Calsequestrin Deletion Facilitates Hippocampal Synaptic Plasticity and Spatial Learning in Post-Natal Development

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
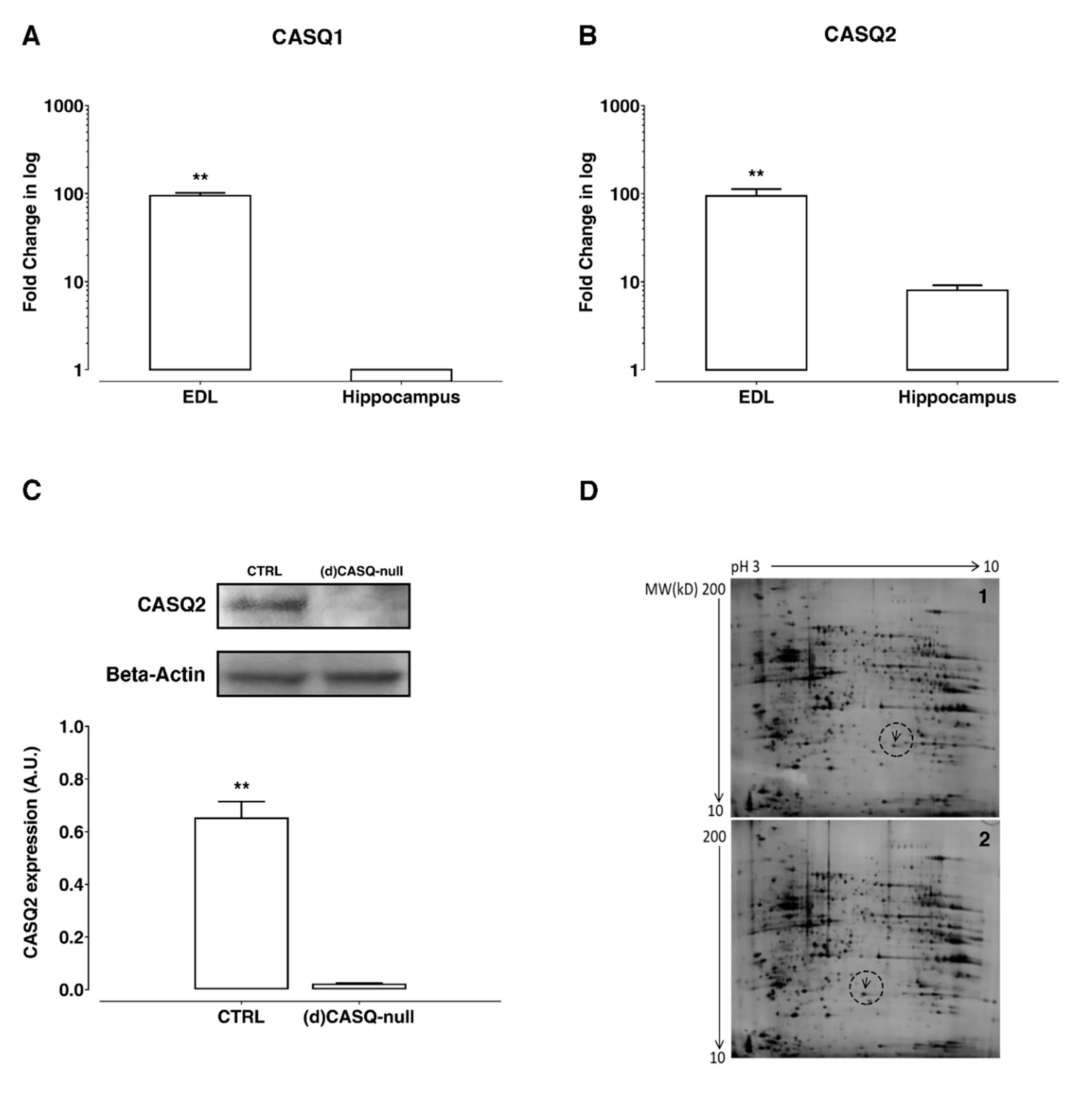
2.1. Biochemical Measurements
2.2. Electrophysiological Analysis
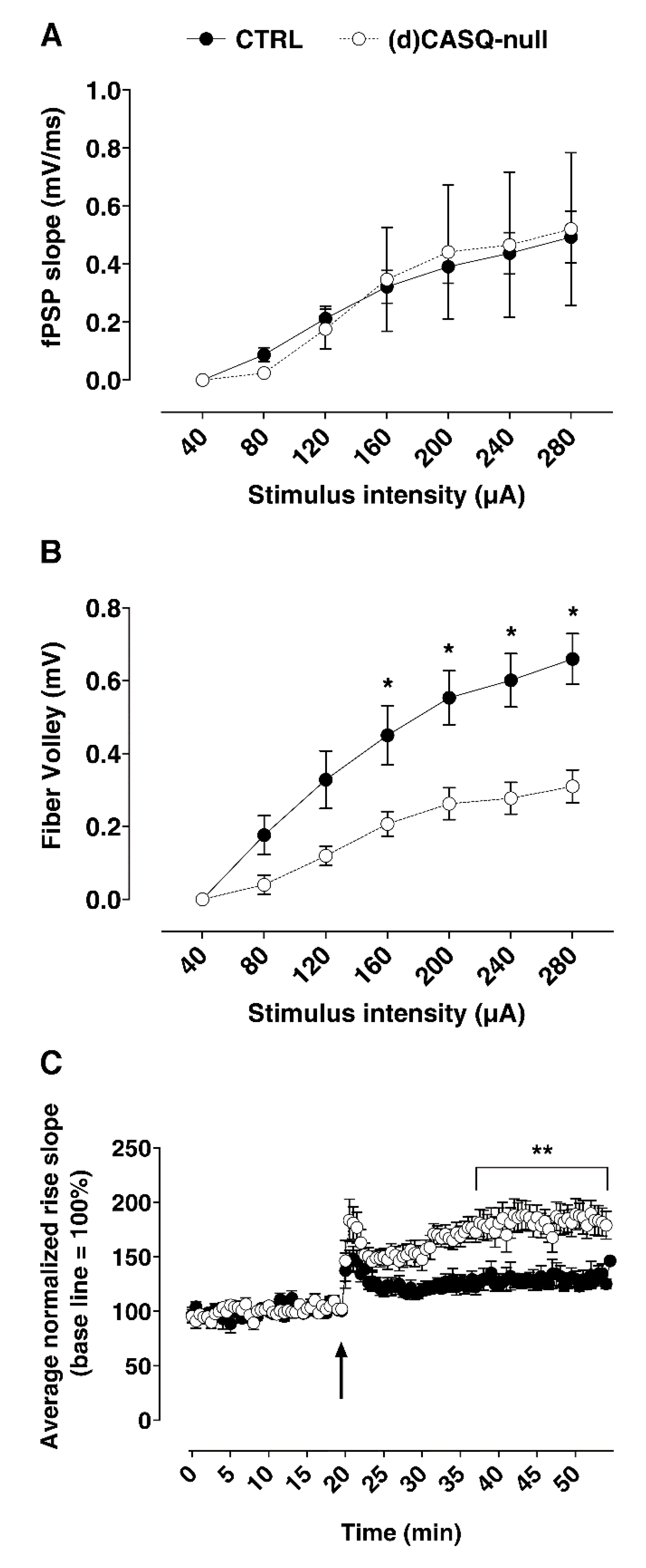
2.2.1. Field Synaptic Responses
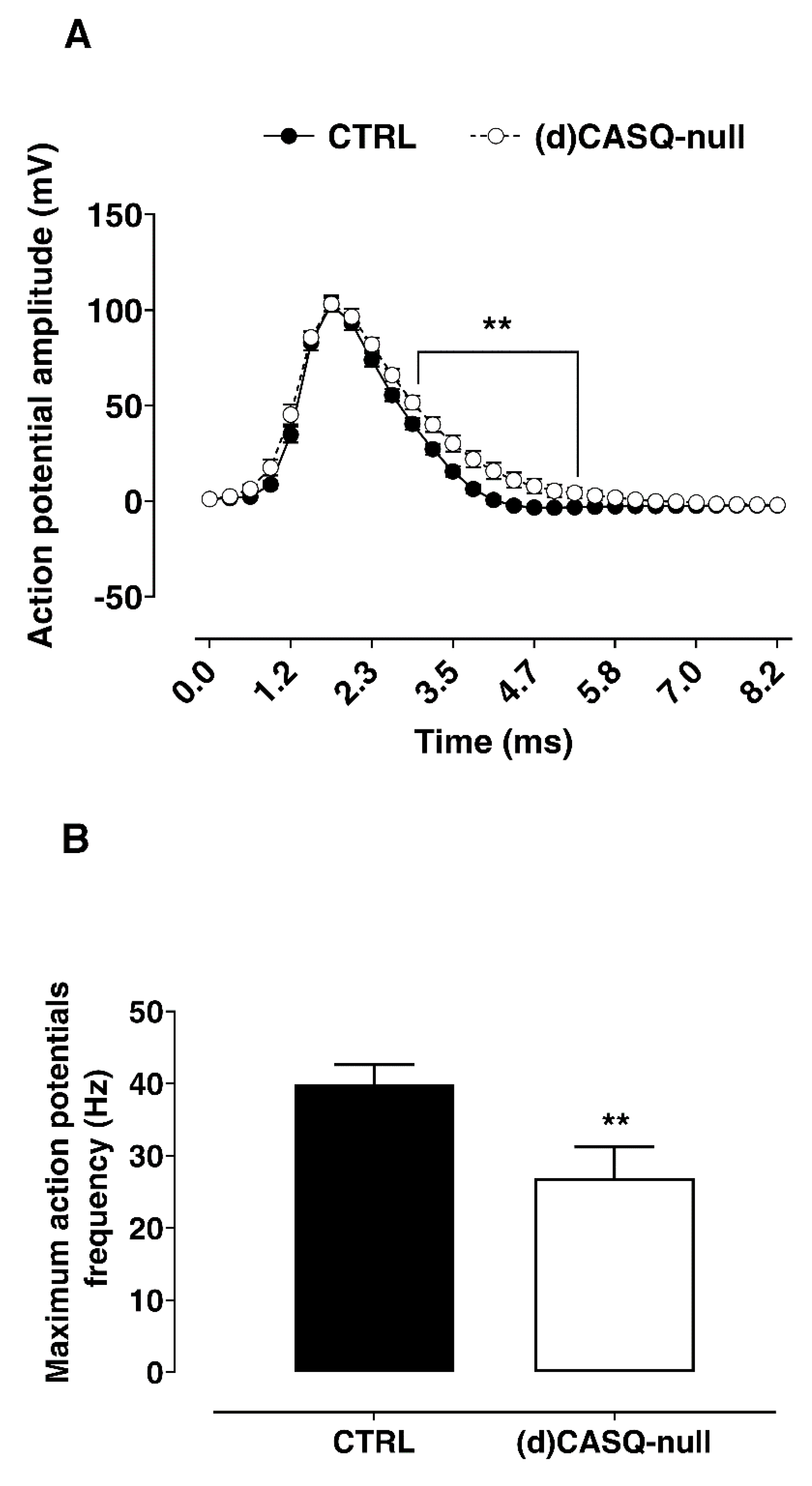
2.2.2. Whole-Cell Patch Clamp
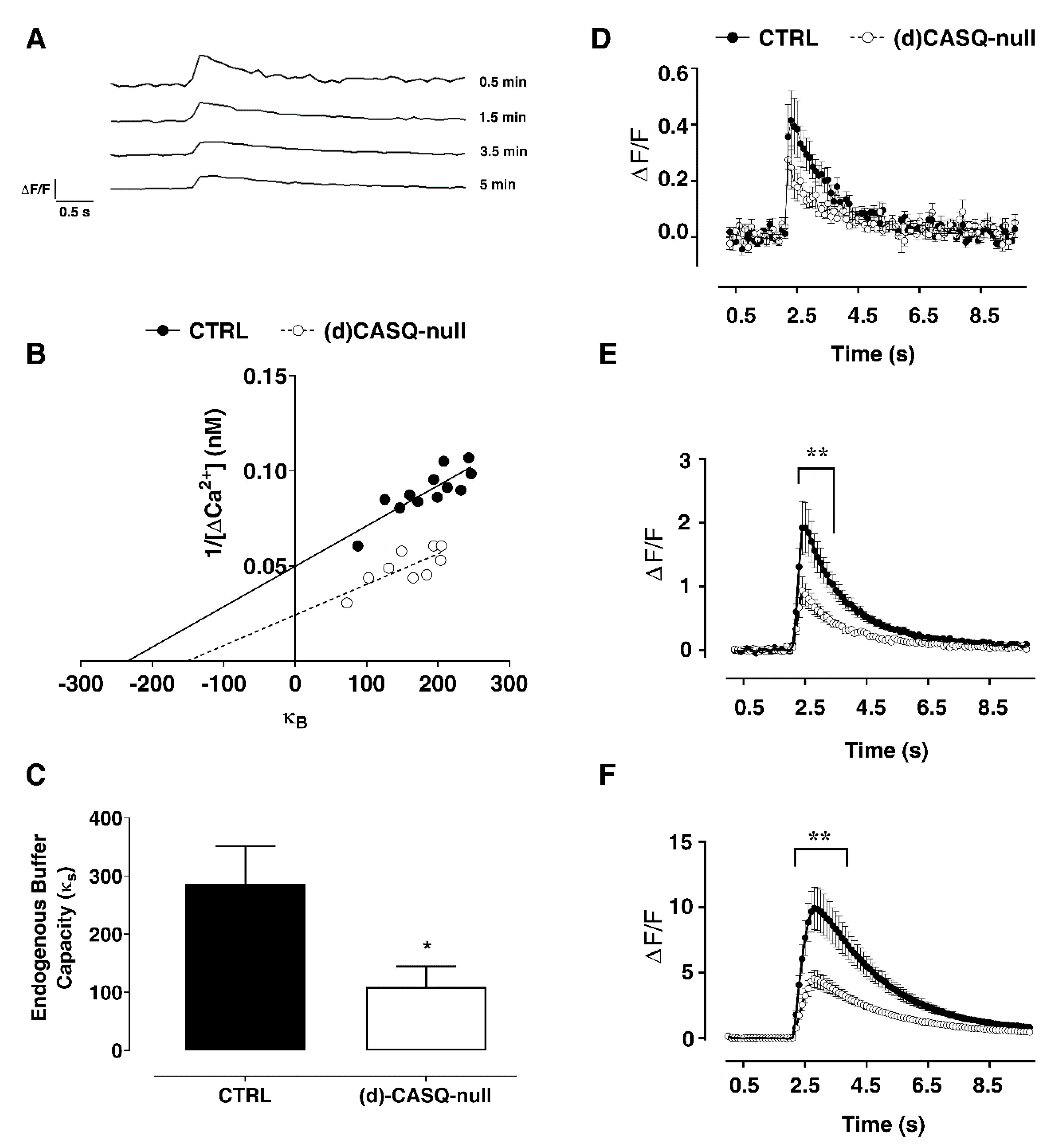
2.2.3. Calcium Imaging
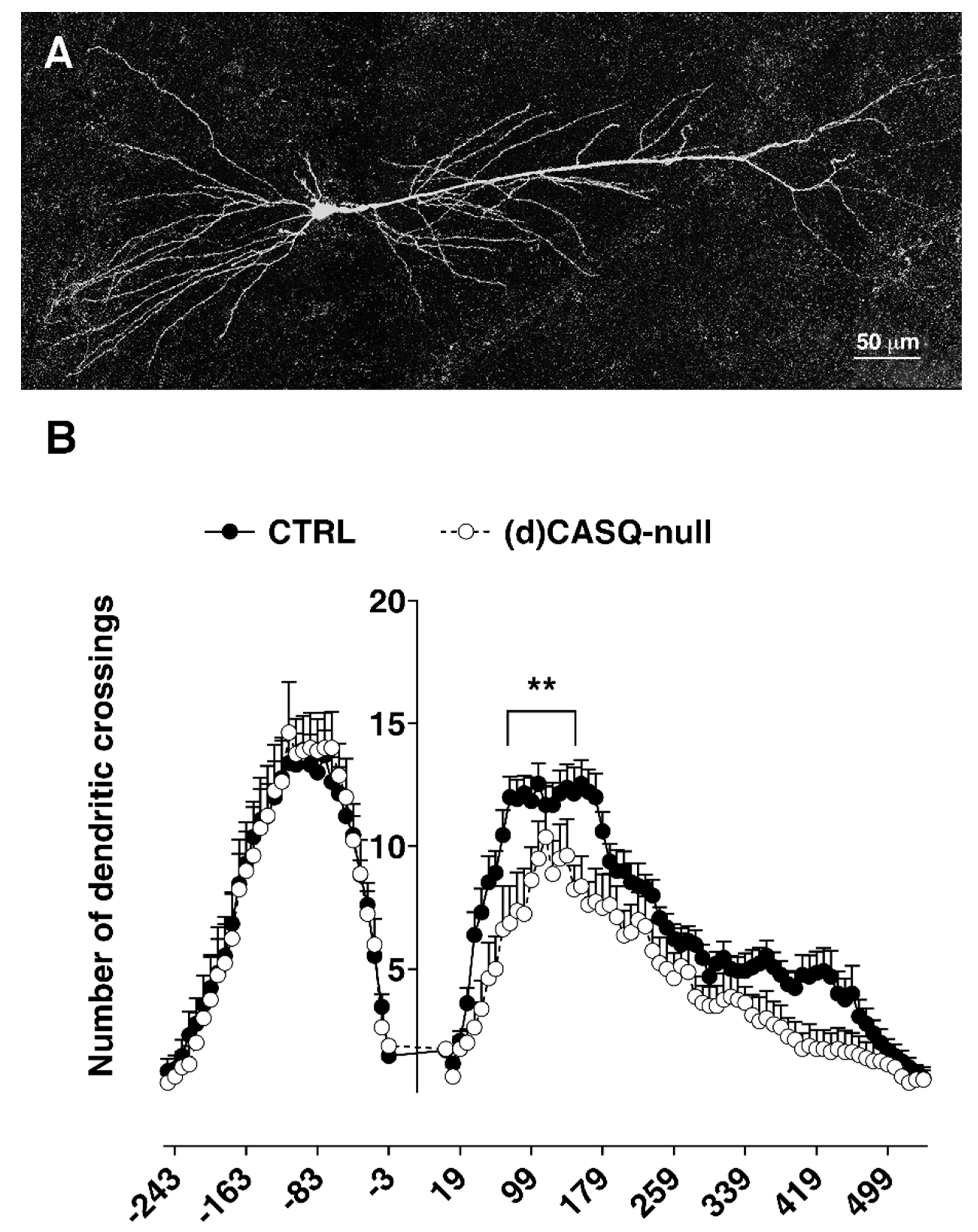
2.3. Morphological Analysis
2.4. Spatial Learning Evaluation
3. Discussion
4. Closing Remarks
5. Materials and Methods
5.1. Double (d)CASQ-Null Mice
5.2. Animal Care
5.3. Biochemical Assays
5.3.1. Gene Expression Analysis
5.3.2. Western Blot (WB) Analysis
5.3.3. Proteomic Analysis
5.4. Electrophysiogical Recordings
5.4.1. Field Potential Recordings
5.4.2. Patch Clamp Recordings
5.4.3. Calcium (Ca2+) Imaging
5.5. Morphological Analysis
5.6. Morris Water Maze
5.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AP | action potential |
| AHP | afterhyperpolarization |
| C | capacitance |
| CASQ | Calsequestrin |
| CASQ1 and CASQ2 | CASQ type-1 and type-2 isoforms |
| CICR | Ca2+-induced Ca2+ release |
| CR | calreticulin |
| ER | endoplasmic reticulum |
| fEPSP | field excitatory postsynaptic potential |
| HFS | high frequency stimulations |
| IR | input resistance |
| LTP | long-term potentiation |
| RMP | resting membrane potential |
| RyR | ryanodine receptor. |
References
- Dolmetsch, R. Excitation-transcription coupling: Signaling by ion channels to the nucleus. Sci. Stke Signal. Transduct. Knowl. Environ. 2003, 2003, Pe4. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Reviews. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- West, A.E.; Chen, W.G.; Dalva, M.B.; Dolmetsch, R.E.; Kornhauser, J.M.; Shaywitz, A.J.; Takasu, M.A.; Tao, X.; Greenberg, M.E. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 11024–11031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, P.L.; Greenberg, M.E. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucker, R.S. Calcium- and activity-dependent synaptic plasticity. Curr. Opin. Neurobiol. 1999, 9, 305–313. [Google Scholar] [CrossRef]
