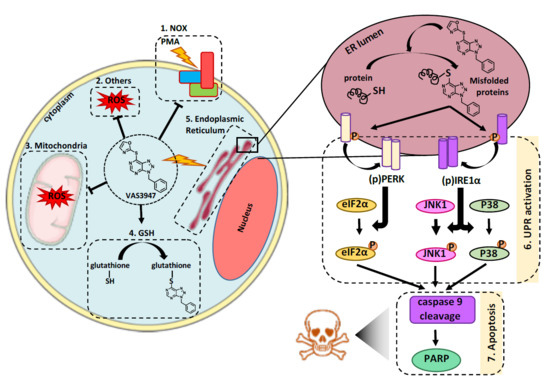
VAS3947 Induces UPR-Mediated Apoptosis through Cysteine Thiol Alkylation in AML Cell Lines

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
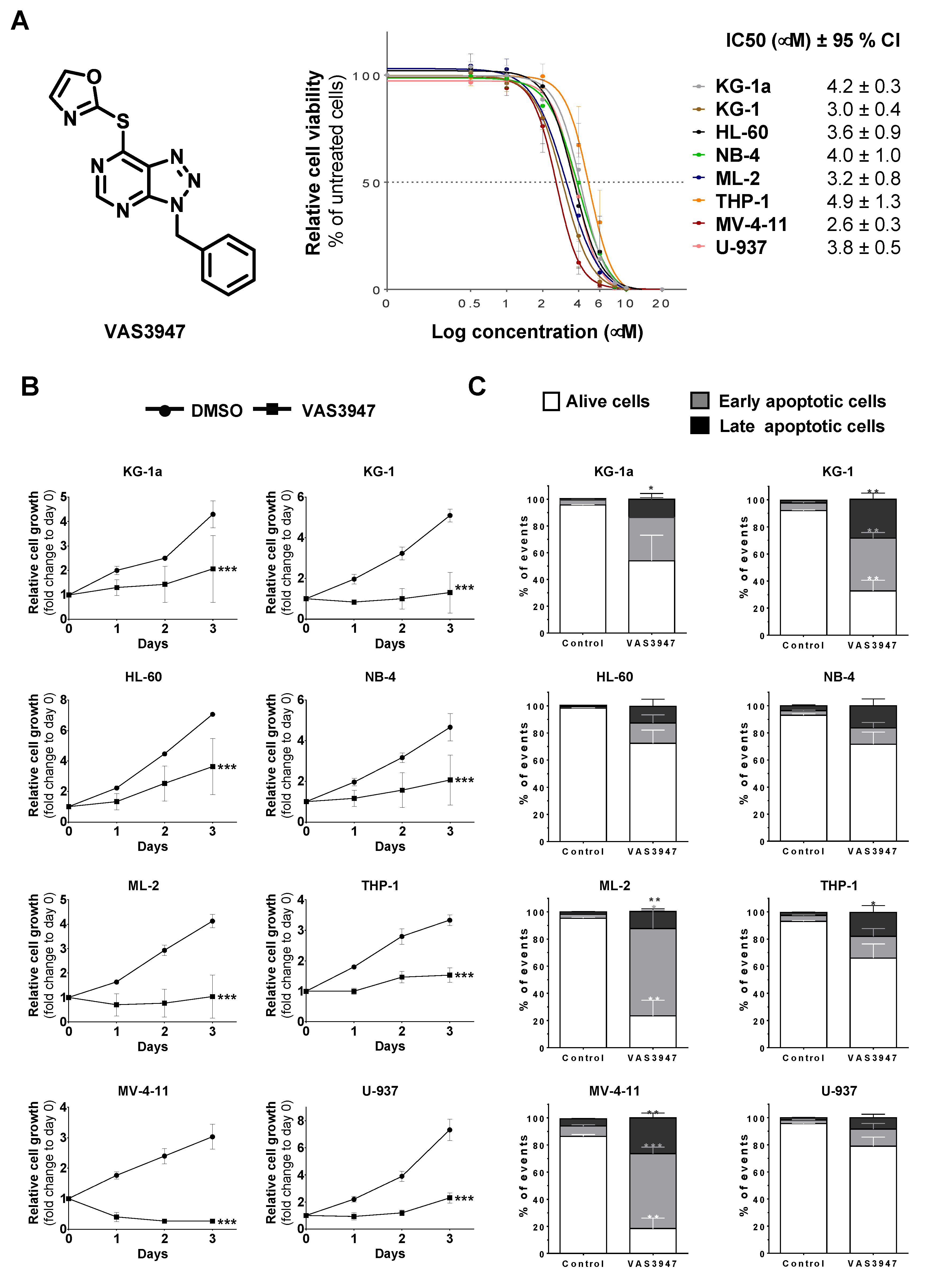
2.1. VAS3947 Triggers Cell Proliferation Arrest and Death Independently of Anti-NOX Activity
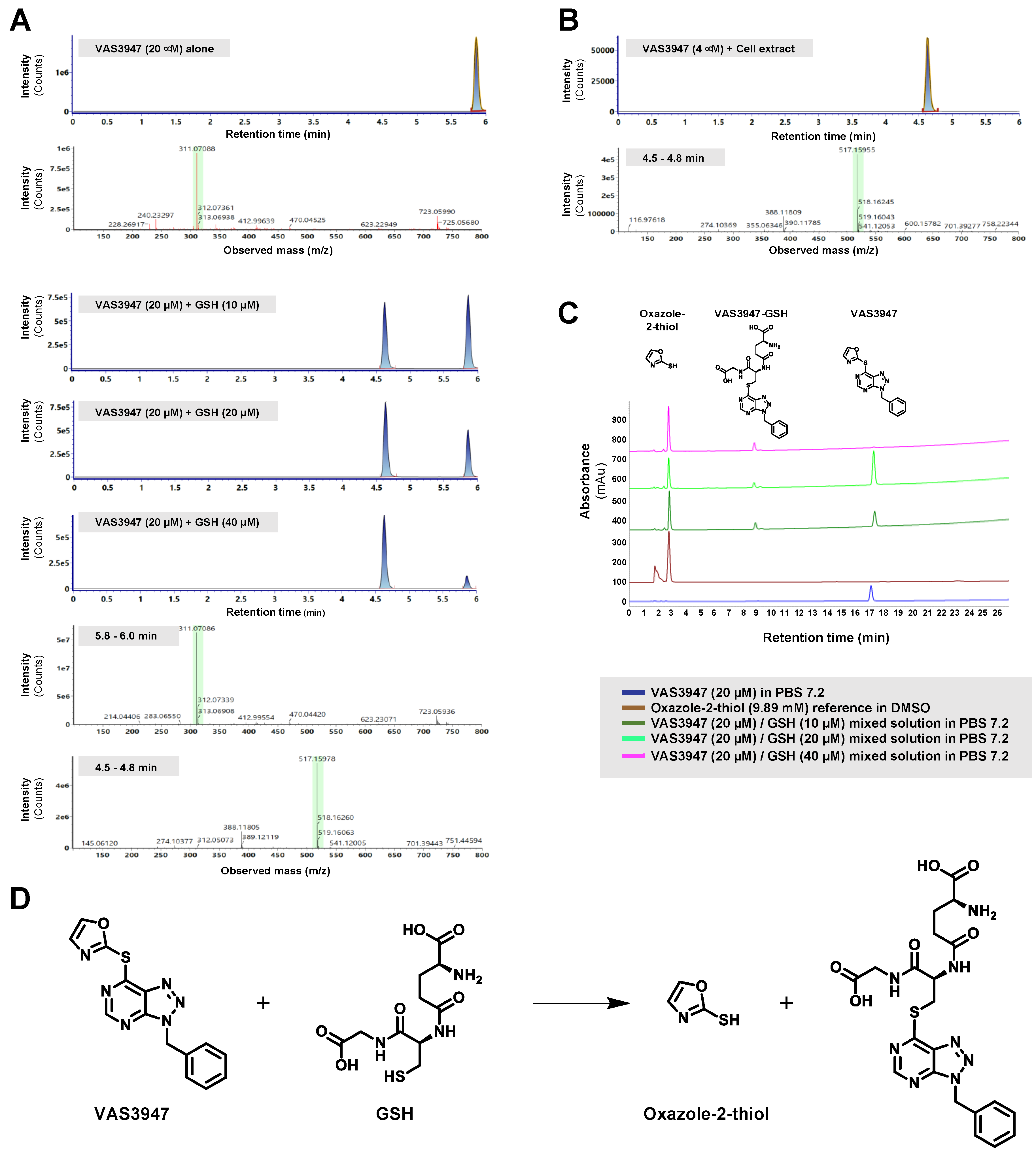
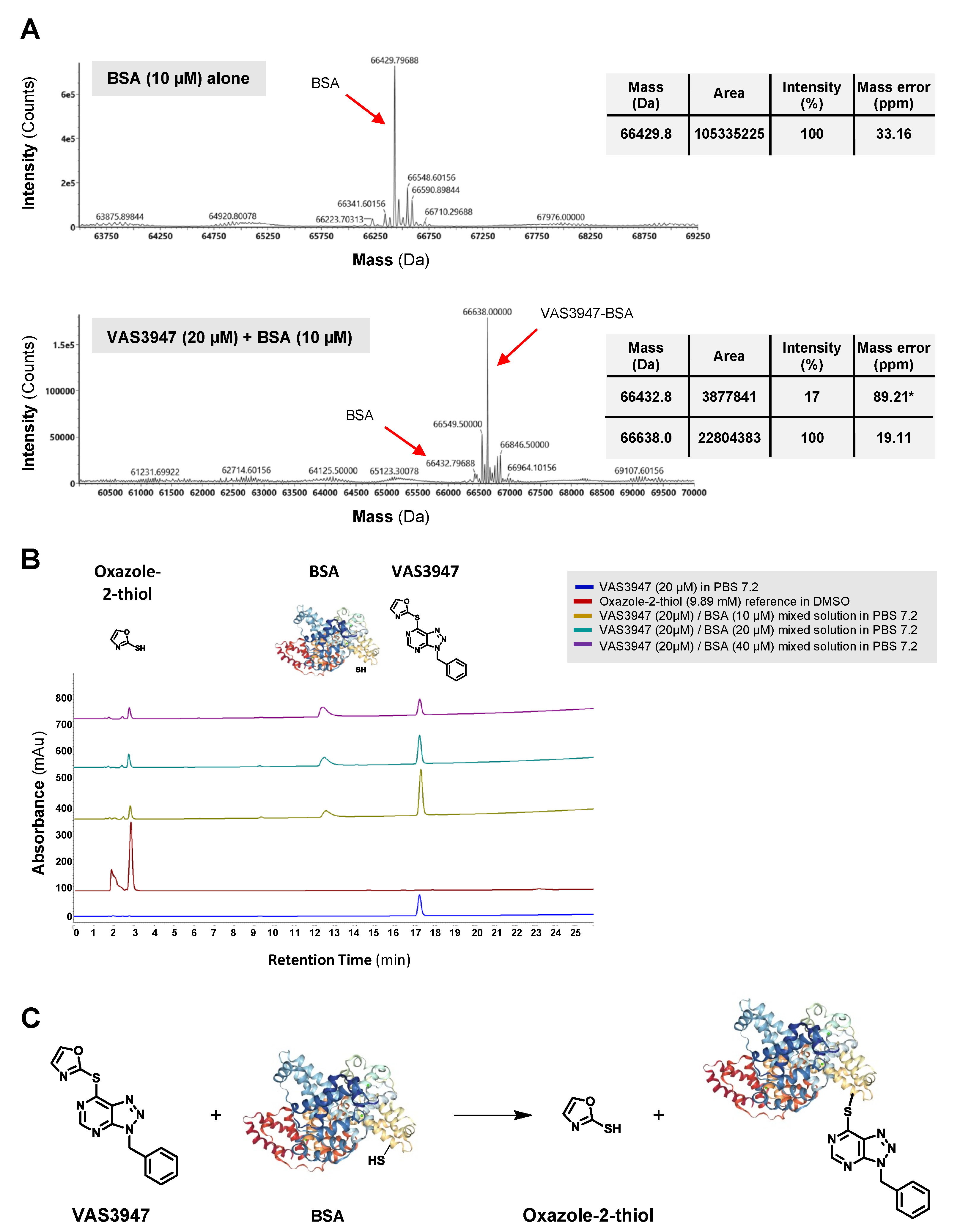
2.2. VAS3947 Alkylates Cys Thiols of Glutathione (GSH) and Bovine Serum Albumin (BSA)
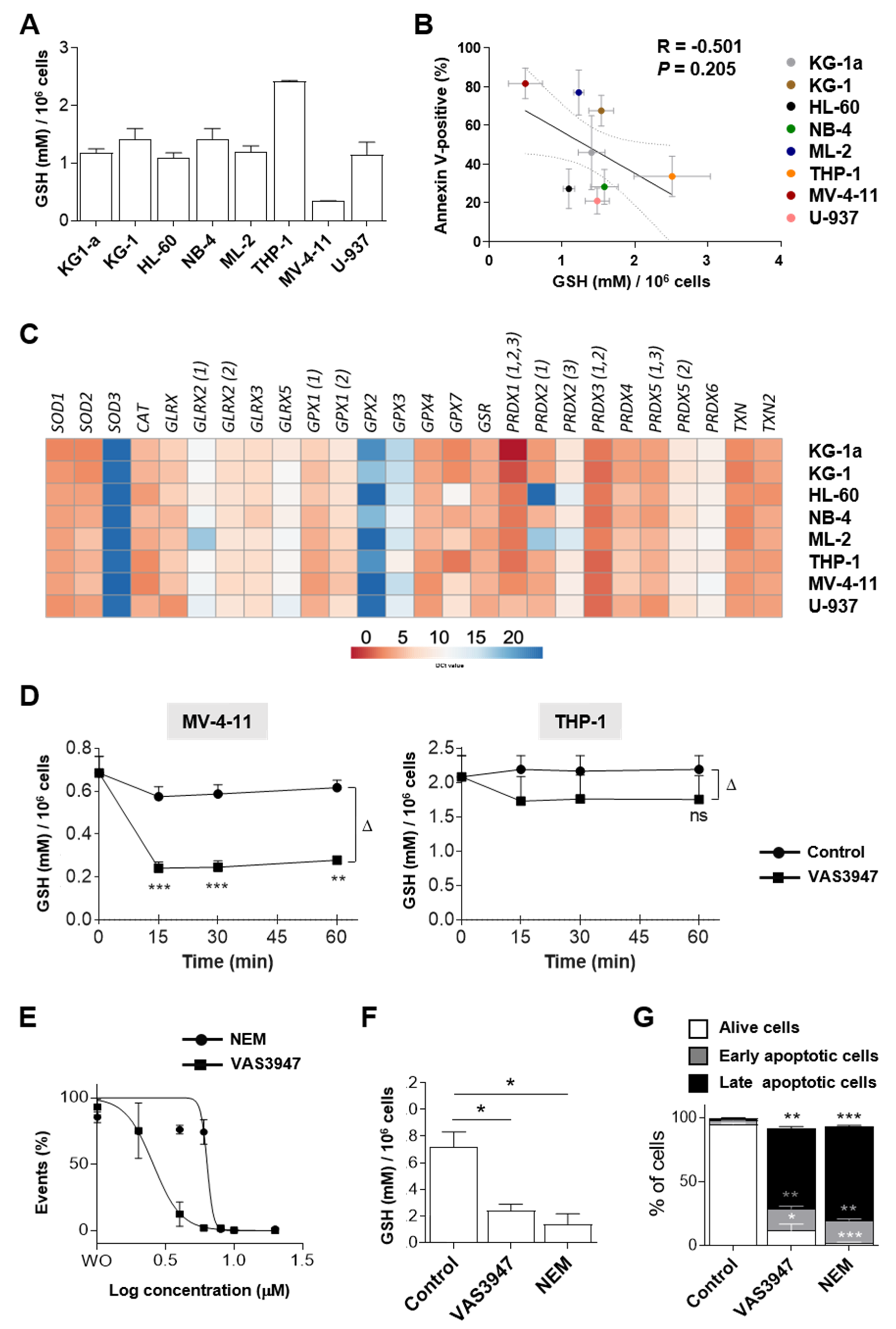
2.3. Sensitivity to VAS3947 Inversely Correlates with the Glutathione Level in AML Cells
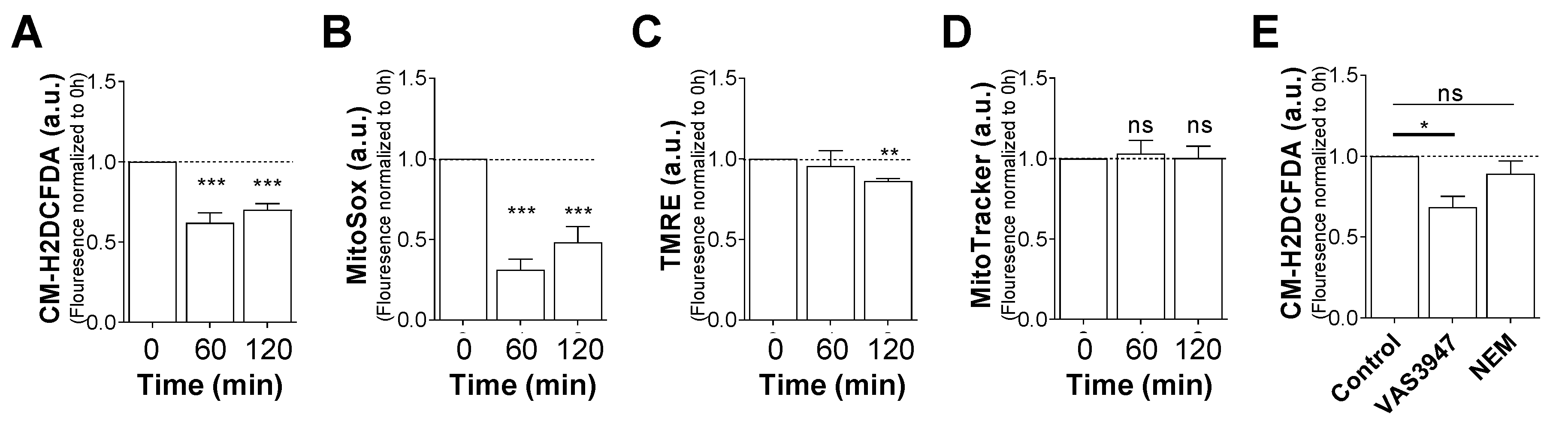
2.4. VAS3947 Decreases ROS Levels
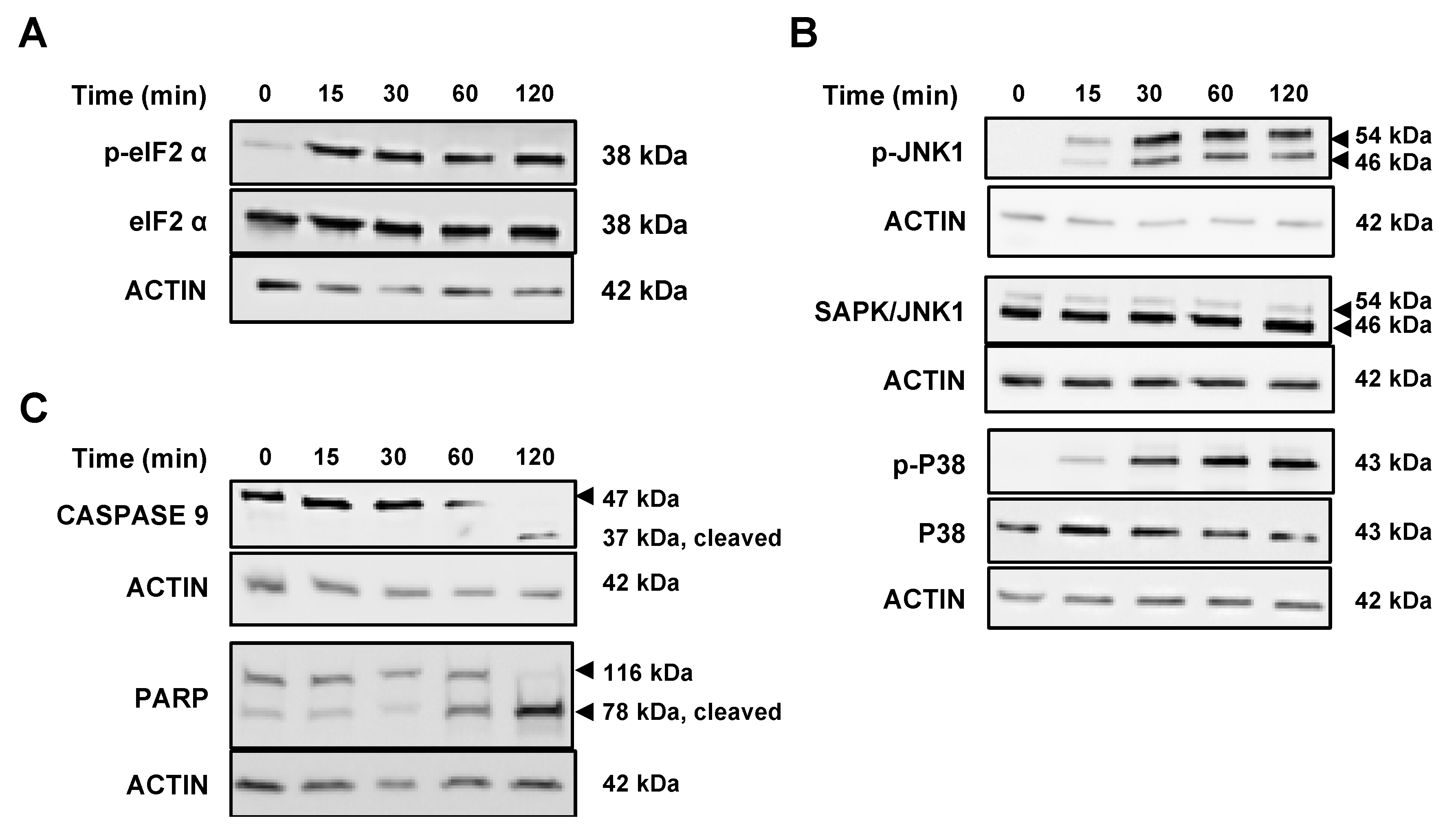
2.5. VAS3947 Triggers Endoplasmic Reticulum (ER) Stress and Consequent Unfolding Protein Response (UPR)
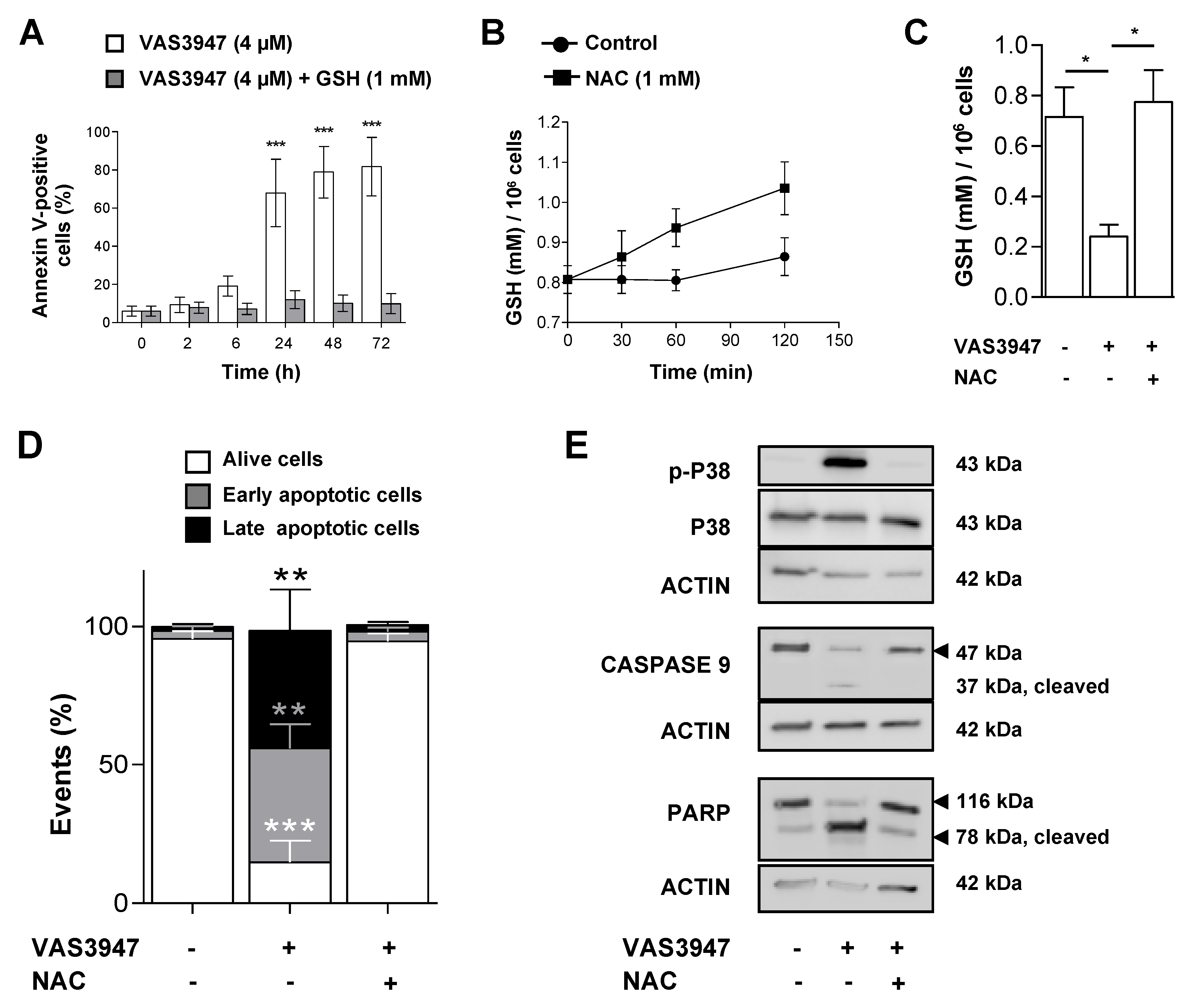
2.6. Supplementation with GSH or N-acetyl Cysteine (NAC) Rescued MV-4-11 Cell Lines from VAS3947 Cytotoxic Effect
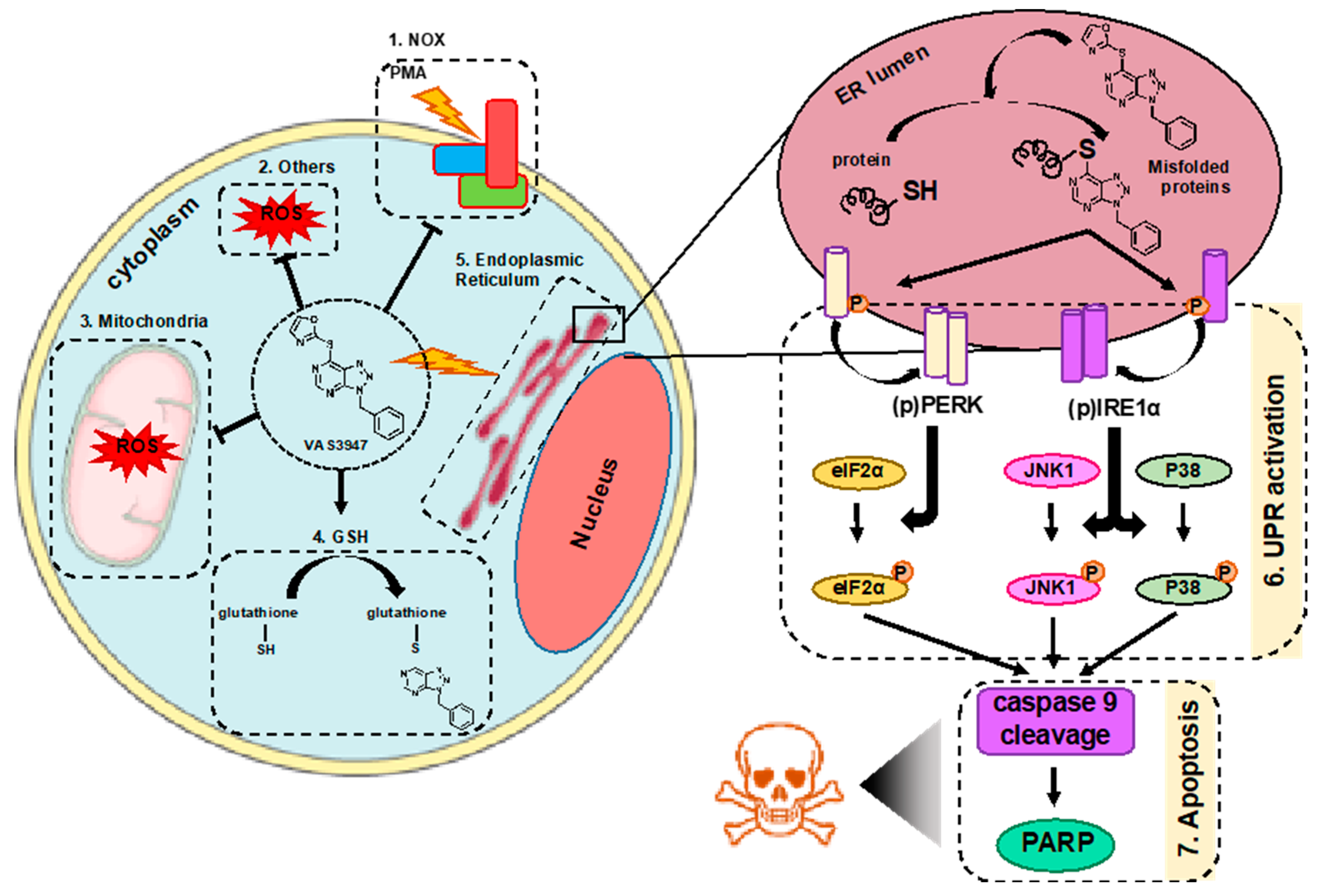
3. Discussion
4. Materials and Methods
4.1. Solution Preparation
4.2. Cell Lines and Culture
4.3. Cell Number Measurement
4.4. Apoptosis Assay
4.5. Enzymatic Activity Assay
4.6. Mass Spectrometry (MS)
4.6.1. For In Vitro VAS3947, GSH and BSA
4.6.2. For Cell Extracts
4.7. High-Performance Liquid Chromatography (HPLC)
4.8. Glutathione Measurement
4.9. Real-Time Reverse Transcription Quantitative PCR (RT-qPCR) Assay
4.10. ROS Measurement
4.11. Mitochondrial Membrane Potential Measurement
4.12. Mitochondrial Biomass Measurement
4.13. Protein Extraction and Western Blotting Assay
