocyte crosstalk: Linking proinflammatory responses to procoagulant state. Thromb. Res. 2013, 131, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Panicker, S.R.; Mehta-D’Souza, P.; Zhang, N.; Klopocki, A.G.; Shao, B.; McEver, R.P. Circulating soluble P-selectin must dimerize to promote inflammation and coagulation in mice. Blood 2017, 130, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelczyk, M.; Glabiński, A.; Kaczorowska, B.; Baj, Z. sP- and sE-selectin in stroke patients with metabolic disorders. Neurol. Neurochir. Polska 2018, 52, 599–605. [Google Scholar] [CrossRef]
- Woollard, K.; Suhartoyo, A.; Harris, E.E.; Eisenhardt, S.; Jackson, S.P.; Peter, K.; Dart, A.; Hickey, M.J.; Chin-Dusting, J.P.F. Pathophysiological Levels of Soluble P-Selectin Mediate Adhesion of Leukocytes to the Endothelium Through Mac-1 Activation. Circ. Res. 2008, 103, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Gross, P.L. Soluble P-selectin is the smoke, not the fire. Blood 2017, 130, 101–102. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, R.; Otero, D.C.; Takahashi, A.A.; Bradley, L.M. PSGL-1: A New Player in the Immune Checkpoint Landscape. Trends Immunol. 2017, 38, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Zarbock, A.; Polanowska-Grabowska, R.K.; Ley, K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2007, 21, 99–111. [Google Scholar] [CrossRef]