- Cavazzini, M.; Bliss, T.; Emptage, N. Ca2+ and synaptic plasticity. Cell Calcium 2005, 38, 355–367. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 2014, 592, 281–293. [Google Scholar] [CrossRef]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 2005, 85, 201–279. [Google Scholar] [CrossRef]
- Bardo, S.; Cavazzini, M.G.; Emptage, N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol. Sci. 2006, 27, 78–84. [Google Scholar] [CrossRef]
- Emptage, N.; Bliss, T.V.; Fine, A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 1999, 22, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Maggio, N.; Vlachos, A. Synaptic plasticity at the interface of health and disease: New insights on the role of endoplasmic reticulum intracellular calcium stores. Neuroscience 2014, 281, 135–146. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Adasme, T.; Hidalgo, C. Contribution of Ca2+ release channels to hippocampal synaptic plasticity and spatial memory: Potential redox modulation. Antioxid. Redox Signal. 2014, 21, 892–914. [Google Scholar] [CrossRef]
- Meldolesi, J.; Grohovaz, F. Total calcium ultrastructure: Advances in excitable cells. Cell Calcium 2001, 30, 1–8. [Google Scholar] [CrossRef]
- Weaver, S.A.; Schaefer, A.L.; Dixon, W.T. The effects of mutated skeletal ryanodine receptors on calreticulin and calsequestrin expression in the brain and pituitary gland of boars. Brain Research. Mol. Brain Res. 2000, 75, 46–53. [Google Scholar] [CrossRef]
- Campbell, K.P.; MacLennan, D.H.; Jorgensen, A.O.; Mintzer, M.C. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53,000 dalton glycoprotein. J. Biol. Chem. 1983, 258, 1197–1204. [Google Scholar]
- Damiani, E.; Volpe, P.; Margreth, A. Coexpression of two isoforms of calsequestrin in rabbit slow-twitch muscle. J. Muscle Res. Cell Motil. 1990, 11, 522–530. [Google Scholar] [CrossRef]
- Fliegel, L.; Ohnishi, M.; Carpenter, M.R.; Khanna, V.K.; Reithmeier, R.A.; MacLennan, D.H. Amino acid sequence of rabbit fast-twitch skeletal muscle calsequestrin deduced from cDNA and peptide sequencing. Proc. Natl. Acad. Sci. USA 1987, 84, 1167–1171. [Google Scholar] [CrossRef] [Green Version]
- Scott, B.T.; Simmerman, H.K.; Collins, J.H.; Nadal-Ginard, B.; Jones, L.R. Complete amino acid sequence of canine cardiac calsequestrin deduced by cDNA cloning. J. Biol. Chem. 1988, 263, 8958–8964. [Google Scholar]