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| amu | Atomic mass unit |
| ATF6α | Activating transcription factor 6 alpha |
| BiP | Binding immunoglobulin protein |
| CM-H2DCFDA | chloromethyl derivative- 2′,7′-dichlorodihydrofluorescein diacetate |
| CYBB | Cytochrome b (-245) beta |
| DPI | Diphenyleneiodonium |
| DUOX | Dual Oxigenase |
| eIF2α | Eukaryotic Initiation Factor 2 alpha |
| ER | Endoplasmic Reticulum |
| ERAD | ER-Associated Degradation |
| FAB | French-American-British |
| FLT3-ITD | Fms-Like Tyrosine Kinase 3—Internal Tandem Repeat |
| GSH | Glutathione (reduced) |
| HPLC | High-Performance Liquid Chromatography |
| IRE1α | Inositol-Requiring Enzyme 1 alpha |
| JNK1 | c-Jun N-Terminal Protein Kinase 1 |
| MAPK | Mitogen-Activated Protein Kinase |
| MS | Mass Spectrometry |
| NEM | N-ethylmaleimide |
| NADPH | Nicotinamide Adénine Dinucléotide Phosphate |
| NOX | Unfolded Protein Response |
| PMA | Phorbol Myristate Acetate |
| PERK | PKR-like ER protein kinase |
| ROS | Reactive Oxygen Species |
| UPR | Directory of open access journals |
| TMRE | Tetramethyl-rhodamine ethyl ester |
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Sillar, J.R.; Germon, Z.P.; De Iuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Shen, Q.; Claret, F.X. Novel Roles of Reactive Oxygen Species in the Pathogenesis of Acute Myeloid Leukemia. J. Leukoc. Biol. 2013, 94, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do Reactive Oxygen Species Play a Role in Myeloid Leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.S.; Wu, J.R.; Hu, C.T. Signal Cross Talks for Sustained MAPK Activation and Cell Migration: The Potential Role of Reactive Oxygen Species. Cancer Metastasis Rev. 2008, 27, 303–314. [Google Scholar] [CrossRef]