- Zimmerman, G.A. Two by two: The pairings of P-selectin and P-selectin glycoprotein ligand 1. Proc. Natl. Acad. Sci. USA 2001, 98, 10023–10024. [Google Scholar] [CrossRef] [Green Version]
- Setiadi, H.; Yago, T.; Liu, Z.; McEver, R.P. Endothelial signaling by neutrophil-released oncostatin M enhances P-selectin–dependent inflammation and thrombosis. Blood Adv. 2019, 3, 168–183. [Google Scholar] [CrossRef] [Green Version]
- Barrionuevo, N.; Gatica, S.; Olivares, P.; Cabello-Verrugio, C.; Simon, F. Endothelial Cells Exhibit Two Waves of P-selectin Surface Aggregation Under Endotoxic and Oxidative Conditions. Protein J. 2019, 38, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Merten, M.; Thiagarajan, P. P-selectin in arterial thrombosis. Zeitschrift für Kardiologie 2004, 93, 855–863. [Google Scholar] [CrossRef]
- Azizova, O.A.; Aseichev, A.V.; Piryazev, A.P.; Roitman, E.V.; Shcheglovitova, O.N. Effects of oxidized fibrinogen on the functions of blood cells, blood clotting, and rheology. Bull. Exp. Biol. Med. 2007, 144, 397–407. [Google Scholar] [CrossRef]
- Morikis, V.A.; Simon, S.I. Neutrophil Mechanosignaling Promotes Integrin Engagement With Endothelial Cells and Motility Within Inflamed Vessels. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Foubert, P.; Varner, J. Integrins in tumor angiogenesis and lymphangiogenesis. Methods Mol. Biol. 2012, 757, 471–486. [Google Scholar] [PubMed] [Green Version]