- Paolini, C.; Quarta, M.; Nori, A.; Boncompagni, S.; Canato, M.; Volpe, P.; Allen, P.D.; Reggiani, C.; Protasi, F. Reorganized stores and impaired calcium handling in skeletal muscle of mice lacking calsequestrin-1. J. Physiol. 2007, 583, 767–784. [Google Scholar] [CrossRef]
- Paolini, C.; Quarta, M.; D’Onofrio, L.; Reggiani, C.; Protasi, F. Differential effect of calsequestrin ablation on structure and function of fast and slow skeletal muscle fibers. J. Biomed. Biotechnol. 2011, 2011, 634075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dainese, M.; Quarta, M.; Lyfenko, A.D.; Paolini, C.; Canato, M.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Anesthetic- and heat-induced sudden death in calsequestrin-1-knockout mice. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protasi, F.; Paolini, C.; Dainese, M. Calsequestrin-1: A new candidate gene for malignant hyperthermia and exertional/environmental heat stroke. J. Physiol. 2009, 587, 3095–3100. [Google Scholar] [CrossRef]
- Canato, M.; Capitanio, P.; Reggiani, C.; Cancellara, L. The disorders of the calcium release unit of skeletal muscles: What have we learned from mouse models? J. Muscle Res. Cell Motil. 2015, 36, 61–69. [Google Scholar] [CrossRef]
- Michelucci, A.; Paolini, C.; Boncompagni, S.; Canato, M.; Reggiani, C.; Protasi, F. Strenuous exercise triggers a life-threatening response in mice susceptible to malignant hyperthermia. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3649–3662. [Google Scholar] [CrossRef] [Green Version]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef] [Green Version]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Vezzani, B.; Galli, L.; Paolini, C.; Toniolo, L.; Pierantozzi, E.; Spinozzi, S.; Barone, V.; Pegoraro, E.; Bello, L.; et al. A mutation in the CASQ1 gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates. Hum. Mutat. 2014, 35, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Di Blasi, C.; Sansanelli, S.; Ruggieri, A.; Moriggi, M.; Vasso, M.; D’Adamo, A.P.; Blasevich, F.; Zanotti, S.; Paolini, C.; Protasi, F.; et al. A CASQ1 founder mutation in three Italian families with protein aggregate myopathy and hyperCKaemia. J. Med. Genet. 2015, 52, 617–626. [Google Scholar] [CrossRef]