- Chen, J. Reactive Oxygen Species and Drug Resistance in Cancer Chemotherapy. Austin J. Clin. Pathol. 2014, 1, 1–7. [Google Scholar]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.M.; Wingler, K.; Schmidt, H.H.H.W. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox Enzymes and New Thinking on Reactive Oxygen: A Double-Edged Sword Revisited. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH Oxidase-2 Derived Superoxide Drives Mitochondrial Transfer from Bone Marrow Stromal Cells to Leukemic Blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, A.K.; Moloney, J.N.; Böhmer, F.D.; Cotter, T.G. NOX-Driven ROS Formation in Cell Transformation of FLT3-ITD-Positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondet, J.; Presti, C.L.; Garrel, C.; Skaare, K.; Mariette, C.; Carras, S.; Park, S.; Carré, M.; Bulabois, C.E.; Molina, L.; et al. Adult Patients with de Novo Acute Myeloid Leukemia Show a Functional Deregulation of Redox Balance at Diagnosis Which Is Correlated with Molecular Subtypes and Overall Survival. Haematologica 2019, 104, E393–E397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.J.; Hopkins, G.L.; Rastogi, N.; Hodges, M.; Doyle, M.; Davies, S.; Hole, P.S.; Omidvar, N.; Darley, R.L.; Tonks, A. Reactive Oxygen Species Drive Proliferation in Acute Myeloid Leukemia via the Glycolytic Regulator PFKFB3. Cancer Res. 2020, 80, 937–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayavelu, A.K.; Müller, J.P.; Bauer, R.; Böhmer, S.A.; Lässig, J.; Cerny-Reiterer, S.; Sperr, W.R.; Valent, P.; Maurer, B.; Moriggl, R.; et