The Background K+ Channel TRESK in Sensory Physiology and Pain

 , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TRESK Identification and Expression
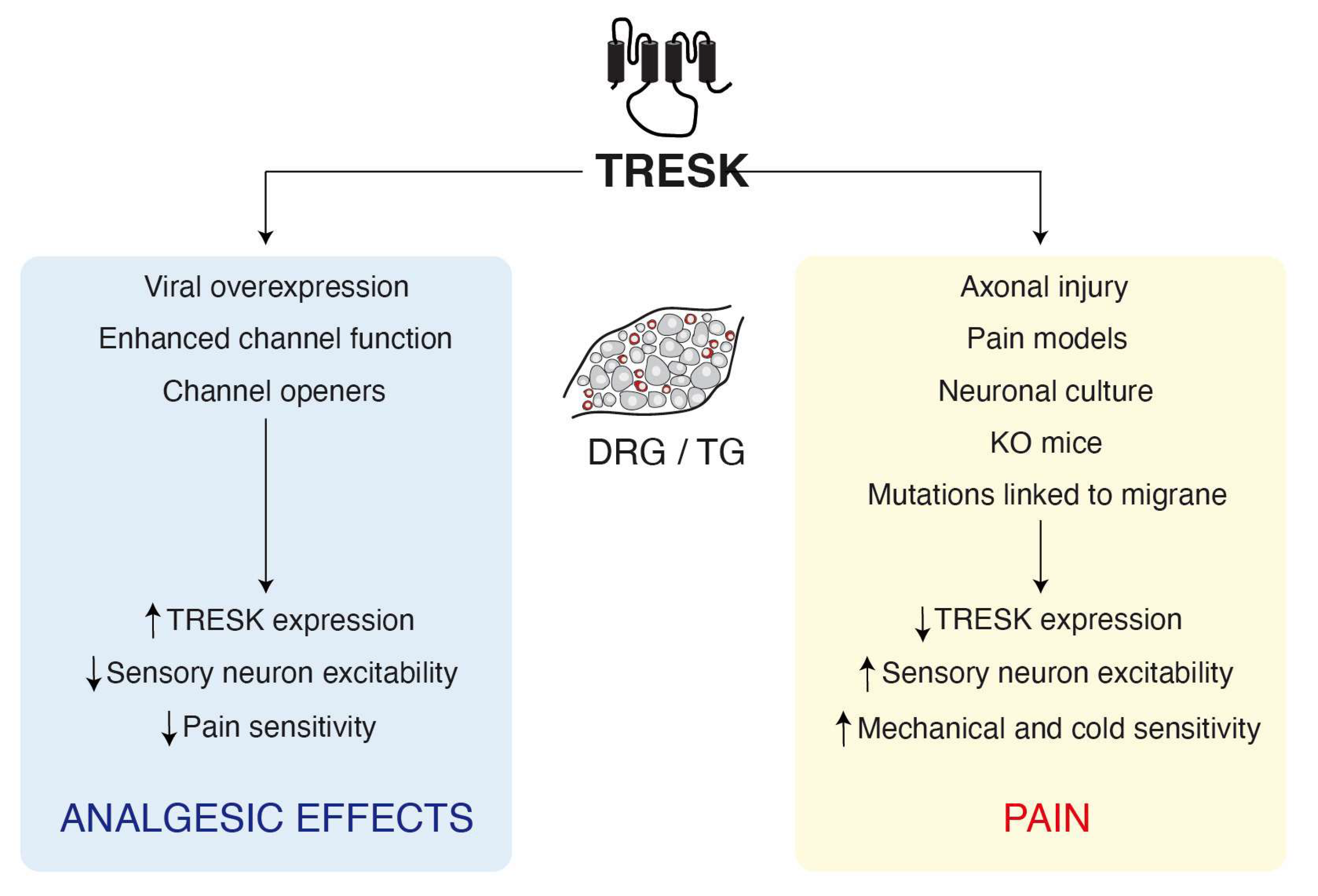
3. Involvement of TRESK in Pain
3.1. TRESK Expression and Sensory Neuron Excitability
3.2. Calcineurin Modulation of TRESK and Pain Sensitivity
3.3. TRESK and Migraine
3.4. TRESK and Mechanosensation
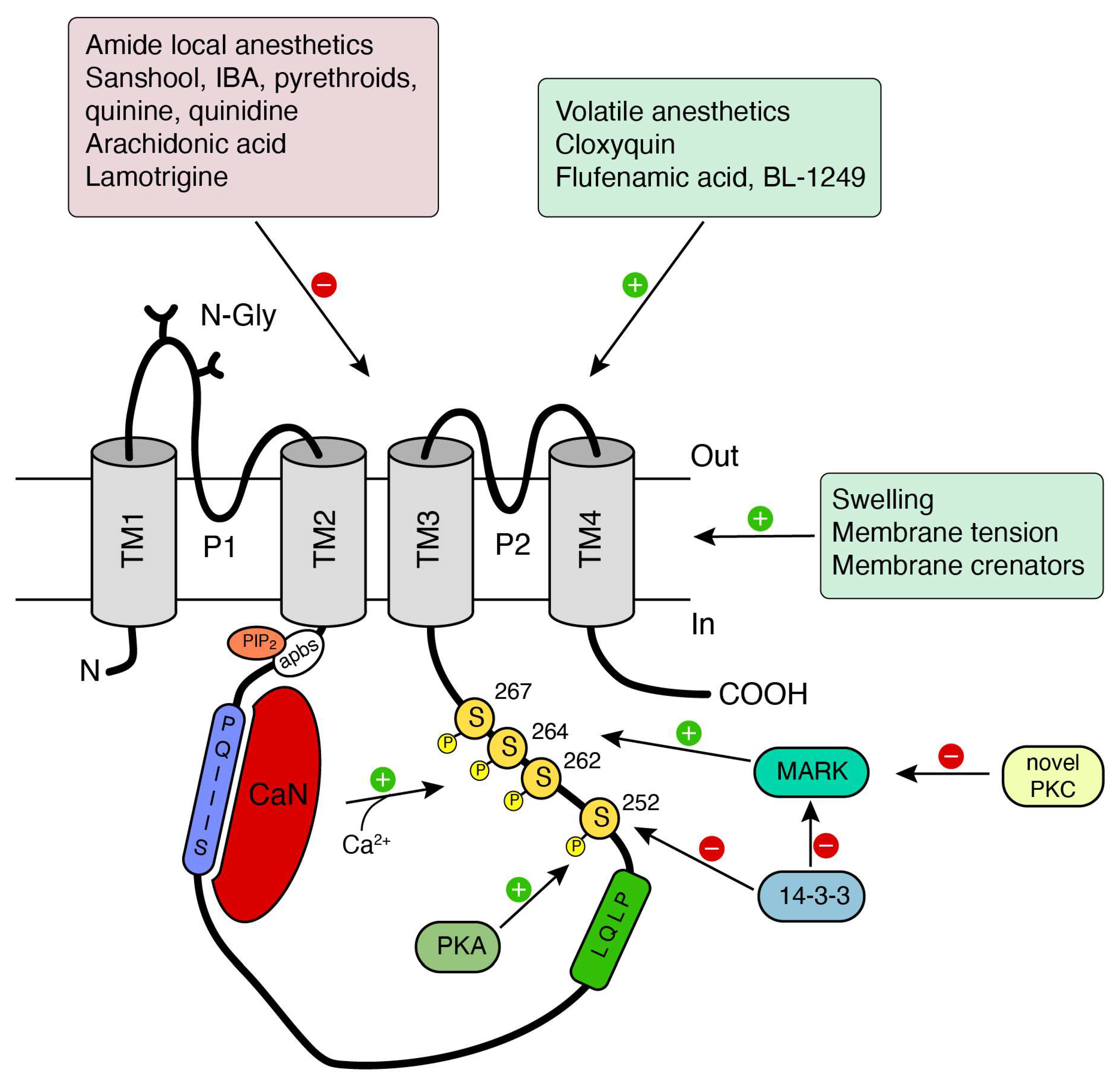
4. TRESK Modulation and Pharmacology: Therapeutics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alexander, S.P.H.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. The Concise Guide to Pharmacology 2019/20: Ion channels. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S142–S228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enyedi, P.; Czirják, G. Molecular Background of Leak K+ Currents: Two-Pore Domain Potassium Channels. Physiol. Rev. 2010, 90, 559–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, M.; Duprat, F.; Lesage, F.; Reyes, R.; Romey, G.; Heurteaux, C.; Lazdunski, M. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 1996, 15, 6854–6862. [Google Scholar] [CrossRef]
- Patel, A.J.; Honore, E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001, 24, 339–346. [Google Scholar] [CrossRef]
- Honore, E. The neuronal background K2P channels: Focus on TREK1. Nat. Rev. Neurosci. 2007, 8, 251–261. [Google Scholar] [CrossRef]
- Renigunta, V.; Schlichthörl, G.; Daut, J. Much more than a leak: Structure and function of K2P-channels. Pflügers Arch. 2015, 467, 867–894. [Google Scholar] [CrossRef]
- Feliciangeli, S.; Chatelain, F.C.; Bichet, D.; Lesage, F. The family of K2Pchannels: Salient structural and functional properties. J. Physiol. 2015, 593, 2587–2603. [Google Scholar] [CrossRef] [Green Version]
- Mathie, A.; Veale, E.L. Two-pore domain potassium channels: Potential therapeutic targets for the treatment of pain. Pflügers. Arch. 2014, 467, 931–943. [Google Scholar] [CrossRef]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; García, J.A.L. Potassium channels in neuropathic pain. Pain 2016, 157, S7–S14. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulas, C.; McMahon, S.B. Opening paths to novel analgesics: The role of potassium channels in chronic pain. Trends Neurosci. 2014, 37, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Busserolles, J.; Gasull, X.; Noël, J. Potassium Channels and Pain; Oxford University Press (OUP): Oxford, UK, 2019; pp. 1–83. [Google Scholar]