- Sabatini, B.L.; Oertner, T.G.; Svoboda, K. The life cycle of Ca(2+) ions in dendritic spines. Neuron 2002, 33, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Sjostrom, P.J.; Nelson, S.B. Spike timing, calcium signals and synaptic plasticity. Curr. Opin. Neurobiol. 2002, 12, 305–314. [Google Scholar] [CrossRef]
- Springer, S.J.; Burkett, B.J.; Schrader, L.A. Modulation of BK channels contributes to activity-dependent increase of excitability through MTORC1 activity in CA1 pyramidal cells of mouse hippocampus. Front. Cell. Neurosci. 2014, 8, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Rakoczy, S.; Brown-Borg, H. Assessment of spatial memory in mice. Life Sci. 2010, 87, 521–536. [Google Scholar] [CrossRef]
- Lisman, J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc. Natl. Acad. Sci. USA 1989, 86, 9574–9578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malenka, R.C.; Lancaster, B.; Zucker, R.S. Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron 1992, 9, 121–128. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Dudek, S.M.; Bear, M.F. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. USA 1992, 89, 4363–4367. [Google Scholar] [CrossRef] [Green Version]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 18–41. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.D.; Edwards, T.M.; Rickard, N.S. The role of intracellular calcium stores in synaptic plasticity and memory consolidation. Neurosci. Biobehav. Rev. 2013, 37, 1211–1239. [Google Scholar] [CrossRef]
- Raymond, C.R.; Redman, S.J. Different calcium sources are narrowly tuned to the induction of different forms of LTP. J. Neurophysiol. 2002, 88, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Mellentin, C.; Jahnsen, H.; Abraham, W.C. Priming of long-term potentiation mediated by ryanodine receptor activation in rat hippocampal slices. Neuropharmacology 2007, 52, 118–125. [Google Scholar] [CrossRef]
- Grigoryan, G.; Korkotian, E.; Segal, M. Selective facilitation of LTP in the ventral hippocampus by calcium stores. Hippocampus 2012, 22, 1635–1644. [Google Scholar] [CrossRef]
- Futatsugi, A.; Kato, K.; Ogura, H.; Li, S.T.; Nagata, E.; Kuwajima, G.; Tanaka, K.; Itohara, S.; Mikoshiba, K. Facilitation of NMDAR-independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron 1999, 24, 701–713. [Google Scholar] [CrossRef] [Green Version]