al. NOX4-Driven ROS Formation Mediates PTP Inactivation and Cell Transformation in FLT3ITD-Positive AML Cells. Leukemia 2016, 30, 473–483. [Google Scholar] [CrossRef]
- Adane, B.; Ye, H.; Khan, N.; Pei, S.; Minhajuddin, M.; Stevens, B.M.; Jones, C.L.; D’Alessandro, A.; Reisz, J.A.; Zaberezhny, V.; et al. The Hematopoietic Oxidase NOX2 Regulates Self-Renewal of Leukemic Stem Cells. Cell Rep. 2019, 27, 238–254. [Google Scholar] [CrossRef] [Green Version]
- Kiffin, R.; Wiktorin, H.G.; Nilsson, M.S.; Aurelius, J.; Aydin, E.; Lenox, B.; Nilsson, J.A.; Ståhlberg, A.; Thorén, F.B.; Hellstrand, K.; et al. Anti-Leukemic Properties of Histamine in Monocytic Leukemia: The Role of NOX2. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Aydin, E.; Hallner, A.; Wiktorin, H.G.; Staffas, A.; Hellstrand, K.; Martner, A. NOX2 Inhibition Reduces Oxidative Stress and Prolongs Survival in Murine KRAS-Induced Myeloproliferative Disease. Oncogene 2019, 38, 1534–1543. [Google Scholar] [CrossRef] [Green Version]
- Wind, S.; Beuerlein, K.; Eucker, T.; Müller, H.; Scheurer, P.; Armitage, M.E.; Ho, H.; Schmidt, H.H.H.W.; Wingler, K. Comparative Pharmacology of Chemically Distinct NADPH Oxidase Inhibitors. Br. J. Pharmacol. 2010, 161, 885–898. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes-Pagano, E.; Meijles, D.N.; Pagano, P.J. The Quest for Selective Nox Inhibitors and Therapeutics: Challenges, Triumphs and Pitfalls. Antioxid. Redox Signal. 2014, 20, 2741–2754. [Google Scholar] [CrossRef] [Green Version]