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Kitada, C.; Yokoi, H.; Nozawa, K.; Okada, H.; Matsushime, H.; Furuichi, K. A Novel Two-pore Domain K+ Channel, TRESK, is Localized in the Spinal Cord. J. Biol. Chem. 2003, 278, 27406–27412. [Google Scholar] [CrossRef] [PubMed]
- Czirják, G.; Tóth, Z.E.; Enyedi, P. The Two-pore Domain K+ Channel, TRESK, Is Activated by the Cytoplasmic Calcium Signal through Calcineurin. J. Biol. Chem. 2004, 279, 18550–18558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.; Mariash, E.; Kim, D.; Meyer, A.N.; Schlaepfer, D.D.; Gastwirt, R.F.; Donoghue, D.J. Functional Expression of TRESK-2, a New Member of the Tandem-pore K+ Channel Family. J. Biol. Chem. 2004, 279, 28063–28070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshavaprasad, B.; Liu, C.; Au, J.D.; Kindler, C.H.; Cotten, J.F.; Yost, C.S. Species-Specific Differences in Response to Anesthetics and Other Modulators by the K2P Channel TRESK. Anesth. Analg. 2005, 101, 1042–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.; Kim, N. TREK-2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am. J. Physiol. Physiol. 2006, 291, C138–C146. [Google Scholar] [CrossRef]
- Cadaveira-Mosquera, A.; Pérez, M.; Reboreda, A.; Rivas-Ramírez, P.; Fernández-Fernández, D.; Lamas, J. Expression of K2P Channels in Sensory and Motor Neurons of the Autonomic Nervous System. J. Mol. Neurosci. 2012, 48, 86–96. [Google Scholar] [CrossRef]
- Tulleuda, A.; Cokic, B.; Callejo, G.; Saiani, B.; Serra, J.; Gasull, X. TRESK channel contribution to nociceptive sensory neurons excitability: Modulation by nerve injury. Mol. Pain 2011, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Dobler, T.; Springauf, A.; Tovornik, S.; Weber, M.; Schmitt, A.; Sedlmeier, R.; Wischmeyer, E.; Döring, F. TRESK two-pore-domain K+channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J. Physiol. 2007, 585, 867–879. [Google Scholar] [CrossRef]
- Lafrenière, R.G.; Cader, M.Z.; Poulin, J.-F.; Andres-Enguix, I.; Simoneau, M.; Gupta, N.; Boisvert, K.; Lafrenière, F.; McLaughlan, S.; Dubé, M.-P.; et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 2010, 16, 1157–1160. [Google Scholar] [CrossRef]
- Marsh, B.; Acosta, C.; Djouhri, L.; Lawson, S.N. Leak K+ channel mRNAs in dorsal root ganglia: Relation to inflammation and spontaneous pain behaviour. Mol. Cell. Neurosci. 2012, 49, 375–386. [Google Scholar] [CrossRef]
- Yoo, S.; Liu, J.; Sabbadini, M.; Au, P.; Xie, G.-X.; Yost, C.S. Regional expression of the anesthetic-activated potassium channel TRESK in the rat nervous system. Neurosci. Lett. 2009, 465, 79–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.A.; Pettingill, P.; Wu, Y.; Duggal, G.; Ilie, A.-S.; Akerman, C.J.; Cader, M.Z. The Role of TRESK in Discrete Sensory Neuron Populations and Somatosensory Processing. Front. Mol. Neurosci. 2019, 12, 170. [Google Scholar] [CrossRef] [Green Version]
- Kollert, S.; Dombert, B.; Döring, F.; Wischmeyer, E. Activation of TRESK channels by the inflammatory mediator lysophosphatidic acid balances nociceptive signalling. Sci. Rep. 2015, 5, 12548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, A.; Pujol-Coma, A.; Andres-Bilbe, A.; Negm, A.; Callejo, G.; Soto, D.; Noel, J.; Comes, N.; Gasull, X. TRESK background K+ channel deletion selectively uncovers enhanced mechanical and cold sensitivity. J. Physiol. 2020, 598, 1017–1038. [Google Scholar] [CrossRef]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; Van Der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef] [Green Version]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggström, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2014, 18, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-L.; Li, K.-C.; Wu, D.; Chen, Y.; Luo, H.; Zhao, J.-R.; Wang, S.-S.; Sun, M.-M.; Lu, Y.-J.; Zhong, Y.-Q.; et al. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res. 2015, 26, 83–102. [Google Scholar] [CrossRef] [Green Version]
- Chiu, I.M.; Barrett, L.B.; Williams, E.K.; Strochlic, D.E.; Lee, S.; Weyer, A.D.; Lou, S.; Bryman, G.; Roberson, D.P.; Ghasemlou, N.; et al. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. Elife 2014, 3, e04660. [Google Scholar] [CrossRef]
- Ray, P.; Torck, A.; Quigley, L.; Wangzhou, A.; Neiman, M.; Rao, C.; Lam, T.; Kim, J.-Y.; Kim, T.H.; Zhang, M.Q.; et al. Comparative transcriptome profiling of the human and mouse dorsal root ganglia. Pain 2018, 159, 1325–1345. [Google Scholar] [CrossRef]
- LaPaglia, D.M.; Sapio, M.R.; Burbelo, P.D.; Thierry-Mieg, J.; Thierry-Mieg, D.; Raithel, S.; Ramsden, C.E.; Iadarola, M.J.; Mannes, A.J. RNA-Seq investigations of human post-mortem trigeminal ganglia. Cephalalgia 2017, 38, 912–932. [Google Scholar] [CrossRef]
- Nguyen, M.Q.; Wu, Y.; Bonilla, L.S.; Von Buchholtz, L.J.; Ryba, N.J.P. Diversity amongst trigeminal neurons revealed by high throughput single cell sequencing. PLoS ONE 2017, 12, e0185543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flegel, C.; Schöbel, N.; Altmüller, J.; Becker, C.; Tannapfel, A.; Hatt, H.; Gisselmann, G. RNA-Seq Analysis of Human Trigeminal and Dorsal Root Ganglia with a Focus on Chemoreceptors. PLoS ONE 2015, 10, e0128951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manteniotis, S.; Lehmann, R.; Flegel, C.; Vogel, F.; Hofreuter, A.; Schreiner, B.S.P.; Altmüller, J.; Becker, C.; Schöbel, N.; Hatt, H.; et al. Comprehensive RNA-Seq Expression Analysis of Sensory Ganglia with a Focus on Ion Channels and GPCRs in Trigeminal Ganglia. PLoS ONE 2013, 8, e79523. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, P.; Bai, L.; Trimmer, J.S.; Bean, B.P.; Ginty, D.D. Deep Sequencing of Somatosensory Neurons Reveals Molecular Determinants of Intrinsic Physiological Properties. Neuron 2019, 103, 598–616.e7. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Kim, G.-T.; Kim, E.-J.; La, J.-H.; Lee, J.-S.; Lee, E.-S.; Park, J.-Y.; Hong, S.-G.; Han, J. Lamotrigine inhibits TRESK regulated by G-protein coupled receptor agonists. Biochem. Biophys. Res. Commun. 2008, 367, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Callejo, G.; Giblin, J.P.; Gasull, X. Modulation of TRESK Background K+ Channel by Membrane Stretch. PLoS ONE 2013, 8, e64471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medhurst, A.D.; Rennie, G.; Chapman, C.G.; Meadows, H.; Duckworth, M.D.; Kelsell, R.E.; Gloger, I.I.; Pangalos, M.N. Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Mol. Brain Res. 2001, 86, 101–114. [Google Scholar] [CrossRef]
- Guo, Z.; Qiu, C.-S.; Jiang, X.; Zhang, J.; Li, F.; Liu, Q.; Dhaka, A.; Cao, Y.-Q.; Jiang, X. TRESK K+ Channel Activity Regulates Trigeminal Nociception and Headache. eNeuro 2019, 6, 0236. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chen, H.; Yang, C.; Zhong, J.; He, W.; Xiong, Q. Reversal of TRESK Downregulation Alleviates Neuropathic Pain by Inhibiting Activation of Gliocytes in the Spinal Cord. Neurochem. Res. 2017, 42, 1288–1298. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Jin, Z.-R.; Jing, H.-B.; Zhao, H.; Liu, B.-H.; Liang, Y.-J.; Liu, L.-Y.; Cai, J.; Wan, Y.; et al. Decreased abundance of TRESK two-pore domain potassium channels in sensory neurons underlies the pain associated with bone metastasis. Sci. Signal. 2018, 11, eaao5150. [Google Scholar] [CrossRef] [Green Version]