- Reddish, F.N.; Miller, C.L.; Gorkhali, R.; Yang, J.J. Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases. Int. J. Mol. Sci. 2017, 18, 1024. [Google Scholar] [CrossRef] [Green Version]
- Beard, N.A.; Wei, L.; Dulhunty, A.F. Control of muscle ryanodine receptor calcium release channels by proteins in the sarcoplasmic reticulum lumen. Clin. Exp. Pharmacol. Physiol. 2009, 36, 340–345. [Google Scholar] [CrossRef]
- Canato, M.; Scorzeto, M.; Giacomello, M.; Protasi, F.; Reggiani, C.; Stienen, G.J. Massive alterations of sarcoplasmic reticulum free calcium in skeletal muscle fibers lacking calsequestrin revealed by a genetically encoded probe. Proc. Natl. Acad. Sci. USA 2010, 107, 22326–22331. [Google Scholar] [CrossRef] [Green Version]
- Michelucci, A.; Paolini, C.; Canato, M.; Wei-Lapierre, L.; Pietrangelo, L.; De Marco, A.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Antioxidants protect calsequestrin-1 knockout mice from halothane- and heat-induced sudden death. Anesthesiology 2015, 123, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Sacchetto, R.; Cliffer, K.D.; Podini, P.; Villa, A.; Christensen, B.N.; Volpe, P. Intracellular Ca2+ stores in chick cerebellum Purkinje neurons: Ontogenetic and functional studies. Am. J. Physiol. 1995, 269, C1219–C1227. [Google Scholar] [CrossRef]
- Treves, S.; De Mattei, M.; Landfredi, M.; Villa, A.; Green, N.M.; MacLennan, D.H.; Meldolesi, J.; Pozzan, T. Calreticulin is a candidate for a calsequestrin-like function in Ca2(+)-storage compartments (calciosomes) of liver and brain. Biochem. J. 1990, 271, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B. Peroxiredoxin 6 in the repair of peroxidized cell membranes and cell signaling. Arch. Biochem. Biophys. 2017, 617, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Foster, T.C. Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J. Neurophysiol. 2004, 91, 2437–2444. [Google Scholar] [CrossRef] [Green Version]
- Borde, M.; Bonansco, C.; Fernandez de Sevilla, D.; Le Ray, D.; Buno, W. Voltage-clamp analysis of the potentiation of the slow Ca2+-activated K+ current in hippocampal pyramidal neurons. Hippocampus 2000, 10, 198–206. [Google Scholar] [CrossRef]