- Freyhaus, H.T.; Huntgeburth, M.; Wingler, K.; Schnitker, J.; Bäumer, A.T.; Vantler, M.; Bekhite, M.M.; Wartenberg, M.; Sauer, H.; Rosenkranz, S. Novel Nox Inhibitor VAS2870 Attenuates PDGF-Dependent Smooth Muscle Cell Chemotaxis, but Not Proliferation. Cardiovasc. Res. 2006, 71, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.A.; Hess, D.T.; Wang, B.; Miyagi, M.; Stamler, J.S. Off-Target Thiol Alkylation by the NADPH Oxidase Inhibitor 3-Benzyl-7-(2-Benzoxazolyl)Thio-1,2,3-Triazolo[4,5-d]Pyrimidine (VAS2870). Free Radic. Biol. Med. 2012, 52, 1897–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, S.; Minhajuddin, M.; Callahan, K.P.; Balys, M.; Ashton, J.M.; Neering, S.J.; Lagadinou, E.D.; Corbett, C.; Ye, H.; Liesveld, J.L.; et al. Targeting Aberrant Glutathione Metabolism to Eradicate Human Acute Myelogenous Leukemia Cells. J. Boil. Chem. 2013, 288, 33542–33558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Culp-Hill, R.; Reisz, J.A.; Pei, S.; Gustafson, A.; Khan, N.; DeGregori, J.; Pollyea, D.A.; et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 2019, 134, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J.; et al. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [Green Version]
- Hérault, O.; Hope, K.J.; Deneault, E.; Mayotte, N.; Chagraoui, J.; Wilhelm, B.T.; Cellot, S.; Sauvageau, M.; Andrade-Navarro, M.A.; Hébert, J.; et al. A role for GPx3 in activity of normal and leukemia stem cells. J. Exp. Med. 2012, 209, 895–901. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-Stroke Inhibition of Induced NADPH Oxidase Type 4 Prevents Oxidative Stress and Neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Fabregat, I. The NADPH Oxidase Inhibitor VAS2870 Impairs Cell Growth and Enhances TGF-β-Induced Apoptosis of Liver Tumor Cells. Biochem. Pharmacol. 