- Wangzhou, A.; McIlvried, L.A.; Paige, C.; Barragan-Iglesias, P.; Shiers, S.; Ahmad, A.; Guzman, C.A.; Dussor, G.; Ray, P.R.; Gereau, R.W.; et al. Pharmacological target-focused transcriptomic analysis of native versus cultured human and mouse dorsal root ganglia. Pain 2020. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Cao, Y.-Q. Over-Expression of TRESK K+ Channels Reduces the Excitability of Trigeminal Ganglion Nociceptors. PLoS ONE 2014, 9, e87029. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, C.-X.; Zhong, J.-Y.; Wang, H.-B. Intrathecal TRESK gene recombinant adenovirus attenuates spared nerve injury-induced neuropathic pain in rats. NeuroReport 2013, 24, 131–136. [Google Scholar] [CrossRef]
- Alloui, A.; Zimmermann, K.; Mamet, J.; Duprat, F.; Noel, J.; Chemin, J.; Guy, N.; Blondeau, N.; Voilley, N.; Rubat-Coudert, C.; et al. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 2006, 25, 2368–2376. [Google Scholar] [CrossRef] [Green Version]
- Noel, J.; Zimmermann, K.; Busserolles, J.; Deval, E.; Alloui, A.; Diochot, S.; Guy, N.; Borsotto, M.; Reeh, P.; Eschalier, A.; et al. The mechano-activated K+ channels TRAAK and TREK-1 control both warm and cold perception. EMBO J. 2009, 28, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Pereira, V.; Busserolles, J.; Christin, M.; Devilliers, M.; Poupon, L.; Legha, W.; Alloui, A.; Aisouni, Y.; Bourinet, E.; Lesage, F.; et al. Role of the TREK2 potassium channel in cold and warm thermosensation and in pain perception. Pain 2014, 155, 2534–2544. [Google Scholar] [CrossRef]
- Hwang, H.Y.; Zhang, E.; Park, S.; Chung, W.; Lee, S.Y.; Kim, D.W.; Ko, Y.; Lee, W. TWIK-Related Spinal Cord K+ Channel Expression Is Increased in the Spinal Dorsal Horn after Spinal Nerve Ligation. Yonsei Med. J. 2015, 56, 1307–1315. [Google Scholar] [CrossRef] [Green Version]
- Czirják, G.; Enyedi, P. Targeting of Calcineurin to an NFAT-like Docking Site Is Required for the Calcium-dependent Activation of the Background K+ Channel, TRESK. J. Biol. Chem. 2006, 281, 14677–14682. [Google Scholar] [CrossRef] [Green Version]
- Czirják, G.; Enyedi, P. TRESK Background K+ Channel is Inhibited by Phosphorylation via Two Distinct Pathways. J. Biol. Chem. 2010, 285, 14549–14557. [Google Scholar] [CrossRef] [Green Version]
- Enyedi, P.; Veres, I.; Braun, G.; Czirják, G. Tubulin Binds to the Cytoplasmic Loop of TRESK Background K+ Channel In Vitro. PLoS ONE 2014, 9, e97854. [Google Scholar] [CrossRef] [Green Version]
- Braun, G.; Nemcsics, B.; Enyedi, P.; Czirják, G. TRESK background K(+) channel is inhibited by PAR-1/MARK microtubule affinity-regulating kinases in Xenopus oocytes. PLoS ONE 2011, 6, e28119. [Google Scholar] [CrossRef]
- Rahm, A.-K.; Gierten, J.; Kisselbach, J.; Staudacher, I.; Staudacher, K.; Schweizer, P.A.; Becker, R.; Katus, H.A.; Thomas, D. PKC-dependent activation of human K2P18.1 K+ channels. Br. J. Pharmacol. 2012, 166, 764–773. [Google Scholar] [CrossRef] [Green Version]
- Pergel, E.; Lengyel, M.; Enyedi, P.; Czirják, G. TRESK (K2P18.1) Background Potassium Channel is Activated by Novel-Type Protein Kinase C via Dephosphorylation. Mol. Pharmacol. 2019, 95, 661–672. [Google Scholar] [CrossRef]
- Smith, H. Calcineurin as a nociceptor modulator. Pain Physician 2009, 12, 309–318. [Google Scholar]
- Liu, P.; Xiao, Z.; Ren, F.; Guo, Z.; Chen, Z.; Zhao, H.; Cao, Y.-Q. Functional analysis of a migraine-associated TRESK K+ channel mutation. J. Neurosci. 2013, 33, 12810–12824. [Google Scholar] [CrossRef] [Green Version]
- Andres-Enguix, I.; Shang, L.; Stansfeld, P.J.; Morahan, J.M.; Sansom, M.S.; Lafrenière, R.G.; Roy, B.; Griffiths, L.R.; Rouleau, G.A.; Ebers, G.C.; et al. Functional analysis of missense variants in the TRESK (KCNK18) K+ channel. Sci. Rep. 2012, 2, 237. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Liu, P.; Ren, F.; Cao, Y.-Q. Non-migraine associated TRESK K+ channel variant C110R does not increase the excitability of trigeminal ganglion neurons. J. Neurophysiol. 2014, 112, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Rainero, I.; Rubino, E.; Gallone, S.; Zavarise, P.; Carli, D.; Boschi, S.; Fenoglio, P.; Savi, L.; Gentile, S.; Benna, P.; et al. KCNK 18 (TRESK) Genetic Variants in Italian Patients With Migraine. Headache J. Head Face Pain 2014, 54, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Royal, P.; Andres-Bilbe, A.; Prado, P.A.; Verkest, C.; Wdziekonski, B.; Schaub, S.; Baron, A.; Lesage, F.; Gasull, X.; Levitz, J.; et al. Migraine-Associated TRESK Mutations Increase Neuronal Excitability through Alternative Translation Initiation and Inhibition of TREK. Neuron 2019, 101, 232–245.e6. [Google Scholar] [CrossRef] [Green Version]
- Pettingill, P.; Weir, G.A.; Wei, T.; Wu, Y.; Flower, G.; Lalic, T.; Handel, A.; Duggal, G.; Chintawar, S.; Cheung, J.; et al. A causal role for TRESK loss of function in migraine mechanisms. Brain 2019, 142, 3852–3867. [Google Scholar] [CrossRef]
- Imbrici, P.; Nematian-Ardestani, E.; Hasan, S.; Pessia, M.; Tucker, S.J.; D’Adamo, M.C. Altered functional properties of a missense variant in the TRESK K+ channel (KCNK18) associated with migraine and intellectual disability. Pflügers Arch. 2020, 2, 1–8. [Google Scholar] [CrossRef]
- Bautista, D.M.; Sigal, Y.M.; Milstein, A.D.; Garrison, J.L.; Zorn, J.A.; Tsuruda, P.R.; Nicoll, R.A.; Julius, D. Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two-pore potassium channels. Nat. Neurosci. 2008, 11, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Lennertz, R.C.; Tsunozaki, M.; Bautista, D.M.; Stucky, C.L. Physiological Basis of Tingling Paresthesia Evokedby Hydroxy-α-Sanshool. J. Neurosci. 2010, 30, 4353–4361. [Google Scholar] [CrossRef]
- Tsunozaki, M.; Lennertz, R.C.; Vilceanu, D.; Katta, S.; Stucky, C.L.; Bautista, D.M. A ‘toothache tree’ alkylamide inhibits Aδ mechanonociceptors to alleviate mechanical pain. J. Physiol. 2013, 591, 3325–3340. [Google Scholar] [CrossRef]
- Albin, K.C.; Simons, C.T. Psychophysical Evaluation of a Sanshool Derivative (Alkylamide) and the Elucidation of Mechanisms Subserving Tingle. PLoS ONE 2010, 5, e9520. [Google Scholar] [CrossRef]
- Klein, A.H.; Sawyer, C.M.; Zanotto, K.L.; Ivanov, M.A.; Cheung, S.; Carstens, M.I.; Furrer, S.; Simons, C.T.; Slack, J.P.; Carstens, E. A tingling sanshool derivative excites primary sensory neurons and elicits nocifensive behavior in rats. J. Neurophysiol. 2011, 105, 1701–1710. [Google Scholar] [CrossRef]
- Castellanos, A.; Andres, A.; Bernal, L.; Callejo, G.; Comes, N.; Gual, A.; Giblin, J.P.; Roza, C.; Gasull, X. Pyrethroids inhibit K2P channels and activate sensory neurons. Pain 2018, 159, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, M.R.C.; Bautista, D.M.; Wu, K.; Haeberle, H.; Lumpkin, E.A.; Julius, D. Radial stretch reveals distinct populations of mechanosensitive mammalian somatosensory neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 20015–20020. [Google Scholar] [CrossRef] [Green Version]
- Brohawn, S.G.; Su, Z.; MacKinnon, R. Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc. Natl. Acad. Sci. USA 2014, 111, 3614–3619. [Google Scholar] [CrossRef] [Green Version]
- Enyedi, P.; Czirják, G. Properties, regulation, pharmacology, and functions of the K₂p channel, TRESK. Pflügers Arch. 2015, 467, 945–958. [Google Scholar] [CrossRef]
- Enyedi, P.; Braun, G.; Czirják, G. TRESK: The lone ranger of two-pore domain potassium channels. Mol. Cell. Endocrinol. 2012, 353, 75–81. [Google Scholar] [CrossRef]
- Monteillier, A.; Loucif, A.; Omoto, K.; Stevens, E.B.; Lainez, S.; Saintot, P.-P.; Cao, L.; Pryde, D.C.; Vicente, S.L. Investigation of the structure activity relationship of flufenamic acid derivatives at the human TRESK channel K2P18.1. Bioorg. Med. Chem. Lett. 2016, 26, 4919–4924. [Google Scholar] [CrossRef]
- Wright, P.D.; Weir, G.; Cartland, J.; Tickle, D.; Kettleborough, C.; Cader, M.Z.; Jerman, J. Cloxyquin (5-chloroquinolin-8-ol) is an activator of the two-pore domain potassium channel TRESK. Biochem. Biophys. Res. Commun. 2013, 441, 463–468. [Google Scholar] [CrossRef]
- Lengyel, M.; Dobolyi, A.; Czirják, G.; Enyedi, P. Selective and state-dependent activation of TRESK (K2P18.1) background potassium channel by cloxyquin. Br. J. Pharmacol. 2017, 174, 2102–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Au, J.D.; Zou, H.L.; Cotten, J.F.; Yost, C.S. Potent Activation of the Human Tandem Pore Domain K Channel TRESK with Clinical Concentrations of Volatile Anesthetics. Anesth. Analg. 2004, 99, 1715–1722. [Google Scholar] [CrossRef]
- Giblin, J.P.; Etayo, I.; Castellanos, A.; Andres-Bilbe, A.; Gasull, X. Anionic Phospholipids Bind to and Modulate the Activity of Human TRESK Background K+ Channel. Mol. Neurobiol. 2018, 56, 2524–2541. [Google Scholar] [CrossRef]
- Rahm, A.-K.; Wiedmann, F.; Gierten, J.; Schmidt, C.; Schweizer, P.A.; Becker, R.; Katus, H.A.; Thomas, D. Functional characterization of zebrafish K2P18.1 (TRESK) two-pore-domain K+ channels. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2013, 387, 291–300. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andres-Bilbe, A.; Castellanos, A.; Pujol-Coma, A.; Callejo, G.; Comes, N.; Gasull, X. The Background K+ Channel TRESK in Sensory Physiology and Pain. Int. J. Mol. Sci. 2020, 21, 5206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155206
Andres-Bilbe A, Castellanos A, Pujol-Coma A, Callejo G, Comes N, Gasull X. The Background K+ Channel TRESK in Sensory Physiology and Pain. International Journal of Molecular Sciences. 2020; 21(15):5206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155206
Chicago/Turabian StyleAndres-Bilbe, Alba, Aida Castellanos, Anna Pujol-Coma, Gerard Callejo, Nuria Comes, and Xavier Gasull. 2020. "The Background K+ Channel TRESK in Sensory Physiology and Pain" International Journal of Molecular Sciences 21, no. 15: 5206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155206