- Kumar, A.; Foster, T.C. 17beta-estradiol benzoate decreases the AHP amplitude in CA1 pyramidal neurons. J. Neurophysiol. 2002, 88, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Landfield, P.W.; Pitler, T.A. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science 1984, 226, 1089–1092. [Google Scholar] [CrossRef]
- Power, J.M.; Wu, W.W.; Sametsky, E.; Oh, M.M.; Disterhoft, J.F. Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 7234–7243. [Google Scholar] [CrossRef] [Green Version]
- Sah, P.; Faber, E.S. Channels underlying neuronal calcium-activated potassium currents. Prog. Neurobiol. 2002, 66, 345–353. [Google Scholar] [CrossRef]
- Lancaster, B.; Nicoll, R.A. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J. Physiol. 1987, 389, 187–203. [Google Scholar] [CrossRef]
- Storm, J.F. Intracellular injection of a Ca2+ chelator inhibits spike repolarization in hippocampal neurons. Brain Res. 1987, 435, 387–392. [Google Scholar] [CrossRef]
- Faber, E.S.; Sah, P. Ca2+-activated K+ (BK) channel inactivation contributes to spike broadening during repetitive firing in the rat lateral amygdala. J. Physiol. 2003, 552, 483–497. [Google Scholar] [CrossRef]
- Norris, C.M.; Halpain, S.; Foster, T.C. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 3171–3179. [Google Scholar]
- Foster, T.C.; Norris, C.M. Age-associated changes in Ca(2+)-dependent processes: Relation to hippocampal synaptic plasticity. Hippocampus 1997, 7, 602–612. [Google Scholar] [CrossRef]
- Foster, T.C. Involvement of hippocampal synaptic plasticity in age-related memory decline. Brain Research. Brain Res. Rev. 1999, 30, 236–249. [Google Scholar] [CrossRef]
- Sah, P.; Bekkers, J.M. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: Implications for the integration of long-term potentiation. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 4537–4542. [Google Scholar]
- Cohen, A.S.; Coussens, C.M.; Raymond, C.R.; Abraham, W.C. Long-lasting increase in cellular excitability associated with the priming of LTP induction in rat hippocampus. J. Neurophysiol. 1999, 82, 3139–3148. [Google Scholar] [CrossRef] [Green Version]
- Sourdet, V.; Russier, M.; Daoudal, G.; Ankri, N.; Debanne, D. Long-term enhancement of neuronal excitability and temporal fidelity mediated by metabotropic glutamate receptor subtype 5. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 10238–10248. [Google Scholar]