2011, 81, 917–924. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Lin, B.; Tan, X.; Liang, J.; Wu, S.; Liu, J.; Zhang, Q.; Zhu, R. A Reduction in Reactive Oxygen Species Contributes to Dihydromyricetin-Induced Apoptosis in Human Hepatocellular Carcinoma Cells. Sci. Rep. 2014, 4, 7041. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER Stress: Mutual Crosstalk between Autophagy, Oxidative Stress and Inflammatory Response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef] [PubMed]
- Hendershot, L.M. The ER Chaperone BiP Is a Master Regulator of ER Function. Mt. Sinai J. Med. 2004, 71, 289–297. [Google Scholar]
- Lu, W.J.; Li, J.Y.; Chen, R.J.; Huang, L.T.; Lee, T.Y.; Lin, K.H. VAS2870 and VAS3947 Attenuate Platelet Activation and Thrombus Formation via a NOX-Independent Pathway Downstream of PKC. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schardt, J.A.; Mueller, B.U.; Pabst, T. Activation of the Unfolded Protein Response in Human Acute Myeloid Leukemia. Methods Enzymol. 2011, 489, 227–243. [Google Scholar] [CrossRef]
- Schardt, J.A.; Weber, D.; Eyholzer, M.; Mueller, B.U.; Pabst, T. Activation of the Unfolded Protein Response Is Associated with Favorable Prognosis in Acute Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 3834–3841. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.M.; Paganelli, F.; Chiarini, F.; Evangelisti, C.; McCubrey, J.A. The Unfolded Protein Response: A Novel Therapeutic Target in Acute Leukemias. Cancers 2020, 12, 333. [Google Scholar] [CrossRef] [Green Version]