- Carman, C.V. High-Resolution Fluorescence Microscopy to Study Transendothelial Migration. Methods Mol. Biol. 2011, 757, 215–245. [Google Scholar]
- Schnoor, M.; Alcaide, P.; Voisin, M.-B.; Van Buul, J.D. Crossing the Vascular Wall: Common and Unique Mechanisms Exploited by Different Leukocyte Subsets during Extravasation. Mediat. Inflamm. 2015, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Matsui, N.M.; Borsig, L.; Rosen, S.D.; Yaghmai, M.; Varki, A.; Embury, S.H. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood 2001, 98, 1955–1962. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Koo, S.; Lin, C.S.; Neu, B. Specific Binding of Red Blood Cells to Endothelial Cells Is Regulated by Nonadsorbing Macromolecules. J. Biol. Chem. 2010, 285, 40489–40495. [Google Scholar] [CrossRef] [Green Version]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020. [Google Scholar] [CrossRef]
- Pillai, R.C.; Fraser, J.F.; Ziegenfuss, M.; Bhaskar, B. The Influence of Circulating Levels of Fibrinogen and Perioperative Coagulation Parameters on Predicting Postoperative Blood Loss in Cardiac Surgery: A Prospective Observational Study. J. Card. Surg. 2013, 29, 189–195. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Der Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J. Am. Coll. Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.D.; Schell, J.C.; Rodgers, G.M. The D-dimer assay. Am. J. Hematol. 2019, 94, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Kyrle, P.A.; Hron, G.; Eichinger, S.; Wagner, O. Circulating P-selectin and the risk of recurrent venous thromboembolism. Thromb. Haemost. 2007, 97, 880–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bath, P.M.; May, J.; Heptinstall, S. Clinical utility of remote platelet function measurement using P-selectin: Assessment of aspirin, clopidogrel, and prasugrel and bleeding disorders. Platelets 2018, 29, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Ayerbe, L.; Risco, C.; Ayis, S. The association between treatment with heparin and survival in patients with Covid-19. J. Thromb. Thrombolysis 2020, 2020, 1–4. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Tavares, J.G.P.; De Oliveira, M.P.; De Carvalho, R.G.; Errante, P.R.; Taha, M.O.; Fagundes, D.J.; Caricati-Neto, A. Anticoagulant and antiarrhythmic effects of heparin in the treatment of COVID-19 patients. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Jesudas, R.; Chaudhury, A.; Laukaitis, C.M. An update on the new classification of Ehlers-Danlos syndrome and review of the causes of bleeding in this population. Haemophilia 2019, 25, 558–566. [Google Scholar] [CrossRef]
- Rubulotta, F.; Soliman-Aboumarie, H.; Filbey, K.; Geldner, G.; Kuck, K.; Ganau, M.; Hemmerling, T.M. Technologies to optimize the care of severe COVID-19 patients for healthcare providers challenged by limited resources. Anesth. Analg. 2020. [Google Scholar] [CrossRef]


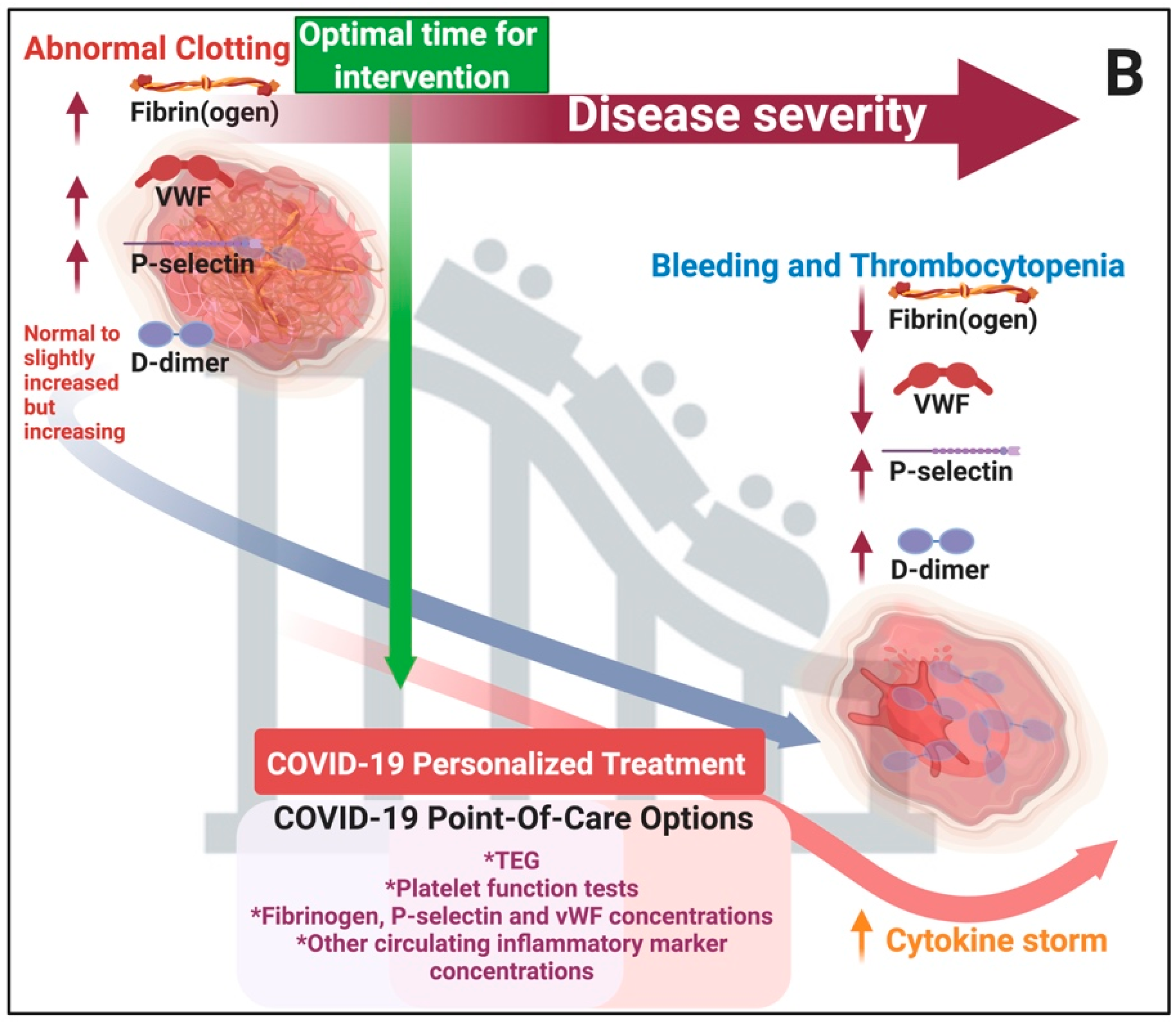




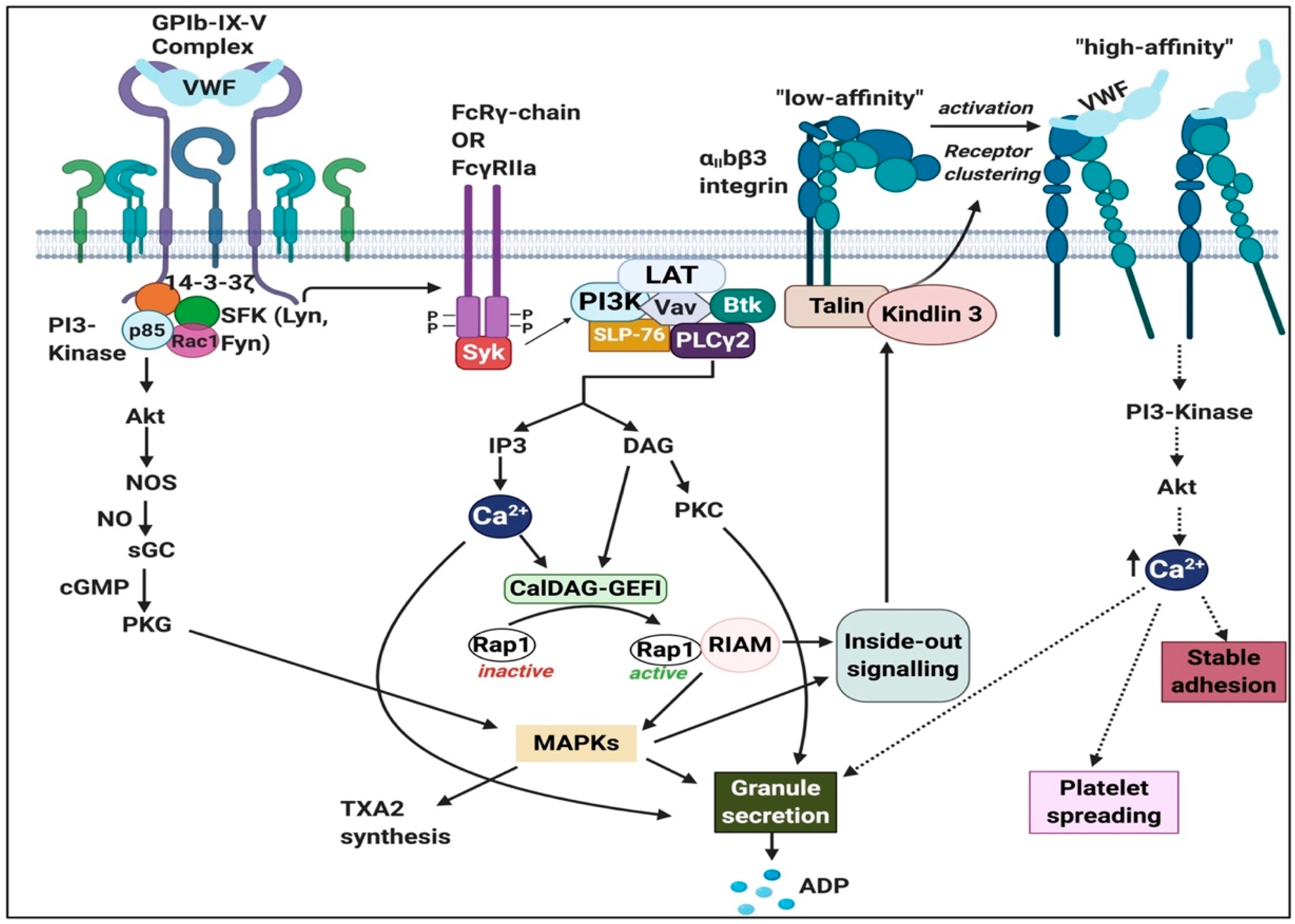



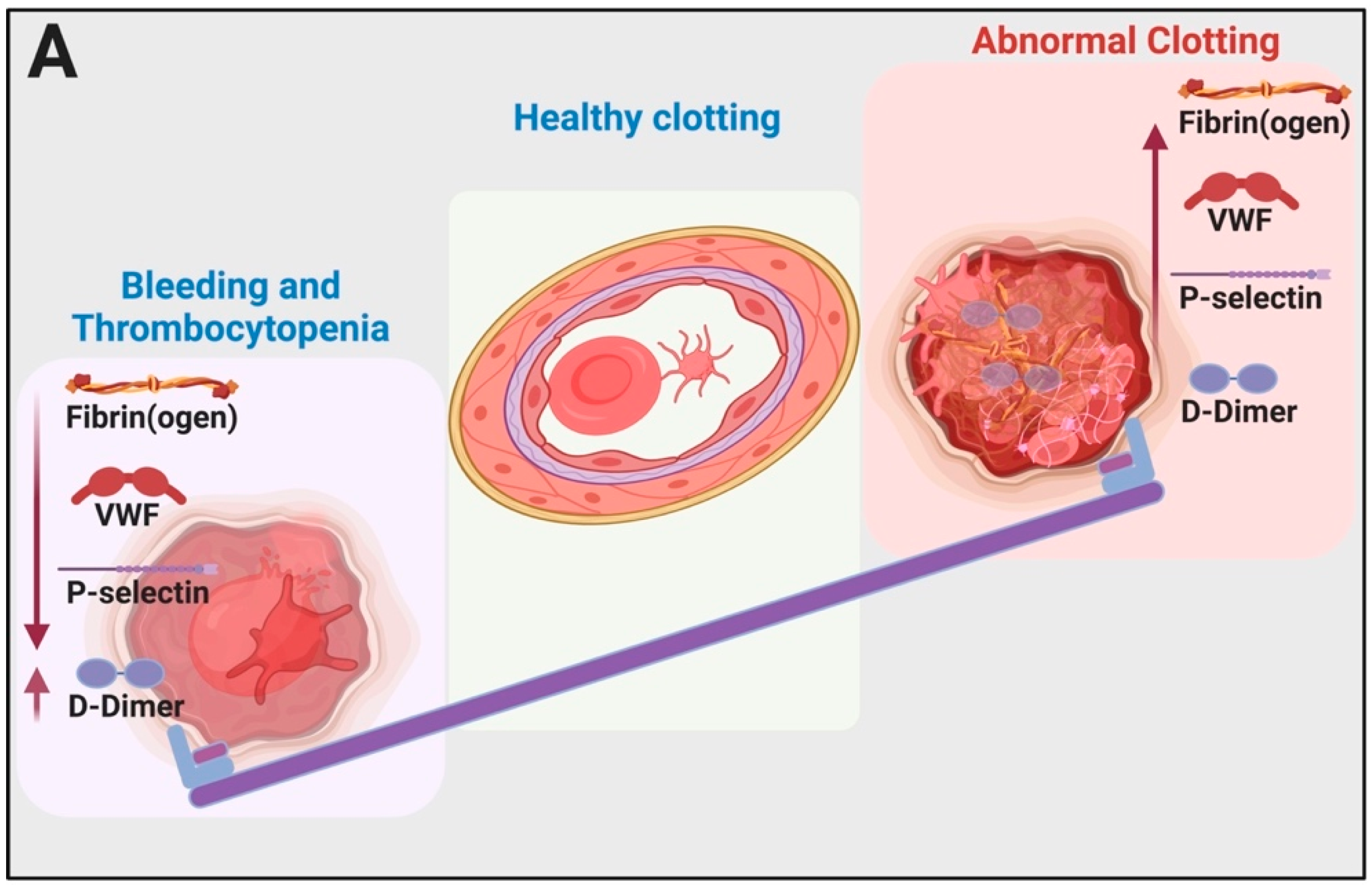
{kind=link}
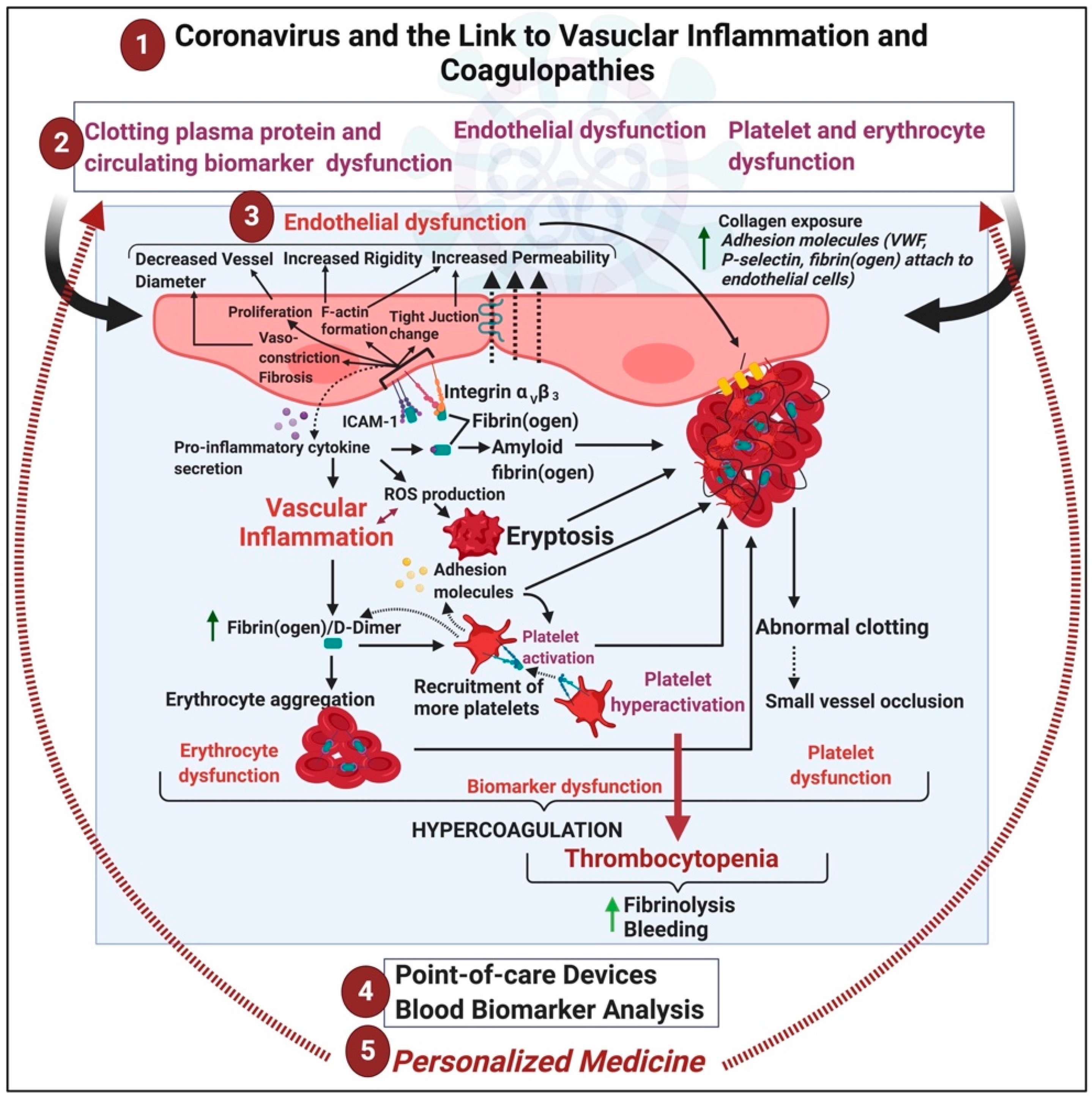
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Receptor | Effect | References |
|---|---|---|---|
| Endothelial cells (EC) | Integrins αVβ3 and α5β1 | Endothelial cell proliferation, endothelial cell activation, angiogenesis, increased EC permeability and vasoconstriction | [34,66,67,68,69] |
| Integrin αMβ2 | Facilitates interaction fibrinogen with ICAM-1 during leukocyte transmigration | [70] | |
| ICAM-1 | Platelet adhesion, leucocyte adhesion and transmigration to site of infection, mitogenesis, angiogenesis, cell survival, release of pro-inflammatory cytokines, ICAM-1 receptor recruitment to EC membrane and vasoconstriction | [70,71,72,73,74,75] | |
| Platelets | Glycoprotein IIb/IIIa | Platelet activation and spreading, integrin activation and granule secretion | [71] |
| Glycoprotein VI (GPVI) | Platelet activation and spreading, integrin activation and granule secretion | [40,48,76,77,78] | |
| Integrins αIIbβ3 αVβ3 | Outside-in signalling in platelets—platelet spreading and granule secretion | [34,40,67,79,80,81] | |
| Erythrocytes | αIIbβ3-related type integrin? CD47 | Erythrocyte aggregation and adhesion | [47,82,83,84] |
| iD-dimer and/or Fragment D | |||
| Endothelial cells | ICAM-1 | Arterial constriction | [47] |
| Platelets | Integrin αIIbβ3 | Platelet spreading and aggregation | [78] |
| GPVI (monomeric/dimeric?) | Platelet spreading | [48,85] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp, J.C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. Covid-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145168
Grobler C, Maphumulo SC, Grobbelaar LM, Bredenkamp JC, Laubscher GJ, Lourens PJ, Steenkamp J, Kell DB, Pretorius E. Covid-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. International Journal of Molecular Sciences. 2020; 21(14):5168. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145168
Chicago/Turabian StyleGrobler, Corlia, Siphosethu C. Maphumulo, L. Mireille Grobbelaar, Jhade C. Bredenkamp, Gert J. Laubscher, Petrus J. Lourens, Janami Steenkamp, Douglas B. Kell, and Etheresia Pretorius. 2020. "Covid-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes" International Journal of Molecular Sciences 21, no. 14: 5168. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145168