Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption

Abstract
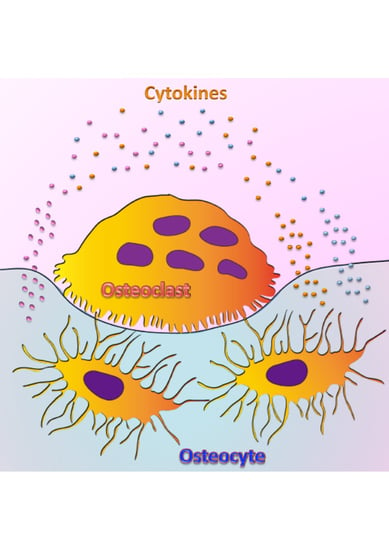
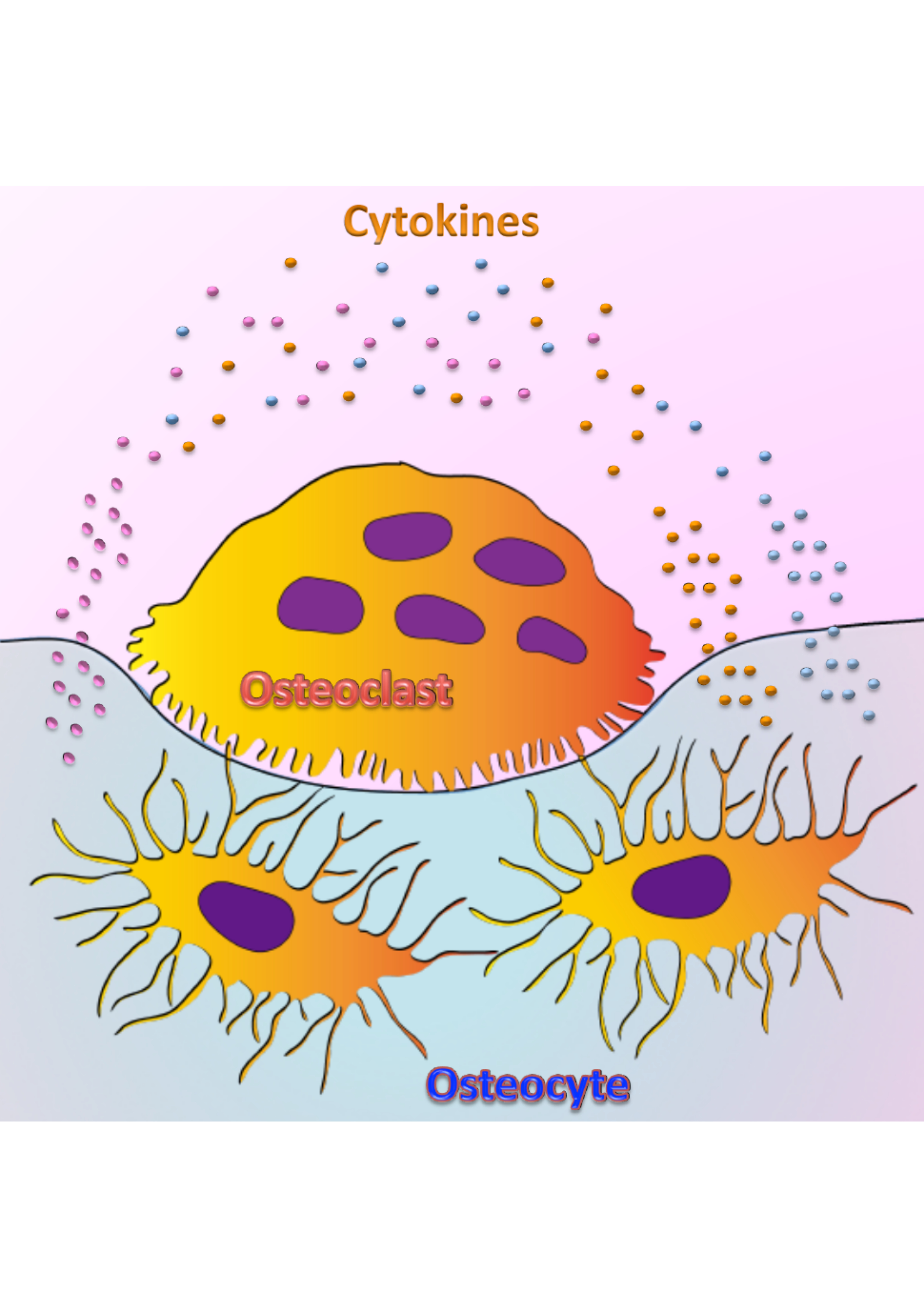
:
{kind=link}
{kind=link}
1. Introduction
2. Osteoclast Regulatory Cytokines
2.1. Osteoclastogenic Cytokines
2.2. Anti-Osteoclastogenic Cytokines
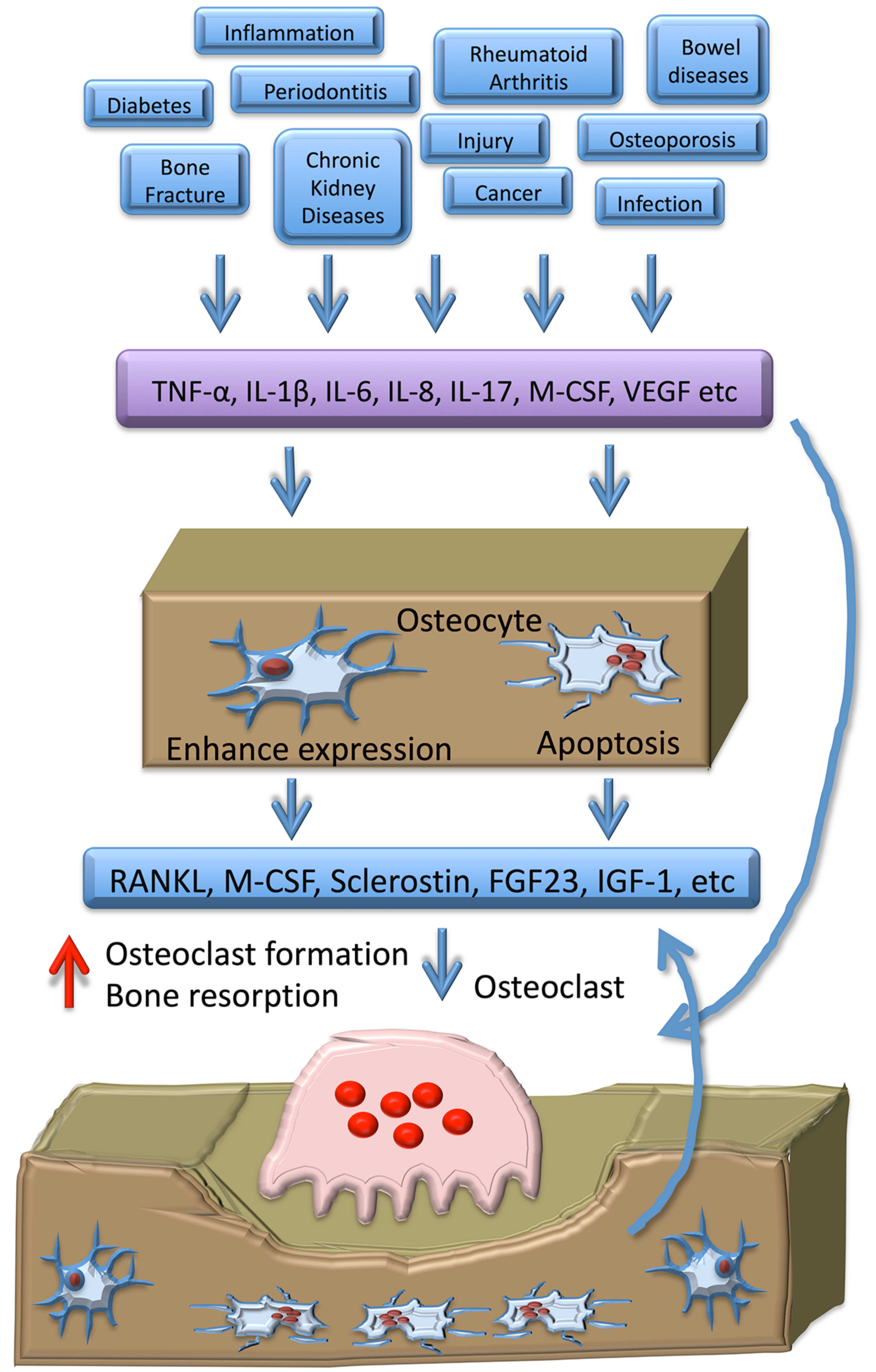
3. Osteocyte-Related Cytokines
3.1. RANKL and OPG
3.2. TNF-α
3.3. IL-1β
3.4. IL-6
3.5. FGF23
3.6. IGF-1
3.7. IL-8
3.8. M-CSF
3.9. VEGF
3.10. Sclerostin
3.11. IL-10
3.12. IL-17
3.13. Parathyroid Hormone
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| M-CSF | macrophage colony-stimulating factor |
| RANKL | receptor activator of nuclear factor κB Ligand |
| PGE2 | prostaglandin E2 |
| 1,25(OH)2D3 | vitamin D |
| TNF-α | tumor necrosis factor-α |
| IL | interleukin |
| LPS | lipopolysaccharide |
| IFN | interferon |
| OPG | osteoprotegerin |
| FGF | fibroblast growth factor |
| IGF | insulin-like growth factor |
| VEGF | vascular endothelial growth factor |
| GH | growth hormone |
| RA | rheumatoid arthritis |
| MCP | monocyte chemotactic protein |
| MAPKs | mitogen-activated protein kinases |
References
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Crotti, T.N.; Dharmapatni, A.A.; Alias, E.; Haynes, D.R. Osteoimmunology: Major and Costimulatory Pathway Expression Associated with Chronic Inflammatory Induced Bone Loss. J. Immunol. Res. 2015, 2015, 281287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.A. CSF-1 signal transduction. J. Leukoc. Biol. 1997, 62, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Takahashi, N.; Udagawa, N.; Jimi, E.; Gillespie, M.T.; Martin, T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999, 20, 345–357. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Feige, U.; Sarosi, I.; Bolon, B.; Tafuri, A.; Morony, S.; Capparelli, C.; Li, J.; Elliott, R.; McCabe, S.; et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 1999, 402, 304–309. [Google Scholar] [CrossRef]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef]
- Bonewald, L.F. Osteocytes as dynamic multifunctional cells. Ann. N. Y. Acad. Sci. 2007, 1116, 281–290. [Google Scholar] [CrossRef]
- Buenzli, P.R.; Sims, N.A. Quantifying the osteocyte network in the human skeleton. Bone 2015, 75, 144–150. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’Brien, C.A. Osteocytes, not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PLoS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Alshabab, A.; Albiero, M.L.; Mattos, M.; Correa, J.D.; Chen, S.; Yang, Y. Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL. J. Clin. Periodontol. 2018, 45, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.M.; Heard-Lipsmeyer, M.E.; Dimori, M.; Thostenson, J.D.; Mannen, E.M.; O’Brien, C.A.; Morello, R. Loss of RANKL in osteocytes dramatically increases cancellous bone mass in the osteogenesis imperfecta mouse (oim). Bone Rep. 2018, 9, 61–73. [Google Scholar] [CrossRef]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-alpha Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [Green Version]
- Ohori, F.; Kitaura, H.; Marahleh, A.; Kishikawa, A.; Ogawa, S.; Qi, J.; Shen, W.R.; Noguchi, T.; Nara, Y.; Mizoguchi, I. Effect of TNF-alpha-Induced Sclerostin on Osteocytes during Orthodontic Tooth Movement. J. Immunol. Res. 2019, 2019, 9716758. [Google Scholar] [CrossRef] [Green Version]
- Qing, H.; Ardeshirpour, L.; Pajevic, P.D.; Dusevich, V.; Jähn, K.; Kato, S.; Wysolmerski, J.; Bonewald, L.F. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res. 2012, 27, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Gruber, H.E.; Stover, S.J. Maternal and weanling bone: The influence of lowered calcium intake and maternal dietary history. Bone 1994, 15, 167–176. [Google Scholar] [CrossRef]
- Kogawa, M.; Khalid, K.A.; Wijenayaka, A.R.; Ormsby, R.T.; Evdokiou, A.; Anderson, P.H.; Findlay, D.M.; Atkins, G.J. Recombinant sclerostin antagonizes effects of ex vivo mechanical loading in trabecular bone and increases osteocyte lacunar size. Am. J. Physiol. Cell Physiol. 2018, 314, C53–C61. [Google Scholar] [CrossRef]
- Rolvien, T.; Krause, M.; Jeschke, A.; Yorgan, T.; Püschel, K.; Schinke, T.; Busse, B.; Demay, M.B.; Amling, M. Vitamin D regulates osteocyte survival and perilacunar remodeling in human and murine bone. Bone 2017, 103, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, K.; Hoshi, K.; Kawamoto, S.; Tanaka, M.; Ejiri, S.; Ozawa, H. Osteocytic osteolysis observed in rats to which parathyroid hormone was continuously administered. J. Bone Miner. Metab. 2004, 22, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Rodionova, N.V.; Oganov, V.S.; Zolotova, N.V. Ultrastructural changes in osteocytes in microgravity conditions. Adv. Space Res. 2002, 30, 765–770. [Google Scholar] [CrossRef]
- Balemans, W.; Van Hul, W. Human genetics of SOST. J. Musculoskelet. Neuronal. Interact. 2006, 6, 355–356. [Google Scholar]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Stegen, S.; Stockmans, I.; Moermans, K.; Thienpont, B.; Maxwell, P.H.; Carmeliet, P.; Carmeliet, G. Osteocytic oxygen sensing controls bone mass through epigenetic regulation of sclerostin. Nat. Commun. 2018, 9, 2557. [Google Scholar] [CrossRef]
- Kim, S.W.; Pajevic, P.D.; Selig, M.; Barry, K.J.; Yang, J.Y.; Shin, C.S.; Baek, W.Y.; Kim, J.E.; Kronenberg, H.M. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J. Bone Miner. Res. 2012, 27, 2075–2084. [Google Scholar] [CrossRef] [Green Version]
- Pathak, J.L.; Bakker, A.D.; Luyten, F.P.; Verschueren, P.; Lems, W.F.; Klein-Nulend, J.; Bravenboer, N. Systemic Inflammation Affects Human Osteocyte-Specific Protein and Cytokine Expression. Calcif. Tissue Int. 2016, 98, 596–608. [Google Scholar] [CrossRef]
- Badran, Z.; Struillou, X.; Verner, C.; Clee, T.; Rakic, M.; Martinez, M.C.; Soueidan, A. Periodontitis as a risk factor for systemic disease: Are microparticles the missing link? Med. Hypotheses 2015, 84, 555–556. [Google Scholar] [CrossRef]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Metzger, C.E.; Narayanan, A.; Zawieja, D.C.; Bloomfield, S.A. Inflammatory Bowel Disease in a Rodent Model Alters Osteocyte Protein Levels Controlling Bone Turnover. J. Bone Miner. Res. 2017, 32, 802–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uluckan, O.; Jimenez, M.; Karbach, S.; Jeschke, A.; Grana, O.; Keller, J.; Busse, B.; Croxford, A.L.; Finzel, S.; Koenders, M.; et al. Chronic skin inflammation leads to bone loss by IL-17-mediated inhibition of Wnt signaling in osteoblasts. Sci. Transl. Med. 2016, 8, 330ra337. [Google Scholar] [CrossRef] [PubMed]
- Pesce Viglietti, A.I.; Arriola Benitez, P.C.; Gentilini, M.V.; Velasquez, L.N.; Fossati, C.A.; Giambartolomei, G.H.; Delpino, M.V. Brucella abortus Invasion of Osteocytes Modulates Connexin 43 and Integrin Expression and Induces Osteoclastogenesis via Receptor Activator of NF-kappaB Ligand and Tumor Necrosis Factor Alpha Secretion. Infect. Immun. 2016, 84, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.X.; Evans, B.A.; Jiang, W.G. New Roles of Osteocytes in Proliferation, Migration and Invasion of Breast and Prostate Cancer Cells. Anticancer Res. 2016, 36, 1193–1201. [Google Scholar] [PubMed]
- Selvarajah, L.; Curtis, A.M.; Kennedy, O.D. Bone Microdamage in Acute Knee Injury. Curr. Rheumatol. Rep. 2018, 20, 89. [Google Scholar] [CrossRef]
- Denis, G.V.; Sebastiani, P.; Bertrand, K.A.; Strissel, K.J.; Tran, A.H.; Slama, J.; Medina, N.D.; Andrieu, G.; Palmer, J.R. Inflammatory signatures distinguish metabolic health in African American women with obesity. PLoS ONE 2018, 13, e0196755. [Google Scholar] [CrossRef] [Green Version]
- Jafaripour, S.; Sedighi, S.; Jokar, M.H.; Aghaei, M.; Moradzadeh, M. Inflammation, diet, and type 2 diabetes: A mini-review. J. Immunoassay Immunochem. 2020, 41, 768–777. [Google Scholar] [CrossRef]
- Schett, G. Review: Immune cells and mediators of inflammatory arthritis. Autoimmunity 2008, 41, 224–229. [Google Scholar] [CrossRef]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Morita, I.; Maruo, N.; Kubota, T.; Murota, S.; Aso, T. Expression of IL-6 receptor and GP130 in mouse bone marrow cells during osteoclast differentiation. Bone 1998, 22, 487–493. [Google Scholar] [CrossRef]
- Kim, J.H.; Sim, J.H.; Lee, S.; Seol, M.A.; Ye, S.K.; Shin, H.M.; Lee, E.B.; Lee, Y.J.; Choi, Y.J.; Yoo, W.H.; et al. Interleukin-7 Induces Osteoclast Formation via STAT5, Independent of Receptor Activator of NF-kappaB Ligand. Front. Immunol. 2017, 8, 1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendre, M.S.; Montague, D.C.; Peery, T.; Akel, N.S.; Gaddy, D.; Suva, L.J. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone 2003, 33, 28–37. [Google Scholar] [CrossRef]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Okabe, I.; Kikuchi, T.; Mogi, M.; Takeda, H.; Aino, M.; Kamiya, Y.; Fujimura, T.; Goto, H.; Okada, K.; Hasegawa, Y.; et al. IL-15 and RANKL Play a Synergistically Important Role in Osteoclastogenesis. J. Cell Biochem 2017, 118, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef]
- Chen, L.; Wei, X.Q.; Evans, B.; Jiang, W.; Aeschlimann, D. IL-23 promotes osteoclast formation by up-regulation of receptor activator of NF-kappaB (RANK) expression in myeloid precursor cells. Eur. J. Immunol. 2008, 38, 2845–2854. [Google Scholar] [CrossRef]
- Chen, Z.; Buki, K.; Vaaraniemi, J.; Gu, G.; Vaananen, H.K. The critical role of IL-34 in osteoclastogenesis. PLoS ONE 2011, 6, e18689. [Google Scholar] [CrossRef]
- Fox, S.W.; Fuller, K.; Bayley, K.E.; Lean, J.M.; Chambers, T.J. TGF-beta 1 and IFN-gamma direct macrophage activation by TNF-alpha to osteoclastic or cytocidal phenotype. J. Immunol. 2000, 165, 4957–4963. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Seong, S.; Kim, J.H.; Kim, K.; Kim, I.; Jeong, B.C.; Nam, K.I.; Kim, K.K.; Hennighausen, L.; Kim, N. STAT5 is a key transcription factor for IL-3-mediated inhibition of RANKL-induced osteoclastogenesis. Sci. Rep. 2016, 6, 30977. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, J.; Shi, Z.; Xu, D.; Luo, S.; Siegal, G.P.; Feng, X.; Wei, S. Interleukin-4 inhibits RANKL-induced NFATc1 expression via STAT6: A novel mechanism mediating its blockade of osteoclastogenesis. J. Cell. Biochem. 2011, 112, 3385–3392. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Wang, M.W.; Teitelbaum, S.L.; Ross, F.P. Interleukin-4 reversibly inhibits osteoclastogenesis via inhibition of NF-kappa B and mitogen-activated protein kinase signaling. J. Biol. Chem. 2002, 277, 6622–6630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaura, H.; Nagata, N.; Fujimura, Y.; Hotokezaka, H.; Tatamiya, M.; Nakao, N.; Yoshida, N.; Nakayama, K. Interleukin-4 directly inhibits tumor necrosis factor-alpha-mediated osteoclast formation in mouse bone marrow macrophages. Immunol. Lett. 2003, 88, 193–198. [Google Scholar] [CrossRef]
- Fujii, T.; Kitaura, H.; Kimura, K.; Hakami, Z.W.; Takano-Yamamoto, T. IL-4 inhibits TNF-alpha-mediated osteoclast formation by inhibition of RANKL expression in TNF-alpha-activated stromal cells and direct inhibition of TNF-alpha-activated osteoclast precursors via a T-cell-independent mechanism in vivo. Bone 2012, 51, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.E.; Fox, S.W. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwood, N.J.; Elliott, J.; Martin, T.J.; Gillespie, M.T. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J. Immunol. 2001, 166, 4915–4921. [Google Scholar] [CrossRef] [Green Version]
- Nagata, N.; Kitaura, H.; Yoshida, N.; Nakayama, K. Inhibition of RANKL-induced osteoclast formation in mouse bone marrow cells by IL-12: Involvement of IFN-gamma possibly induced from non-T cell population. Bone 2003, 33, 721–732. [Google Scholar] [CrossRef]
- Kitaura, H.; Nagata, N.; Fujimura, Y.; Hotokezaka, H.; Yoshida, N.; Nakayama, K. Effect of IL-12 on TNF-alpha-mediated osteoclast formation in bone marrow cells: Apoptosis mediated by Fas/Fas ligand interaction. J. Immunol. 2002, 169, 4732–4738. [Google Scholar] [CrossRef]
- Yoshimatsu, M.; Kitaura, H.; Fujimura, Y.; Eguchi, T.; Kohara, H.; Morita, Y.; Yoshida, N. IL-12 inhibits TNF-alpha induced osteoclastogenesis via a T cell-independent mechanism in vivo. Bone 2009, 45, 1010–1016. [Google Scholar] [CrossRef]
- Udagawa, N.; Horwood, N.J.; Elliott, J.; Mackay, A.; Owens, J.; Okamura, H.; Kurimoto, M.; Chambers, T.J.; Martin, T.J.; Gillespie, M.T. Interleukin-18 (interferon-gamma-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-gamma to inhibit osteoclast formation. J. Exp. Med. 1997, 185, 1005–1012. [Google Scholar] [CrossRef]
- Kitaura, H.; Tatamiya, M.; Nagata, N.; Fujimura, Y.; Eguchi, T.; Yoshida, N.; Nakayama, K. IL-18 induces apoptosis of adherent bone marrow cells in TNF-alpha mediated osteoclast formation in synergy with IL-12. Immunol. Lett. 2006, 107, 22–31. [Google Scholar] [CrossRef]
- Morita, Y.; Kitaura, H.; Yoshimatsu, M.; Fujimura, Y.; Kohara, H.; Eguchi, T.; Yoshida, N. IL-18 inhibits TNF-alpha-induced osteoclastogenesis possibly via a T cell-independent mechanism in synergy with IL-12 in vivo. Calcif. Tissue Int. 2010, 86, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, M.; Kitaura, H.; Fujimura, Y.; Kohara, H.; Morita, Y.; Yoshida, N. IL-12 Inhibits Lipopolysaccharide Stimulated Osteoclastogenesis in Mice. J. Immunol. Res. 2015, 2015, 214878. [Google Scholar] [CrossRef] [PubMed]
- Onoe, Y.; Miyaura, C.; Kaminakayashiki, T.; Nagai, Y.; Noguchi, K.; Chen, Q.R.; Seo, H.; Ohta, H.; Nozawa, S.; Kudo, I.; et al. IL-13 and IL-4 inhibit bone resorption by suppressing cyclooxygenase-2-dependent prostaglandin synthesis in osteoblasts. J. Immunol. 1996, 156, 758–764. [Google Scholar] [PubMed]
- Furukawa, M.; Takaishi, H.; Takito, J.; Yoda, M.; Sakai, S.; Hikata, T.; Hakozaki, A.; Uchikawa, S.; Matsumoto, M.; Chiba, K.; et al. IL-27 abrogates receptor activator of NF-kappa B ligand-mediated osteoclastogenesis of human granulocyte-macrophage colony-forming unit cells through STAT1-dependent inhibition of c-Fos. J. Immunol. 2009, 183, 2397–2406. [Google Scholar] [CrossRef]
- Kiyomiya, H.; Ariyoshi, W.; Okinaga, T.; Kaneuji, T.; Mitsugi, S.; Sakurai, T.; Habu, M.; Yoshioka, I.; Tominaga, K.; Nishihara, T. IL-33 inhibits RANKL-induced osteoclast formation through the regulation of Blimp-1 and IRF-8 expression. Biochem. Biophys. Res. Commun. 2015, 460, 320–326. [Google Scholar] [CrossRef]
- Ohori, F.; Kitaura, H.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Marahleh, A.; Nara, Y.; Pramusita, A.; Mizoguchi, I. IL-33 Inhibits TNF-alpha-Induced Osteoclastogenesis and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 1130. [Google Scholar] [CrossRef] [Green Version]
- Avnet, S.; Cenni, E.; Perut, F.; Granchi, D.; Brandi, M.L.; Giunti, A.; Baldini, N. Interferon-alpha inhibits in vitro osteoclast differentiation and renal cell carcinoma-induced angiogenesis. Int. J. Oncol. 2007, 30, 469–476. [Google Scholar]
- Lee, Y.; Hyung, S.W.; Jung, H.J.; Kim, H.J.; Staerk, J.; Constantinescu, S.N.; Chang, E.J.; Lee, Z.H.; Lee, S.W.; Kim, H.H. The ubiquitin-mediated degradation of Jak1 modulates osteoclastogenesis by limiting interferon-beta-induced inhibitory signaling. Blood 2008, 111, 885–893. [Google Scholar] [CrossRef]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef]
- Ji, J.D.; Park-Min, K.H.; Shen, Z.; Fajardo, R.J.; Goldring, S.R.; McHugh, K.P.; Ivashkiv, L.B. Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-gamma in human osteoclast precursors. J. Immunol. 2009, 183, 7223–7233. [Google Scholar] [CrossRef] [Green Version]
- Kohara, H.; Kitaura, H.; Fujimura, Y.; Yoshimatsu, M.; Morita, Y.; Eguchi, T.; Masuyama, R.; Yoshida, N. IFN-gamma directly inhibits TNF-alpha-induced osteoclastogenesis in vitro and in vivo and induces apoptosis mediated by Fas/Fas ligand interactions. Immunol. Lett. 2011, 137, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Divieti Pajevic, P.; Krause, D.S. Osteocyte regulation of bone and blood. Bone 2019, 119, 13–18. [Google Scholar] [CrossRef]
- Jilka, R.L.; Noble, B.; Weinstein, R.S. Osteocyte apoptosis. Bone 2013, 54, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Halleux, C.; Keller, H.; Pegurri, M.; Gooi, J.H.; Weber, P.B.; Feng, J.Q.; Bonewald, L.F.; Kneissel, M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell. Biol. 2010, 30, 3071–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Kim, A.R.; Choi, Y.H.; Jang, S.; Woo, G.H.; Cha, J.H.; Bak, E.J.; Yoo, Y.J. Tumor necrosis factor-alpha antagonist diminishes osteocytic RANKL and sclerostin expression in diabetes rats with periodontitis. PLoS ONE 2017, 12, e0189702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Han, X.; Shu, R.; Jiang, F.; Xu, L.; Xue, C.; Chen, T.; Bai, D. Effect of sclerostin removal in vivo on experimental periodontitis in mice. J. Oral Sci. 2016, 58, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Kar, R.; Riquelme, M.A.; Werner, S.; Jiang, J.X. Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J. Bone Miner. Res. 2013, 28, 1611–1621. [Google Scholar] [CrossRef] [Green Version]
- Bivi, N.; Condon, K.W.; Allen, M.R.; Farlow, N.; Passeri, G.; Brun, L.R.; Rhee, Y.; Bellido, T.; Plotkin, L.I. Cell autonomous requirement of connexin 43 for osteocyte survival: Consequences for endocortical resorption and periosteal bone formation. J. Bone Miner. Res. 2012, 27, 374–389. [Google Scholar] [CrossRef]
- Davis, H.M.; Pacheco-Costa, R.; Atkinson, E.G.; Brun, L.R.; Gortazar, A.R.; Harris, J.; Hiasa, M.; Bolarinwa, S.A.; Yoneda, T.; Ivan, M.; et al. Disruption of the Cx43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell 2017, 16, 551–563. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Bellido, T.; Roodman, G.D. Role of osteocytes in multiple myeloma bone disease. Curr. Opin. Support. Palliat. Care 2014, 8, 407–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, H.; Cui, Z.; Yang, S.; Ji, D.; Wang, Y.; Yang, Y.; Han, X.; Fan, Q.; Qin, A.; Wang, T.; et al. Targeting Osteocytes to Attenuate Early Breast Cancer Bone Metastasis by Theranostic Upconversion Nanoparticles with Responsive Plumbagin Release. ACS Nano 2017, 11, 7259–7273. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Arora, S.; Li, J.; Rahmani, R.; Sun, L.; Steinlauf, A.F.; Mechanick, J.I.; Zaidi, M. Bone, inflammation, and inflammatory bowel disease. Curr. Osteoporos. Rep. 2011, 9, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Eser, P.; Frotzler, A.; Zehnder, Y.; Wick, L.; Knecht, H.; Denoth, J.; Schiessl, H. Relationship between the duration of paralysis and bone structure: A pQCT study of spinal cord injured individuals. Bone 2004, 34, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Beggs, L.A.; Ye, F.; Ghosh, P.; Beck, D.T.; Conover, C.F.; Balaez, A.; Miller, J.R.; Phillips, E.G.; Zheng, N.; Williams, A.A.; et al. Sclerostin inhibition prevents spinal cord injury-induced cancellous bone loss. J. Bone Miner. Res. 2015, 30, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Metzger, C.E.; Gong, S.; Aceves, M.; Bloomfield, S.A.; Hook, M.A. Osteocytes reflect a pro-inflammatory state following spinal cord injury in a rodent model. Bone 2019, 120, 465–475. [Google Scholar] [CrossRef]
- Kennedy, O.D.; Laudier, D.M.; Majeska, R.J.; Sun, H.B.; Schaffler, M.B. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone 2014, 64, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Hoscheit, M.; Conner, G.; Roemer, J.; Vuckovska, A.; Abbasnia, P.; Vana, P.; Shankar, R.; Kennedy, R.; Callaci, J. Burn Injury Has Skeletal Site-Specific Effects on Bone Integrity and Markers of Bone Remodeling. J. Burn Care Res. 2016, 37, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Takemura, Y.; Moriyama, Y.; Ayukawa, Y.; Kurata, K.; Rakhmatia, Y.D.; Koyano, K. Mechanical loading induced osteocyte apoptosis and connexin 43 expression in three-dimensional cell culture and dental implant model. J. Biomed. Mater. Res. Part A 2019, 107, 815–827. [Google Scholar] [CrossRef]
- Nile, C.J.; Sherrabeh, S.; Ramage, G.; Lappin, D.F. Comparison of circulating tumour necrosis factor superfamily cytokines in periodontitis patients undergoing supportive therapy: A case-controlled cross-sectional study comparing smokers and non-smokers in health and disease. J. Clin. Periodontol. 2013, 40, 875–882. [Google Scholar] [CrossRef]
- Chen, C.H.; Chen, H.A.; Liao, H.T.; Liu, C.H.; Tsai, C.Y.; Chou, C.T. Soluble receptor activator of nuclear factor-kappaB ligand (RANKL) and osteoprotegerin in ankylosing spondylitis: OPG is associated with poor physical mobility and reflects systemic inflammation. Clin. Rheumatol. 2010, 29, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Geusens, P.P.; Landewé, R.B.; Garnero, P.; Chen, D.; Dunstan, C.R.; Lems, W.F.; Stinissen, P.; van der Heijde, D.M.; van der Linden, S.; Boers, M. The ratio of circulating osteoprotegerin to RANKL in early rheumatoid arthritis predicts later joint destruction. Arthritis Rheum. 2006, 54, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Sarink, D.; Schock, H.; Johnson, T.; Overvad, K.; Holm, M.; Tjønneland, A.; Boutron-Ruault, M.-C.; His, M.; Kvaskoff, M.; Boeing, H.; et al. Circulating RANKL and RANKL/OPG and Breast Cancer Risk by ER and PR Subtype: Results from the EPIC Cohort. Cancer Prev. Res. 2017, 10, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Cawley, K.; Piemontese, M.; Fujiwara, Y.; Zhao, H.; Goellner, J.J.; O’Brien, C.A. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat. Commun. 2018, 9, 2909. [Google Scholar] [CrossRef] [PubMed]
- Kurata, K.; Heino, T.J.; Higaki, H.; Väänänen, H.K. Bone marrow cell differentiation induced by mechanically damaged osteocytes in 3D gel-embedded culture. J. Bone Miner. Res. 2006, 21, 616–625. [Google Scholar] [CrossRef]
- Tracey, K.J.; Cerami, A. Tumor necrosis factor, other cytokines and disease. Annu. Rev. Cell Biol. 1993, 9, 317–343. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Baek, K.; Hwang, H.R.; Park, H.J.; Kwon, A.; Qadir, A.S.; Ko, S.H.; Woo, K.M.; Ryoo, H.M.; Kim, G.S.; Baek, J.H. TNF-alpha upregulates sclerostin expression in obese mice fed a high-fat diet. J. Cell. Physiol. 2014, 229, 640–650. [Google Scholar] [CrossRef]
- Garlet, G.P. Destructive and protective roles of cytokines in periodontitis: A re-appraisal from host defense and tissue destruction viewpoints. J. Dent. Res. 2010, 89, 1349–1363. [Google Scholar] [CrossRef]
- Salvi, G.E.; Beck, J.D.; Offenbacher, S. PGE2, IL-1 beta, and TNF-alpha responses in diabetics as modifiers of periodontal disease expression. Ann. Periodontol. 1998, 3, 40–50. [Google Scholar] [CrossRef]
- Kang, J.; Boonanantanasarn, K.; Baek, K.; Woo, K.M.; Ryoo, H.M.; Baek, J.H.; Kim, G.S. Hyperglycemia increases the expression levels of sclerostin in a reactive oxygen species- and tumor necrosis factor-alpha-dependent manner. J. Periodontal Implant Sci. 2015, 45, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidwell, J.P.; Yang, J.; Robling, A.G. Is HMGB1 an osteocyte alarmin? J. Cell. Biochem. 2008, 103, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claude-Taupin, A.; Bissa, B.; Jia, J.; Gu, Y.; Deretic, V. Role of autophagy in IL-1beta export and release from cells. Semin. Cell Dev. Biol. 2018, 83, 36–41. [Google Scholar] [CrossRef]
- Dinarello, C.A. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 612–632. [Google Scholar] [CrossRef]
- Almehmadi, A.H.; Alghamdi, F. Biomarkers of alveolar bone resorption in gingival crevicular fluid: A systematic review. Arch. Oral Biol. 2018, 93, 12–21. [Google Scholar] [CrossRef]
- McLean, R.R. Proinflammatory cytokines and osteoporosis. Curr. Osteoporos. Rep. 2009, 7, 134–139. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Bakker, A.D.; Everts, V.; Klein-Nulend, J. Mechanical loading prevents the stimulating effect of IL-1beta on osteocyte-modulated osteoclastogenesis. Biochem. Biophys. Res. Commun. 2012, 420, 11–16. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, D.E.; Woo, G.H.; Cha, J.H.; Bak, E.J.; Yoo, Y.J. Osteocytic Sclerostin Expression in Alveolar Bone in Rats With Diabetes Mellitus and Ligature-Induced Periodontitis. J. Periodontol. 2015, 86, 1005–1011. [Google Scholar] [CrossRef]
- Qu, X.; Mei, J.; Yu, Z.; Zhai, Z.; Qiao, H.; Dai, K. Lenalidomide regulates osteocytes fate and related osteoclastogenesis via IL-1beta/NF-kappaB/RANKL signaling. Biochem. Biophys. Res. Commun. 2018, 501, 547–555. [Google Scholar] [CrossRef]
- Kushner, I. The acute phase response: From Hippocrates to cytokine biology. Eur. Cytokine Netw. 1991, 2, 75–80. [Google Scholar] [PubMed]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar] [CrossRef]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotz, M. Interleukin-6: A comprehensive review. Cancer Treat. Res. 1995, 80, 209–233. [Google Scholar] [CrossRef]
- Uciechowski, P.; Dempke, W.C.M. Interleukin-6: A Masterplayer in the Cytokine Network. Oncology 2020, 98, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Katagiri, T.; Tamura, T.; Wada, S.; Findlay, D.M.; Martin, T.J.; Hirota, H.; Taga, T.; Kishimoto, T.; et al. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J. Exp. Med. 1995, 182, 1461–1468. [Google Scholar] [CrossRef]
- De Benedetti, F.; Rucci, N.; Del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired skeletal development in interleukin-6-transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006, 54, 3551–3563. [Google Scholar] [CrossRef]
- Devlin, R.D.; Reddy, S.V.; Savino, R.; Ciliberto, G.; Roodman, G.D. IL-6 mediates the effects of IL-1 or TNF, but not PTHrP or 1,25(OH)2D3, on osteoclast-like cell formation in normal human bone marrow cultures. J. Bone Miner. Res. 1998, 13, 393–399. [Google Scholar] [CrossRef]
- Machado, V.; Botelho, J.; Lopes, J.; Patrao, M.; Alves, R.; Chambrone, L.; Alcoforado, G.; Mendes, J.J. Periodontitis Impact in Interleukin-6 Serum Levels in Solid Organ Transplanted Patients: A Systematic Review and Meta-Analysis. Diagnostics 2020, 10, 184. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, M.; Mihara, M. The roles of interleukin-6 in the pathogenesis of rheumatoid arthritis. Arthritis 2011, 2011, 765624. [Google Scholar] [CrossRef]
- Colmenero, J.D.; Ruiz-Mesa, J.D.; Plata, A.; Bermudez, P.; Martin-Rico, P.; Queipo-Ortuno, M.I.; Reguera, J.M. Clinical findings, therapeutic approach, and outcome of brucellar vertebral osteomyelitis. Clin. Infect. Dis. 2008, 46, 426–433. [Google Scholar] [CrossRef]
- Yu, K.; Ma, Y.; Li, X.; Wu, X.; Liu, W.; Li, X.; Shen, J.; Wang, H. Lipopolysaccharide increases IL-6 secretion via activation of the ERK1/2 signaling pathway to up-regulate RANKL gene expression in MLO-Y4 cells. Cell Biol. Int. 2017, 41, 84–92. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell. Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, W.; Wu, X.; Gou, M.; Shen, J.; Wang, H. Advanced glycation end products induced IL-6 and VEGF-A production and apoptosis in osteocyte-like MLO-Y4 cells by activating RAGE and ERK1/2, P38 and STAT3 signalling pathways. Int. Immunopharmacol. 2017, 52, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Yamaguchi, T.; Kanazawa, I.; Sugimoto, T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochem. Biophys. Res. Commun. 2015, 461, 193–199. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F.; Wacker, M.J. FGF23 production by osteocytes. Pediatr. Nephrol. 2013, 28, 563–568. [Google Scholar] [CrossRef]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiko, Y.; Wang, H.; Minamizaki, T.; Ijuin, C.; Yamamoto, R.; Suemune, S.; Kozai, K.; Tanne, K.; Aubin, J.E.; Maeda, N. Mineralized tissue cells are a principal source of FGF23. Bone 2007, 40, 1565–1573. [Google Scholar] [CrossRef]
- Consortium, A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef] [Green Version]
- The HYP Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat. Genet. 1995, 11, 130–136. [Google Scholar] [CrossRef]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Laurence, J.S.; Schwarz, P.M.; Lafer, E.M.; Hedge, A.M.; Rowe, P.S. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology 2008, 149, 1757–1772. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Guo, R.; Simpson, L.G.; Xiao, Z.S.; Burnham, C.E.; Quarles, L.D. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J. Biol. Chem. 2003, 278, 37419–37426. [Google Scholar] [CrossRef] [Green Version]
- Rowe, P.S.; Kumagai, Y.; Gutierrez, G.; Garrett, I.R.; Blacher, R.; Rosen, D.; Cundy, J.; Navvab, S.; Chen, D.; Drezner, M.K.; et al. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 2004, 34, 303–319. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Rowe, P.S.N.; Vierthaler, L.; Zhou, J.; Quarles, L.D. Phosphorylated acidic serine-aspartate-rich MEPE-associated motif peptide from matrix extracellular phosphoglycoprotein inhibits phosphate regulating gene with homologies to endopeptidases on the X-chromosome enzyme activity. J. Endocrinol. 2007, 192, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Allard, L.; Demoncheaux, N.; Machuca-Gayet, I.; Georgess, D.; Coury-Lucas, F.; Jurdic, P.; Bacchetta, J. Biphasic Effects of Vitamin D and FGF23 on Human Osteoclast Biology. Calcif. Tissue Int. 2015, 97, 69–79. [Google Scholar] [CrossRef]
- Hayashibara, T.; Hiraga, T.; Sugita, A.; Wang, L.; Hata, K.; Ooshima, T.; Yoneda, T. Regulation of osteoclast differentiation and function by phosphate: Potential role of osteoclasts in the skeletal abnormalities in hypophosphatemic conditions. J. Bone Miner. Res. 2007, 22, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Hollberg, K.; Marsell, R.; Norgard, M.; Larsson, T.; Jonsson, K.B.; Andersson, G. Osteoclast polarization is not required for degradation of bone matrix in rachitic FGF23 transgenic mice. Bone 2008, 42, 1111–1121. [Google Scholar] [CrossRef]
- Morena, M.; Jaussent, I.; Dupuy, A.M.; Bargnoux, A.S.; Kuster, N.; Chenine, L.; Leray-Moragues, H.; Klouche, K.; Vernhet, H.; Canaud, B.; et al. Osteoprotegerin and sclerostin in chronic kidney disease prior to dialysis: Potential partners in vascular calcifications. Nephrol. Dial. Transplant. 2015, 30, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, N.; Findlay, D.M.; Anderson, P.H.; Bonewald, L.F.; Atkins, G.J. Extracellular phosphate modulates the effect of 1alpha,25-dihydroxy vitamin D3 (1,25D) on osteocyte like cells. J. Steroid Biochem. Mol. Biol. 2013, 136, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Wijenayaka, A.R.; Prideaux, M.; Kogawa, M.; Ormsby, R.T.; Evdokiou, A.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol. Cell. Endocrinol. 2015, 399, 208–218. [Google Scholar] [CrossRef]
- Sato, H.; Kazama, J.J.; Murasawa, A.; Otani, H.; Abe, A.; Ito, S.; Ishikawa, H.; Nakazono, K.; Kuroda, T.; Nakano, M.; et al. Serum Fibroblast Growth Factor 23 (FGF23) in Patients with Rheumatoid Arthritis. Intern. Med. 2016, 55, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Ohlsson, C.; Mohan, S.; Sjogren, K.; Tivesten, A.; Isgaard, J.; Isaksson, O.; Jansson, J.O.; Svensson, J. The role of liver-derived insulin-like growth factor-I. Endocr. Rev. 2009, 30, 494–535. [Google Scholar] [CrossRef] [Green Version]
- Yakar, S.; Isaksson, O. Regulation of skeletal growth and mineral acquisition by the GH/IGF-1 axis: Lessons from mouse models. Growth Horm. IGF Res. 2016, 28, 26–42. [Google Scholar] [CrossRef] [Green Version]
- Lean, J.M.; Mackay, A.G.; Chow, J.W.; Chambers, T.J. Osteocytic expression of mRNA for c-fos and IGF-I: An immediate early gene response to an osteogenic stimulus. Am. J. Physiol. 1996, 270, E937–E945. [Google Scholar] [CrossRef]
- Sheng, M.H.; Zhou, X.D.; Bonewald, L.F.; Baylink, D.J.; Lau, K.H. Disruption of the insulin-like growth factor-1 gene in osteocytes impairs developmental bone growth in mice. Bone 2013, 52, 133–144. [Google Scholar] [CrossRef]
- Lean, J.M.; Jagger, C.J.; Chambers, T.J.; Chow, J.W. Increased insulin-like growth factor I mRNA expression in rat osteocytes in response to mechanical stimulation. Am. J. Physiol. 1995, 268, E318–E327. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Wang, Y.; Bikle, D.D. IGF-1 signaling mediated cell-specific skeletal mechano-transduction. J. Orthop. Res. 2018, 36, 576–583. [Google Scholar] [CrossRef]
- Zhang, W.; Shen, X.; Wan, C.; Zhao, Q.; Zhang, L.; Zhou, Q.; Deng, L. Effects of insulin and insulin-like growth factor 1 on osteoblast proliferation and differentiation: Differential signalling via Akt and ERK. Cell Biochem. Funct. 2012, 30, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Mu, J.; Fan, Z.; Lei, G.; Yan, M.; Wang, S.; Tang, C.; Wang, Z.; Yu, J.; Zhang, G. Insulin-like growth factor 1 enhances the proliferation and osteogenic differentiation of human periodontal ligament stem cells via ERK and JNK MAPK pathways. Histochem. Cell Biol 2012, 137, 513–525. [Google Scholar] [CrossRef]
- Elis, S.; Courtland, H.W.; Wu, Y.; Fritton, J.C.; Sun, H.; Rosen, C.J.; Yakar, S. Elevated serum IGF-1 levels synergize PTH action on the skeleton only when the tissue IGF-1 axis is intact. J. Bone Miner. Res. 2010, 25, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Crane, J.L.; Xie, L.; Xian, L.; Xie, H.; Cao, X. IGF-I induced phosphorylation of PTH receptor enhances osteoblast to osteocyte transition. Bone Res. 2018, 6, 5. [Google Scholar] [CrossRef]
- Hill, P.A.; Reynolds, J.J.; Meikle, M.C. Osteoblasts mediate insulin-like growth factor-I and -II stimulation of osteoclast formation and function. Endocrinology 1995, 136, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nishida, S.; Elalieh, H.Z.; Long, R.K.; Halloran, B.P.; Bikle, D.D. Role of IGF-I signaling in regulating osteoclastogenesis. J. Bone Miner. Res. 2006, 21, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Menendez, A.; Fong, C.; ElAlieh, H.Z.; Chang, W.; Bikle, D.D. Ephrin B2/EphB4 mediates the actions of IGF-I signaling in regulating endochondral bone formation. J. Bone Miner. Res. 2014, 29, 1900–1913. [Google Scholar] [CrossRef] [Green Version]
- Holmes, W.E.; Lee, J.; Kuang, W.J.; Rice, G.C.; Wood, W.I. Structure and functional expression of a human interleukin-8 receptor. Science 1991, 253, 1278–1280. [Google Scholar] [CrossRef]
- Waugh, D.J.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osiri, M.; Wongpiyabovorn, J.; Sattayasomboon, Y.; Thammacharoenrach, N. Inflammatory cytokine levels, disease activity, and function of patients with rheumatoid arthritis treated with combined conventional disease-modifying antirheumatic drugs or biologics. Clin. Rheumatol. 2016, 35, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Bendre, M.S.; Margulies, A.G.; Walser, B.; Akel, N.S.; Bhattacharrya, S.; Skinner, R.A.; Swain, F.; Ramani, V.; Mohammad, K.S.; Wessner, L.L.; et al. Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res. 2005, 65, 11001–11009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopesky, P.; Tiedemann, K.; Alkekhia, D.; Zechner, C.; Millard, B.; Schoeberl, B.; Komarova, S.V. Autocrine signaling is a key regulatory element during osteoclastogenesis. Biol. Open 2014, 3, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.B.; Garcia-Gomez, A.; Garayoa, M.; Corchete, L.A.; Hernandez, J.M.; San Miguel, J.; Gutierrez, N.C. Effects of IL-8 Up-Regulation on Cell Survival and Osteoclastogenesis in Multiple Myeloma. Am. J. Pathol. 2016, 186, 2171–2182. [Google Scholar] [CrossRef] [Green Version]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Xu, G.; Shi, Y.; Leith, C.P.; Kim, K.; Trivedi, P.; Kim, J.; Hematti, P.; et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol. Cancer 2010, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Catrina, A.I.; Svensson, C.I.; Malmstrom, V.; Schett, G.; Klareskog, L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 79–86. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Z.; Lan, T.; Liang, P.; Tao, Q. Upregulation of interleukin-8 and activin A induces osteoclastogenesis in ameloblastoma. Int. J. Mol. Med. 2019, 43, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Sosnoski, D.M.; Mastro, A.M. Breast cancer metastasis to the bone: Mechanisms of bone loss. Breast Cancer Res. 2010, 12, 215. [Google Scholar] [CrossRef] [Green Version]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef]
- Kodama, H.; Nose, M.; Niida, S.; Yamasaki, A. Essential role of macrophage colony-stimulating factor in the osteoclast differentiation supported by stromal cells. J. Exp. Med. 1991, 173, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Begg, S.K.; Radley, J.M.; Pollard, J.W.; Chisholm, O.T.; Stanley, E.R.; Bertoncello, I. Delayed hematopoietic development in osteopetrotic (op/op) mice. J. Exp. Med. 1993, 177, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Barleon, B.; Siemeister, G.; Martiny-Baron, G.; Weindel, K.; Herzog, C.; Marme, D. Vascular endothelial growth factor up-regulates its receptor fms-like tyrosine kinase 1 (FLT-1) and a soluble variant of FLT-1 in human vascular endothelial cells. Cancer Res. 1997, 57, 5421–5425. [Google Scholar]
- Yang, Q.; McHugh, K.P.; Patntirapong, S.; Gu, X.; Wunderlich, L.; Hauschka, P.V. VEGF enhancement of osteoclast survival and bone resorption involves VEGF receptor-2 signaling and beta3-integrin. Matrix Biol. 2008, 27, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Niida, S.; Kaku, M.; Amano, H.; Yoshida, H.; Kataoka, H.; Nishikawa, S.; Tanne, K.; Maeda, N.; Nishikawa, S.; Kodama, H. Vascular endothelial growth factor can substitute for macrophage colony-stimulating factor in the support of osteoclastic bone resorption. J. Exp. Med. 1999, 190, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, M.; Kaneda, T.; Arakawa, T.; Morita, S.; Sato, T.; Yomada, T.; Hanada, K.; Kumegawa, M.; Hakeda, Y. Vascular endothelial growth factor (VEGF) directly enhances osteoclastic bone resorption and survival of mature osteoclasts. FEBS Lett. 2000, 473, 161–164. [Google Scholar] [CrossRef]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bezooijen, R.L.; Roelen, B.A.; Visser, A.; van der Wee-Pals, L.; de Wilt, E.; Karperien, M.; Hamersma, H.; Papapoulos, S.E.; ten Dijke, P.; Lowik, C.W. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 2004, 199, 805–814. [Google Scholar] [CrossRef]
- Kusu, N.; Laurikkala, J.; Imanishi, M.; Usui, H.; Konishi, M.; Miyake, A.; Thesleff, I.; Itoh, N. Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J. Biol. Chem. 2003, 278, 24113–24117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odagaki, N.; Ishihara, Y.; Wang, Z.; Ei Hsu Hlaing, E.; Nakamura, M.; Hoshijima, M.; Hayano, S.; Kawanabe, N.; Kamioka, H. Role of Osteocyte-PDL Crosstalk in Tooth Movement via SOST/Sclerostin. J. Dent. Res. 2018, 97, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol Chem 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robling, A.G.; Bonewald, L.F. The Osteocyte: New Insights. Annu. Rev. Physiol. 2020, 82, 485–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehmeyer, C.; Frank, S.; Beckmann, D.; Bottcher, M.; Cromme, C.; Konig, U.; Fennen, M.; Held, A.; Paruzel, P.; Hartmann, C.; et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci. Transl. Med. 2016, 8, 330ra335. [Google Scholar] [CrossRef]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrzyk, B.; Wyskida, K.; Ficek, J.; Kolonko, A.; Ficek, R.; Wiecek, A.; Olszanecka-Glinianowicz, M.; Chudek, J. Relationship between plasma levels of sclerostin, calcium-phosphate disturbances, established markers of bone turnover, and inflammation in haemodialysis patients. Int. Urol. Nephrol. 2019, 51, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Naylor, K.L.; McArthur, E.; Leslie, W.D.; Fraser, L.A.; Jamal, S.A.; Cadarette, S.M.; Pouget, J.G.; Lok, C.E.; Hodsman, A.B.; Adachi, J.D.; et al. The three-year incidence of fracture in chronic kidney disease. Kidney Int. 2014, 86, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Chandra, A.; Lin, T.; Young, T.; Tong, W.; Ma, X.; Tseng, W.J.; Kramer, I.; Kneissel, M.; Levine, M.A.; Zhang, Y.; et al. Suppression of Sclerostin Alleviates Radiation-Induced Bone Loss by Protecting Bone-Forming Cells and Their Progenitors Through Distinct Mechanisms. J. Bone Miner. Res. 2017, 32, 360–372. [Google Scholar] [CrossRef]
- O’Garra, A.; Barrat, F.J.; Castro, A.G.; Vicari, A.; Hawrylowicz, C. Strategies for use of IL-10 or its antagonists in human disease. Immunol. Rev. 2008, 223, 114–131. [Google Scholar] [CrossRef]
- Mosser, D.M.; Zhang, X. Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.X.; Kukita, T.; Kukita, A.; Otsuka, T.; Niho, Y.; Iijima, T. Interleukin-10 selectively inhibits osteoclastogenesis by inhibiting differentiation of osteoclast progenitors into preosteoclast-like cells in rat bone marrow culture system. J. Cell. Physiol. 1995, 165, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasheed, A.; Scheerens, H.; Srivastava, A.K.; Rennick, D.M.; Tatakis, D.N. Accelerated alveolar bone loss in mice lacking interleukin-10: Late onset. J. Periodontal. Res. 2004, 39, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Kakuta, S.; Shimizu, K.; Kadoki, M.; Kamiya, T.; Shimazu, T.; Kubo, S.; Saijo, S.; Ishigame, H.; Nakae, S.; et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Nat. Immunol. 2018, 19, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.F.; Pan, H.Y.; Ying, X.H.; Lou, J.; Ji, J.S.; Zou, H. Mast Cells Comprise the Major of Interleukin 17-Producing Cells and Predict a Poor Prognosis in Hepatocellular Carcinoma. Medicine 2016, 95, e3220. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Zhou, K.; Liang, Q.L.; Lin, S.; Wang, Y.C.; Xiong, X.Y.; Meng, Z.Y.; Zhao, T.; Zhu, W.Y.; Yang, Y.R.; et al. Interleukin-23 Secreted by Activated Macrophages Drives gammadeltaT Cell Production of Interleukin-17 to Aggravate Secondary Injury After Intracerebral Hemorrhage. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Milosavljevic, N.; Gazdic, M.; Simovic Markovic, B.; Arsenijevic, A.; Nurkovic, J.; Dolicanin, Z.; Djonov, V.; Lukic, M.L.; Volarevic, V. Mesenchymal stem cells attenuate acute liver injury by altering ratio between interleukin 17 producing and regulatory natural killer T cells. Liver Transpl. 2017, 23, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Li, T.J.; Jiang, Y.M.; Hu, Y.F.; Huang, L.; Yu, J.; Zhao, L.Y.; Deng, H.J.; Mou, T.Y.; Liu, H.; Yang, Y.; et al. Interleukin-17-Producing Neutrophils Link Inflammatory Stimuli to Disease Progression by Promoting Angiogenesis in Gastric Cancer. Clin. Cancer Res. 2017, 23, 1575–1585. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Liu, M.; Xiong, H.; Peng, B. Up-regulation of IL-23 expression in human dental pulp fibroblasts by IL-17 via activation of the NF-kappaB and MAPK pathways. Int. Endod. J. 2018, 51, 622–631. [Google Scholar] [CrossRef]
- Hu, Z.; Luo, D.; Wang, D.; Ma, L.; Zhao, Y.; Li, L. IL-17 Activates the IL-6/STAT3 Signal Pathway in the Proliferation of Hepatitis B Virus-Related Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2017, 43, 2379–2390. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, S.; Song, X.; Hu, Q.; Pan, W. IL-17 enhances oxidative stress in hepatocytes through Nrf2/keap1 signal pathway activation. Int. J. Clin. Exp. Pathol. 2018, 11, 3318–3323. [Google Scholar] [PubMed]
- Hot, A.; Lavocat, F.; Lenief, V.; Miossec, P. Simvastatin inhibits the pro-inflammatory and pro-thrombotic effects of IL-17 and TNF-alpha on endothelial cells. Ann. Rheum. Dis. 2013, 72, 754–760. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, J.; Chen, L.; Liu, T.; Xu, G.; Li, C.; Yuan, W.; Xu, H.; Su, Z. IL-17 contributed to the neuropathic pain following peripheral nerve injury by promoting astrocyte proliferation and secretion of proinflammatory cytokines. Mol. Med. Rep. 2017, 15, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Jia, S.; Gao, T.; Zhang, R.; Liu, Z.; Wang, Y. Dexmedetomidine mitigate acute lung injury by inhibiting IL-17-induced inflammatory reaction. Immunobiology 2018, 223, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, S.; Martini, E.; Bergh, K.; Chang, D.; Rethi, B.; Stahle, M.; Eidsmo, L. Granzyme A potentiates chemokine production in IL-17-stimulated keratinocytes. Exp. Dermatol. 2017, 26, 824–827. [Google Scholar] [CrossRef] [Green Version]
- Adamopoulos, I.E.; Chao, C.C.; Geissler, R.; Laface, D.; Blumenschein, W.; Iwakura, Y.; McClanahan, T.; Bowman, E.P. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res. Ther. 2010, 12, R29. [Google Scholar] [CrossRef] [Green Version]
- Yago, T.; Nanke, Y.; Ichikawa, N.; Kobashigawa, T.; Mogi, M.; Kamatani, N.; Kotake, S. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: A novel mechanism of osteoclastogenesis by IL-17. J. Cell. Biochem. 2009, 108, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Rasool, M. Interleukin 17 regulates SHP-2 and IL-17RA/STAT-3 dependent Cyr61, IL-23 and GM-CSF expression and RANKL mediated osteoclastogenesis by fibroblast-like synoviocytes in rheumatoid arthritis. Mol. Immunol. 2017, 91, 134–144. [Google Scholar] [CrossRef]
- Zhang, F.; Tanaka, H.; Kawato, T.; Kitami, S.; Nakai, K.; Motohashi, M.; Suzuki, N.; Wang, C.L.; Ochiai, K.; Isokawa, K.; et al. Interleukin-17A induces cathepsin K and MMP-9 expression in osteoclasts via celecoxib-blocked prostaglandin E2 in osteoblasts. Biochimie 2011, 93, 296–305. [Google Scholar] [CrossRef]
- Liao, C.; Cheng, T.; Wang, S.; Zhang, C.; Jin, L.; Yang, Y. Shear stress inhibits IL-17A-mediated induction of osteoclastogenesis via osteocyte pathways. Bone 2017, 101, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein-Nulend, J.; Bakker, A.D.; Bacabac, R.G.; Vatsa, A.; Weinbaum, S. Mechanosensation and transduction in osteocytes. Bone 2013, 54, 182–190. [Google Scholar] [CrossRef]
- Li, J.Y.; Yu, M.; Tyagi, A.M.; Vaccaro, C.; Hsu, E.; Adams, J.; Bellido, T.; Weitzmann, M.N.; Pacifici, R. IL-17 Receptor Signaling in Osteoblasts/Osteocytes Mediates PTH-Induced Bone Loss and Enhances Osteocytic RANKL Production. J. Bone Miner. Res. 2019, 34, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arican, O.; Kurutas, E.B.; Sasmaz, S. Oxidative stress in patients with acne vulgaris. Mediators Inflamm. 2005, 2005, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Gensure, R.C.; Gardella, T.J.; Jüppner, H. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem. Biophys. Res. Commun. 2005, 328, 666–678. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Plotkin, L.I.; Galli, C.; Goellner, J.J.; Gortazar, A.R.; Allen, M.R.; Robling, A.G.; Bouxsein, M.; Schipani, E.; Turner, C.H.; et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 2008, 3, e2942. [Google Scholar] [CrossRef] [Green Version]
- Powell, W.F., Jr.; Barry, K.J.; Tulum, I.; Kobayashi, T.; Harris, S.E.; Bringhurst, F.R.; Pajevic, P.D. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J. Endocrinol. 2011, 209, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Bellido, T.; Saini, V.; Pajevic, P.D. Effects of PTH on osteocyte function. Bone 2013, 54, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Bellido, T.; Ali, A.A.; Gubrij, I.; Plotkin, L.I.; Fu, Q.; O’Brien, C.A.; Manolagas, S.C.; Jilka, R.L. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: A novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 2005, 146, 4577–4583. [Google Scholar] [CrossRef]
- Van Lierop, A.H.; Witteveen, J.E.; Hamdy, N.A.; Papapoulos, S.E. Patients with primary hyperparathyroidism have lower circulating sclerostin levels than euparathyroid controls. Eur. J. Endocrinol. 2010, 163, 833–837. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.-R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145169
Kitaura H, Marahleh A, Ohori F, Noguchi T, Shen W-R, Qi J, Nara Y, Pramusita A, Kinjo R, Mizoguchi I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. International Journal of Molecular Sciences. 2020; 21(14):5169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145169
Chicago/Turabian StyleKitaura, Hideki, Aseel Marahleh, Fumitoshi Ohori, Takahiro Noguchi, Wei-Ren Shen, Jiawei Qi, Yasuhiko Nara, Adya Pramusita, Ria Kinjo, and Itaru Mizoguchi. 2020. "Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption" International Journal of Molecular Sciences 21, no. 14: 5169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145169