- Masouleh, B.K.; Chevet, E.; Panse, J.; Jost, E.; O’Dwyer, M.; Bruemmendorf, T.H.; Samali, A. Drugging the unfolded protein response in acute leukemias. J. Hematol. Oncol. 2015, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Lin, D.-C.; Guo, X.; Masouleh, B.K.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef] [PubMed]
- Rouault-Pierre, K.; Lopez-Onieva, L.; Foster, K.; Anjos-Afonso, F.; Lamrissi-Garcia, I.; Serrano-Sanchez, M.; Mitter, R.; Ivanovic, Z.; De Verneuil, H.; Gribben, J.; et al. HIF-2α Protects Human Hematopoietic Stem/Progenitors and Acute Myeloid Leukemic Cells from Apoptosis Induced by Endoplasmic Reticulum Stress. Cell Stem Cell 2013, 13, 549–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 Induction through an Atypical Integrated Stress Response to ONC201 Triggers P53-Independent Apoptosis in Hematological Malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masciarelli, S.; Capuano, E.; Ottone, T.; Divona, M.; Lavorgna, S.; Liccardo, F.; Śniegocka, M.; Travaglini, S.; Noguera, N.I.; Picardi, A.; et al. Retinoic Acid Synergizes with the Unfolded Protein Response and Oxidative Stress to Induce Cell Death in FLT3-ITD1 AML. Blood Adv. 2019, 3, 4155–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safiedeen, Z.; Rodríguez-Gómez, I.; Vergori, L.; Soleti, R.; Vaithilingam, D.; Douma, I.; Agouni, A.; Leiber, D.; Dubois, S.; Simard, G.; et al. Temporal Cross Talk between Endoplasmic Reticulum and Mitochondria Regulates Oxidative Stress and Mediates Microparticle-Induced Endothelial Dysfunction. Antioxid. Redox Signal. 2017, 26, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Tietze, F. Enzymic Method for Quantitative Determination of Nanogram Amounts of Total and Oxidized Glutathione: Applications to Mammalian Blood and Other Tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Picou, F.; Vignon, C.; Debeissat, C.; Lachot, S.; Kosmider, O.; Gallay, N.; Foucault, A.; Estienne, M.H.; Ravalet, N.; Bene, M.C.; et al. Bone Marrow Oxidative Stress and Specific Antioxidant Signatures in Myelodysplastic Syndromes. Blood Adv. 2019, 3, 4271–4279. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Dor, M.; Dakik, H.; Polomski, M.; Haudebourg, E.; Brachet, M.; Gouilleux, F.; Prié, G.; Zibara, K.; Mazurier, F. VAS3947 Induces UPR-Mediated Apoptosis through Cysteine Thiol Alkylation in AML Cell Lines. Int. J. Mol. Sci. 2020, 21, 5470. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155470
El Dor M, Dakik H, Polomski M, Haudebourg E, Brachet M, Gouilleux F, Prié G, Zibara K, Mazurier F. VAS3947 Induces UPR-Mediated Apoptosis through Cysteine Thiol Alkylation in AML Cell Lines. International Journal of Molecular Sciences. 2020; 21(15):5470. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155470
Chicago/Turabian StyleEl Dor, Maya, Hassan Dakik, Marion Polomski, Eloi Haudebourg, Marie Brachet, Fabrice Gouilleux, Gildas Prié, Kazem Zibara, and Frédéric Mazurier. 2020. "VAS3947 Induces UPR-Mediated Apoptosis through Cysteine Thiol Alkylation in AML Cell Lines" International Journal of Molecular Sciences 21, no. 15: 5470. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155470