- Kramar, E.A.; Lin, B.; Lin, C.Y.; Arai, A.C.; Gall, C.M.; Lynch, G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 5151–5161. [Google Scholar] [CrossRef]
- Le Ray, D.; Fernandez De Sevilla, D.; Belen Porto, A.; Fuenzalida, M.; Buno, W. Heterosynaptic metaplastic regulation of synaptic efficacy in CA1 pyramidal neurons of rat hippocampus. Hippocampus 2004, 14, 1011–1025. [Google Scholar] [CrossRef]
- Murphy, G.G.; Fedorov, N.B.; Giese, K.P.; Ohno, M.; Friedman, E.; Chen, R.; Silva, A.J. Increased neuronal excitability, synaptic plasticity, and learning in aged Kvbeta1.1 knockout mice. Curr. Biol. Cb 2004, 14, 1907–1915. [Google Scholar] [CrossRef] [Green Version]
- Fuenzalida, M.; Fernandez de Sevilla, D.; Buno, W. Changes of the EPSP waveform regulate the temporal window for spike-timing-dependent plasticity. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 11940–11948. [Google Scholar] [CrossRef]
- Xu, J.; Kang, J. The mechanisms and functions of activity-dependent long-term potentiation of intrinsic excitability. Rev. Neurosci. 2005, 16, 311–323. [Google Scholar] [CrossRef]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Abel, T.; Nguyen, P.V.; Barad, M.; Deuel, T.A.; Kandel, E.R.; Bourtchouladze, R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 1997, 88, 615–626. [Google Scholar] [CrossRef] [Green Version]
- Malenka, R.C.; Nicoll, R.A. Long-term potentiation--a decade of progress? Science 1999, 285, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.A. Long-term potentiation and memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Whitlock, J.R.; Heynen, A.J.; Shuler, M.G.; Bear, M.F. Learning induces long-term potentiation in the hippocampus. Science 2006, 313, 1093–1097. [Google Scholar] [CrossRef] [Green Version]
- van Strien, N.M.; Cappaert, N.L.; Witter, M.P. The anatomy of memory: An interactive overview of the parahippocampal-hippocampal network. Nat. Reviews. Neurosci. 2009, 10, 272–282. [Google Scholar] [CrossRef]
- Davis, S.; Butcher, S.P.; Morris, R.G. The NMDA receptor antagonist D-2-amino-5-phosphonopentanoate (D-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro. J. Neurosci. Off. J. Soc. Neurosci. 1992, 12, 21–34. [Google Scholar]
- Nicoll, R.A. A Brief History of Long-Term Potentiation. Neuron 2017, 93, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, J.D. Dynamic DNA methylation controls glutamate receptor trafficking and synaptic scaling. J. Neurochem. 2016, 137, 312–330. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Betti, M.; Ambrogini, P.; Minelli, A.; Floridi, A.; Lattanzi, D.; Ciuffoli, S.; Bucherelli, C.; Prospero, E.; Frontini, A.; Santarelli, L.; et al. Maternal dietary loads of alpha-tocopherol depress protein kinase C signaling and synaptic plasticity in rat postnatal developing hippocampus and promote permanent deficits in adult offspring. J. Nutr. Biochem. 2011, 22, 60–70. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sestili, P.; Barbieri, E.; Martinelli, C.; Battistelli, M.; Guescini, M.; Vallorani, L.; Casadei, L.; D’Emilio, A.; Falcieri, E.; Piccoli, G.; et al. Creatine supplementation prevents the inhibition of myogenic differentiation in oxidatively injured C2C12 murine myoblasts. Mol. Nutr. Food Res. 2009, 53, 1187–1204. [Google Scholar] [CrossRef]
- Sinha, P.; Poland, J.; Schnolzer, M.; Rabilloud, T. A new silver staining apparatus and procedure for matrix-assisted laser desorption/ionization-time of flight analysis of proteins after two-dimensional electrophoresis. Proteomics 2001, 1, 835–840. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef]
- Guescini, M.; Guidolin, D.; Vallorani, L.; Casadei, L.; Gioacchini, A.M.; Tibollo, P.; Battistelli, M.; Falcieri, E.; Battistin, L.; Agnati, L.F.; et al. C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp. Cell Res. 2010, 316, 1977–1984. [Google Scholar] [CrossRef]
- Barbieri, E.; Battistelli, M.; Casadei, L.; Vallorani, L.; Piccoli, G.; Guescini, M.; Gioacchini, A.M.; Polidori, E.; Zeppa, S.; Ceccaroli, P.; et al. Morphofunctional and Biochemical Approaches for Studying Mitochondrial Changes during Myoblasts Differentiation. J. Aging Res. 2011, 2011, 845379. [Google Scholar] [CrossRef] [Green Version]
- Ambrogini, P.; Lattanzi, D.; Ciuffoli, S.; Agostini, D.; Bertini, L.; Stocchi, V.; Santi, S.; Cuppini, R. Morpho-functional characterization of neuronal cells at different stages of maturation in granule cell layer of adult rat dentate gyrus. Brain Res. 2004, 1017, 21–31. [Google Scholar] [CrossRef]
- Lattanzi, D.; