ORAI2 Down-Regulation Potentiates SOCE and Decreases Aβ42 Accumulation in Human Neuroglioma Cells

 , and
, and 
Abstract
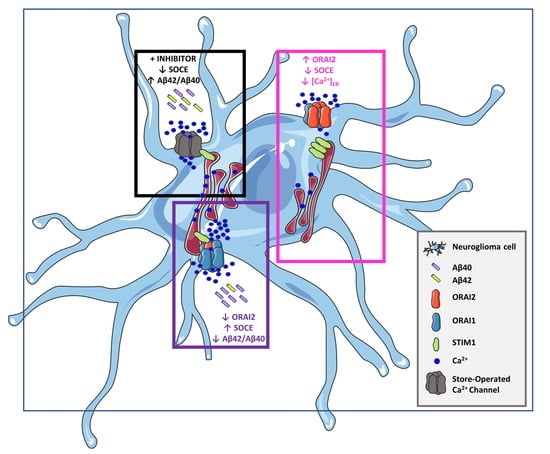
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
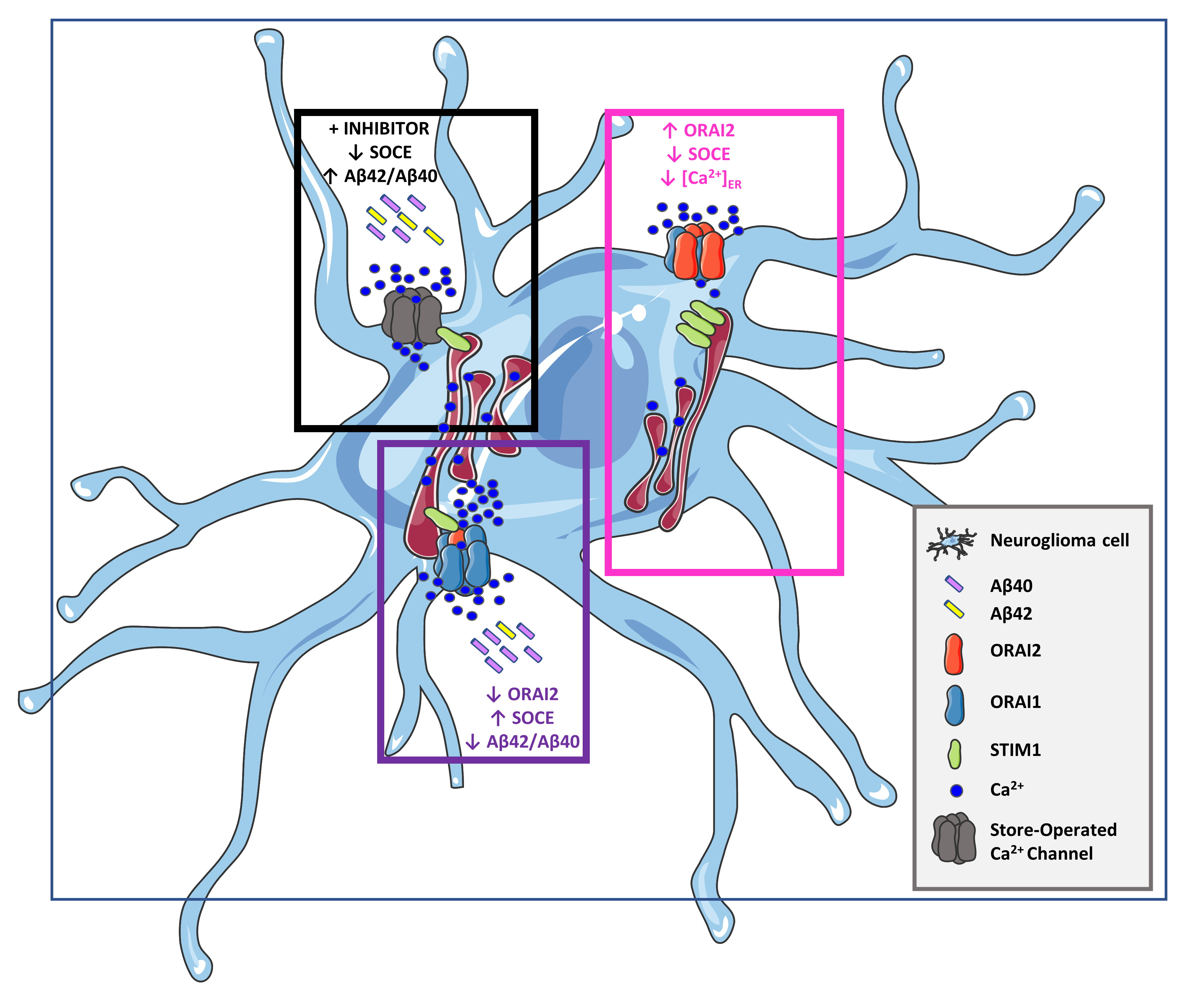
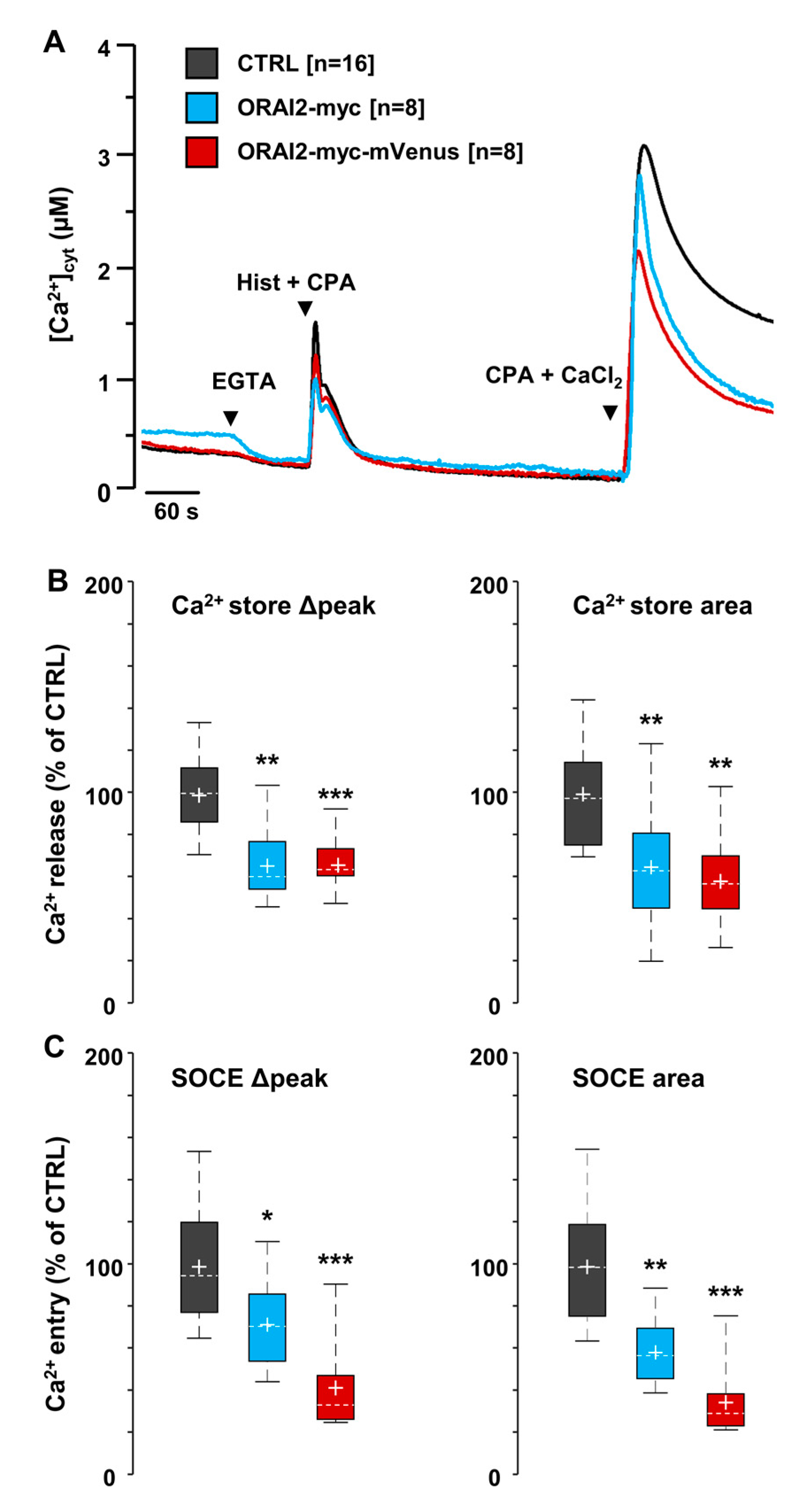
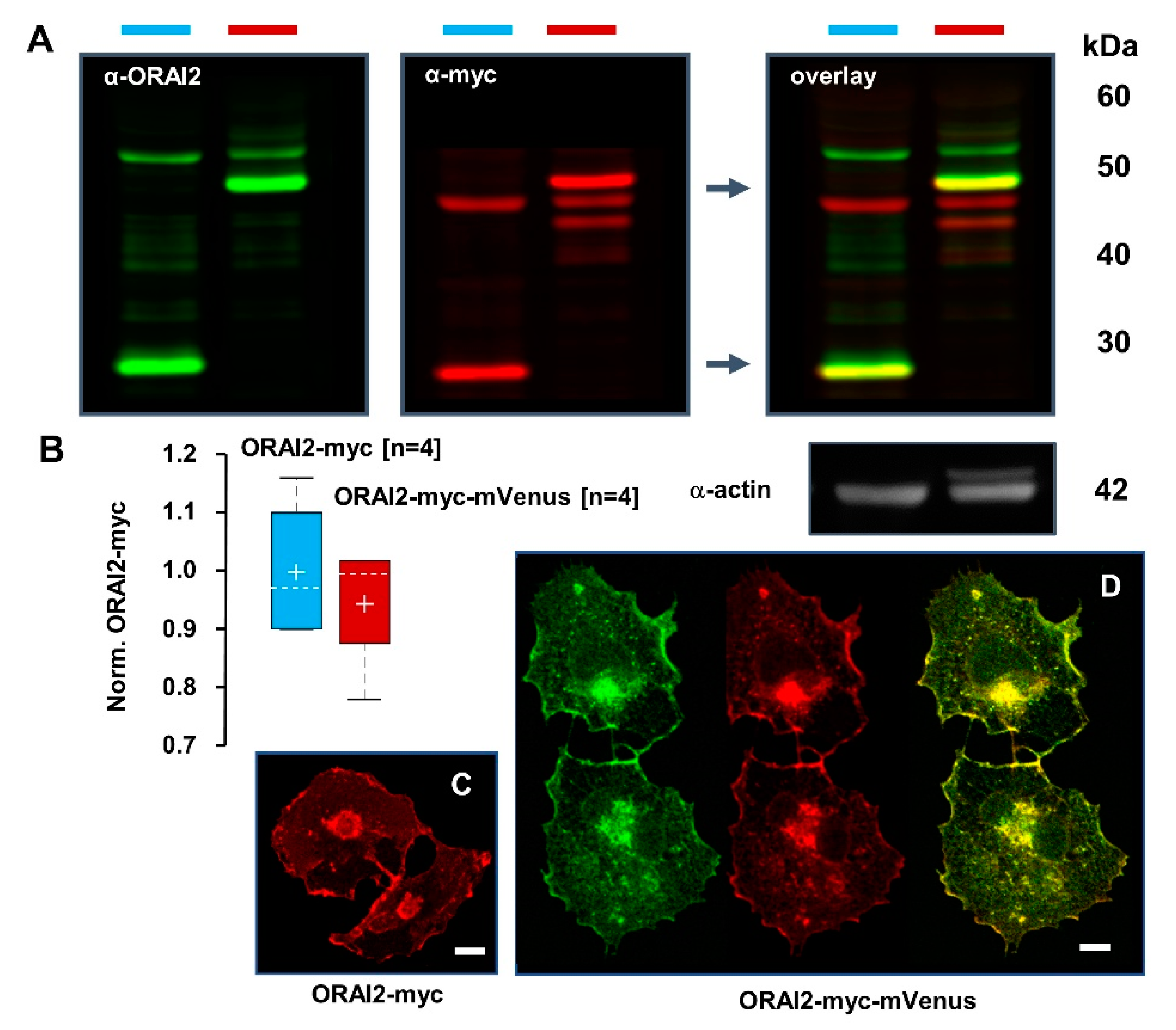
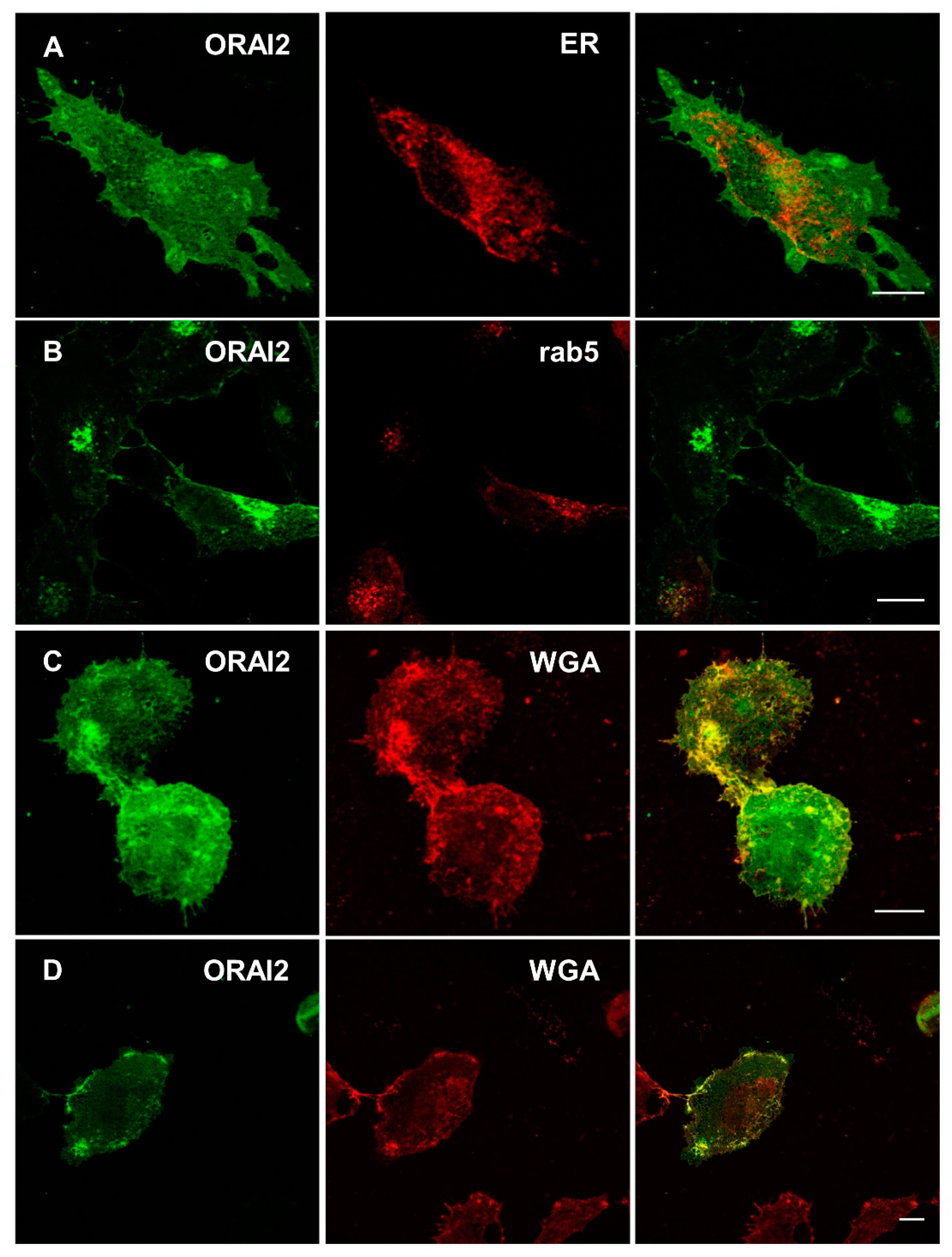
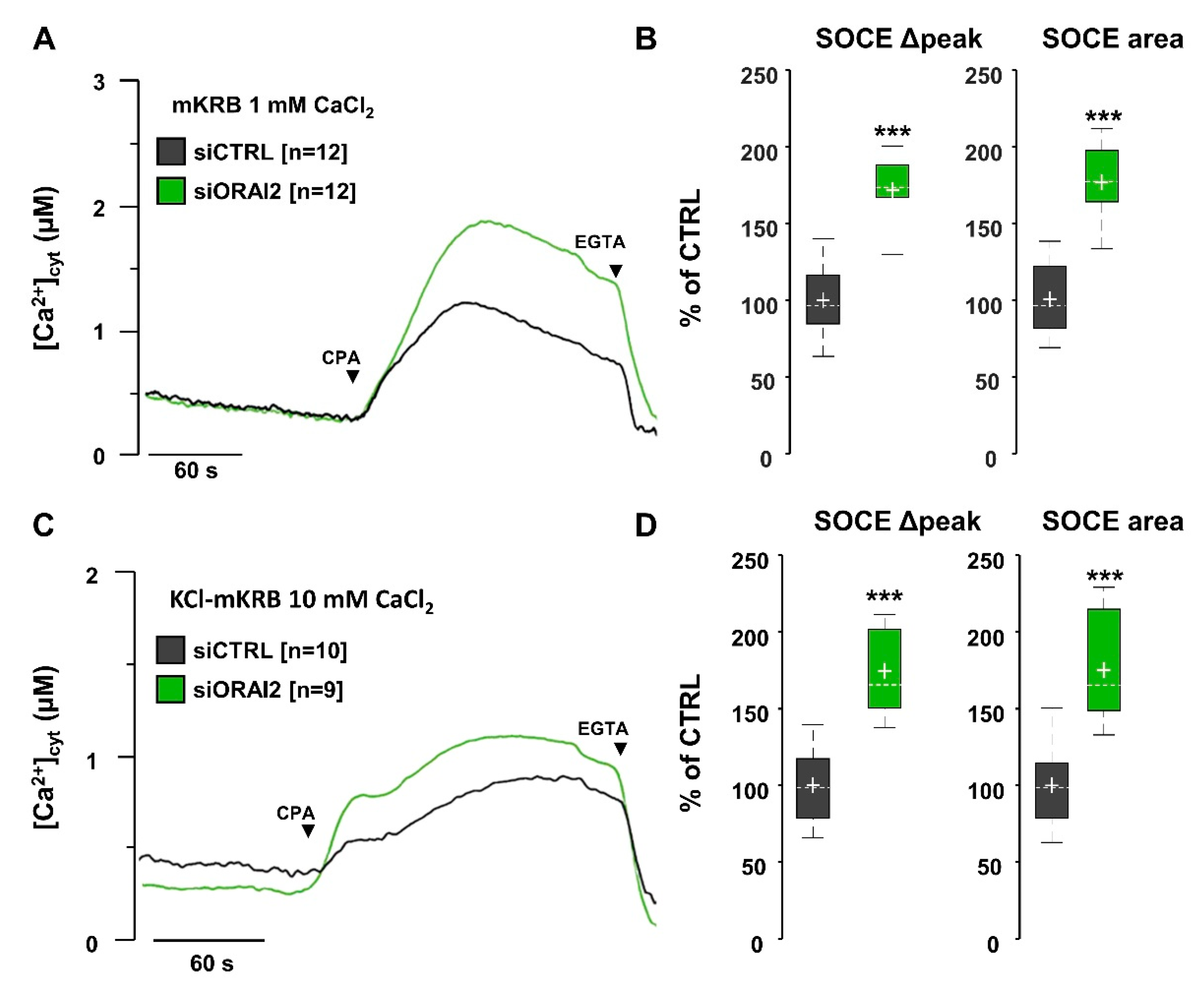
2.1. ORAI2 Modulates SOCE in Human Neuroglioma Cells
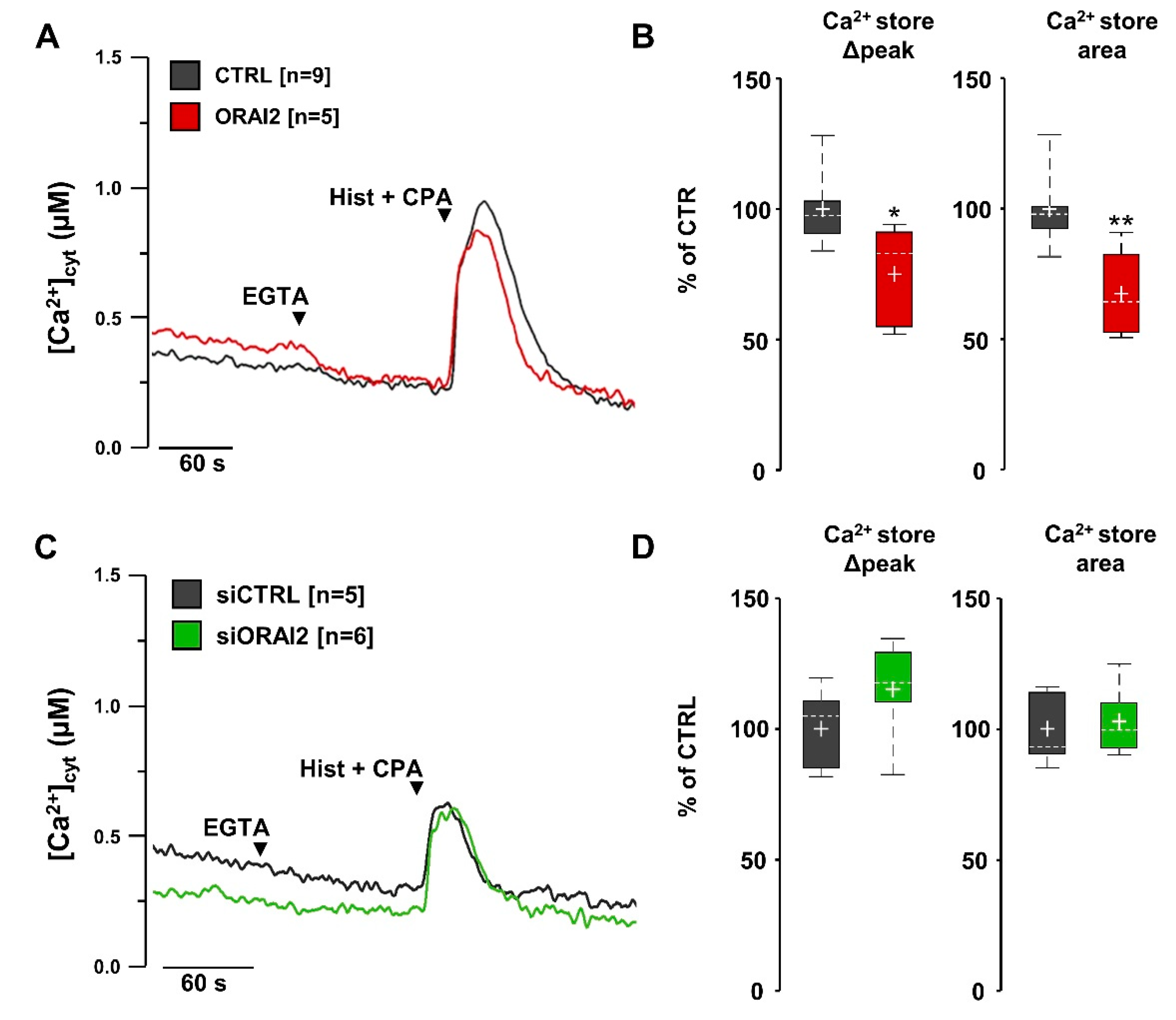
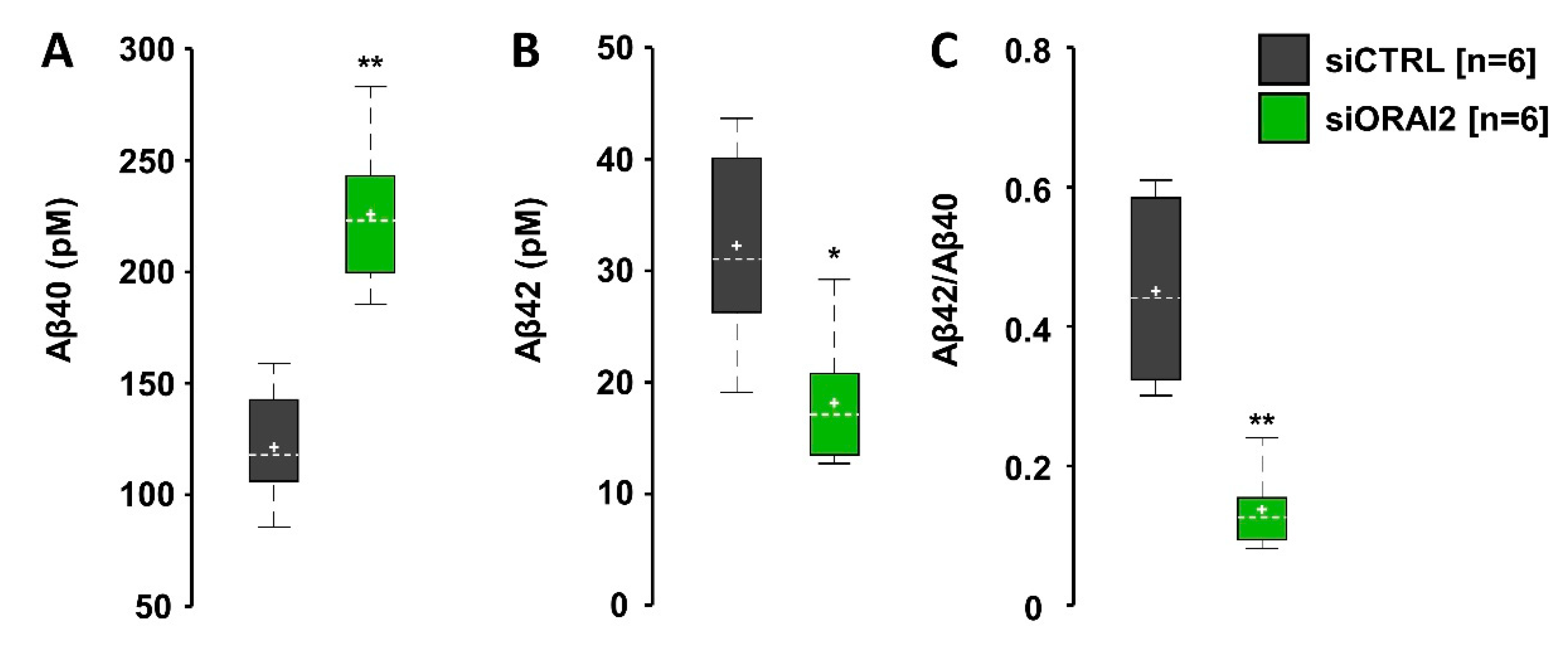
2.2. SOCE Levels and Aβ-Secretion in Neuroglioma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Aequorin Measurements
4.3. Ca2+ Imaging
4.4. Materials
4.5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Hardy, J.; Higgins, G. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. Embo. Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Gutiérrez, L.; Szaruga, M. Mechanisms of neurodegeneration — Insights from familial Alzheimer’s disease. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell. Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-Mediated Modulation of Capacitative Calcium Entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.P.; Hettiarachchi, N.T.; Wilkinson, J.A.; Pearson, H.A.; Scragg, J.L.; Lendon, C.; Al-Owais, M.M.; Kim, C.B.; Myers, D.M.; Warburton, P.; et al. Cellular consequences of the expression of Alzheimer’s disease-causing presenilin 1 mutations in human neuroblastoma (SH-SY5Y) cells. Brain Res. 2012, 1443, 75–88. [Google Scholar] [CrossRef]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 74, 44–56. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.-J.; Feske, S.; White, C.L.; Bezprozvanny, I. Reduced Synaptic STIM2 Expression and Impaired Store-Operated Calcium Entry Cause Destabilization of Mature Spines in Mutant Presenilin Mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, B.C.-K.; Lee, C.S.-K.; Cheng, W.-H.; Lai, K.-O.; Kevin Foskett, J.; Cheung, K.-H. Familial Alzheimer’s disease–associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojarski, L.; Pomorski, P.; Szybinska, A.; Drab, M.; Skibinska-Kijek, A.; Gruszczynska-Biegala, J.; Kuznicki, J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim. Biophys. Acta 2009, 1793, 1050–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Lim, D. Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef]
- Herms, J.; Schneider, I.; Dewachter, I.; Caluwaerts, N.; Kretzschmar, H.; Van Leuven, F. Capacitive Calcium Entry Is Directly Attenuated by Mutant Presenilin-1, Independent of the Expression of the Amyloid Precursor Protein. J. Biol. Chem. 2003, 278, 2484–2489. [Google Scholar] [CrossRef] [Green Version]
- Bezprozvanny, I. Presenilins and Calcium Signaling--Systems Biology to the Rescue. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Honarnejad, K.; Herms, J. Presenilins: Role in calcium homeostasis. Int. J. Biochem. Cell Biol. 2012, 44, 1983–1986. [Google Scholar] [CrossRef]
- Shilling, D.; Mak, D.-O.D.; Kang, D.E.; Foskett, J.K. Lack of Evidence for Presenilins as Endoplasmic Reticulum Ca2+ Leak Channels. J. Biol. Chem. 2012, 287, 10933–10944. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Barbiero, L.; Zatti, G.; Squitti, R.; Binetti, G.; Pozzan, T.; Fasolato, C.; Ghidoni, R.; Pizzo, P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol. Dis. 2005, 18, 638–648. [Google Scholar] [CrossRef]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J. Cell. Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiger, W.; Vetrivel, K.S.; Buggia-Prévot, V.; Nguyen, P.D.; Wagner, S.L.; Villereal, M.L.; Thinakaran, G. Ca2+ Influx through Store-operated Ca2+ Channels Reduces Alzheimer Disease β-Amyloid Peptide Secretion. J. Biol. Chem. 2013, 288, 26955–26966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierrot, N.; Ghisdal, P.; Caumont, A.-S.; Octave, J.-N. Intraneuronal amyloid-β1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J. Neurochem. 2004, 88, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Crestini, A.; Piscopo, P.; Iazeolla, M.; Albani, D.; Rivabene, R.; Forloni, G.; Confaloni, A. Rosuvastatin and Thapsigargin Modulate γ-Secretase Gene Expression and APP Processing in a Human Neuroglioma Model. J. Mol. Neurosci. 2011, 43, 461–469. [Google Scholar] [CrossRef]
- Al-Mousa, F.; Michelangeli, F. Some Commonly Used Brominated Flame Retardants Cause Ca Ca2+-ATPase Inhibition, Beta-Amyloid Peptide Release and Apoptosis in SH-SY5Y Neuronal Cells. PLoS ONE 2012, 7, e33059. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W. Forms and functions of store-operated calcium entry mediators, STIM and Orai. Adv. Biol. Regul. 2018, 68, 88–96. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, Y.; Song, R.; Xu, J.; Yuan, Y.; Liu, J.; Li, J.; Zheng, S.; Liu, T.; Lu, B.; et al. Toward a Model for Activation of Orai Channel. iScience 2019, 16, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Barak, P.; Parekh, A.B. Signaling through Ca2+ Microdomains from Store-Operated CRAC Channels. Cold Spring Harb. Perspect. Biol. 2020, 12, a035097. [Google Scholar] [CrossRef] [PubMed]
- Hoth, M.; Niemeyer, B.A. The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr Top Membr. 2013, 71, 237–271. [Google Scholar] [PubMed]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J.A. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim. Biophys. Acta 2017, 1864, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsvilovskyy, V.; Solís-López, A.; Schumacher, D.; Medert, R.; Roers, A.; Kriebs, U.; Freichel, M. Deletion of Orai2 augments endogenous CRAC currents and degranulation in mast cells leading to enhanced anaphylaxis. Cell Calcium 2018, 71, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Stegner, D.; Hofmann, S.; Schuhmann, M.K.; Kraft, P.; Herrmann, A.M.; Popp, S.; Höhn, M.; Popp, M.; Klaus, V.; Post, A.; et al. Loss of Orai2-Mediated Capacitative Ca2+ Entry Is Neuroprotective in Acute Ischemic Stroke. Stroke 2019, 50, 3238–3245. [Google Scholar] [CrossRef]
- Clemens, R.A.; Lowell, C.A. CRAC channel regulation of innate immune cells in health and disease. Cell Calcium 2019, 78, 56–65. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, W.; Gao, C.; Liu, X.; Zhao, X.; Wei, T.; Gao, A.Z. Leucine-rich repeat kinase 2 regulates mouse dendritic cell migration by ORAI2. FASEB J. 2019, 33, 9775–9784. [Google Scholar] [CrossRef] [Green Version]
- Feske, S. CRAC channels and disease–From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef]
- Lu, B.; Fivaz, M. Neuronal SOCE: Myth or Reality? Trends Cell Biol. 2016, 26, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 Regulates Capacitive Ca2+ Entry in Neurons and Plays a Key Role in Hypoxic Neuronal Cell Death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Fon Tacer, K.; Bezprozvanny, I. Store-Operated Calcium Channel Complex in Postsynaptic Spines: A New Therapeutic Target for Alzheimer’s Disease Treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; An, H.; Sa, M.; Won, J.; Shin, J.I.; Lee, C.J. Orai1 and Orai3 in Combination with Stim1 Mediate the Majority of Store-operated Calcium Entry in Astrocytes. Exp. Neurobiol. 2017, 26, 42–54. [Google Scholar] [CrossRef]
- Dou, Y.; Xia, J.; Gao, R.; Gao, X.; Munoz, F.M.; Wei, D.; Tian, Y.; Barrett, J.E.; Ajit, S.; Meucci, O.; et al. Orai1 Plays a Crucial Role in Central Sensitization by Modulating Neuronal Excitability. J. Neurosci. 2018, 38, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, P.; Burgo, A.; Pozzan, T.; Fasolato, C. Role of capacitative calcium entry on glutamate-induced calcium influx in type-I rat cortical astrocytes. J. Neurochem. 2001, 79, 98–109. [Google Scholar] [CrossRef]
- Okubo, Y.; Iino, M.; Hirose, K. Store-operated Ca2+ entry-dependent Ca2+ refilling in the endoplasmic reticulum in astrocytes. Biochem. Biophys. Res. Commun. 2020, 522, 1003–1008. [Google Scholar] [CrossRef]
- Toth, A.B.; Hori, K.; Novakovic, M.M.; Bernstein, N.G.; Lambot, L.; Prakriya, M. CRAC channels regulate astrocyte Ca2+ signaling and gliotransmitter release to modulate hippocampal GABAergic transmission. Sci. Signal. 2019, 12, eaaw5450. [Google Scholar] [CrossRef] [PubMed]
- Bandara, S.; Malmersjo, S.; Meyer, T. Regulators of Calcium Homeostasis Identified by Inference of Kinetic Model Parameters from Live Single Cells Perturbed by siRNA. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert Olivé, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Oberstein, T.J.; Spitzer, P.; Klafki, H.-W.; Linning, P.; Neff, F.; Knölker, H.-J.; Lewczuk, P.; Wiltfang, J.; Kornhuber, J.; Maler, J.M. Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Aβ peptides. Neurobiol. Dis. 2015, 73, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, G.R.; Li, Y.-M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [Green Version]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.-U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Ourdev, D.; Schmaus, A.; Kar, S. Kainate Receptor Activation Enhances Amyloidogenic Processing of APP in Astrocytes. Mol. Neurobiol. 2019, 56, 5095–5110. [Google Scholar] [CrossRef]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Sil, S.; Hu, G.; Liao, K.; Niu, F.; Callen, S.; Periyasamy, P.; Fox, H.S.; Buch, S. HIV-1 Tat-mediated astrocytic amyloidosis involves the HIF-1α/lncRNA BACE1-AS axis. PloS Biol. 2020, 18, e3000660. [Google Scholar] [CrossRef]
- Hoover, P.J.; Lewis, R.S. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2011, 108, 13299–13304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alansary, D.; Kilch, T.; Holzmann, C.; Peinelt, C.; Hoth, M.; Lis, A. The Minimal Requirements to Use Calcium Imaging to Analyze ICRAC. Cold Spring Harb. Protoc. 2014, 2014, pdb.prot073262. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kimura, T.; Shinoda, H.; Bai, G.; Daniels, M.J.; Arai, Y.; Nakano, M.; Nagai, T. Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat. Commun. 2016, 7, 13718. [Google Scholar] [CrossRef] [PubMed]
- Haugabook, S.; Yager, D.; Eckman, E.; Golde, T.; Younkin, S.; Eckman, C. High throughput screens for the identification of compounds that alter the accumulation of the Alzheimer’s amyloid β peptide (Aβ). J. Neurosci. Methods 2001, 108, 171–179. [Google Scholar] [CrossRef]
- Chen, T.-W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Ciscato, F.; Filadi, R.; Masgras, I.; Pizzi, M.; Marin, O.; Damiano, N.; Pizzo, P.; Gori, A.; Frezzato, F.; Chiara, F.; et al. Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca2+-dependent death of cancer cells. EMBO Rep. 2020, 21, e49117. [Google Scholar] [CrossRef]
- Gross, S.A.; Wissenbach, U.; Philipp, S.E.; Freichel, M.; Cavalié, A.; Flockerzi, V. Murine ORAI2 Splice Variants Form Functional Ca2+ Release-activated Ca2+ (CRAC) Channels. J. Biol. Chem. 2007, 282, 19375–19384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworska, A.; Dzbek, J.; Styczynska, M.; Kuznicki, J. Analysis of calcium homeostasis in fresh lymphocytes from patients with sporadic Alzheimer’s disease or mild cognitive impairment. Biochim. Biophys. Acta 2013, 1833, 1692–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greotti, E.; Fortunati, I.; Pendin, D.; Ferrante, C.; Galla, L.; Zentilin, L.; Giacca, M.; Kaludercic, N.; Di Sante, M.; Mariotti, L.; et al. mCerulean3-Based Cameleon Sensor to Explore Mitochondrial Ca2+ Dynamics In Vivo. iScience 2019, 19, 161. [Google Scholar] [CrossRef]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.M.; Martin-Romero, F.J. STIM1 deficiency is linked to Alzheimer’s disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca2+ entry. J. Mol. Med. 2018, 96, 1061–1079. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Parpura, V. Store-operated calcium entry in neuroglia. Neurosci. Bull. 2014, 30, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolev, I.; Fogel, H.; Milshtein, H.; Berdichevsky, Y.; Lipstein, N.; Brose, N.; Gazit, N.; Slutsky, I. Spike bursts increase amyloid-β 40/42 ratio by inducing a presenilin-1 conformational change. Nat. Neurosci. 2013, 16, 587–595. [Google Scholar] [CrossRef]
- Zoltowska, K.M.; Maesako, M.; Berezovska, O. Interrelationship between Changes in the Amyloid β 42/40 Ratio and Presenilin 1 Conformation. Mol. Med. 2016, 22, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2. Int. J. Mol. Sci. 2020, 21, 770. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Marsault, R.; Bastianutto, C.; Alvarez, J.; Pozzan, T.; Rizzuto, R. Transfected Aequorin in the Measurement of Cytosolic Ca2+ Concentration ([Ca2+]c). J. Biol. Chem. 1995, 270, 9896–9903. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scremin, E.; Agostini, M.; Leparulo, A.; Pozzan, T.; Greotti, E.; Fasolato, C. ORAI2 Down-Regulation Potentiates SOCE and Decreases Aβ42 Accumulation in Human Neuroglioma Cells. Int. J. Mol. Sci. 2020, 21, 5288. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155288
Scremin E, Agostini M, Leparulo A, Pozzan T, Greotti E, Fasolato C. ORAI2 Down-Regulation Potentiates SOCE and Decreases Aβ42 Accumulation in Human Neuroglioma Cells. International Journal of Molecular Sciences. 2020; 21(15):5288. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155288
Chicago/Turabian StyleScremin, Elena, Mario Agostini, Alessandro Leparulo, Tullio Pozzan, Elisa Greotti, and Cristina Fasolato. 2020. "ORAI2 Down-Regulation Potentiates SOCE and Decreases Aβ42 Accumulation in Human Neuroglioma Cells" International Journal of Molecular Sciences 21, no. 15: 5288. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155288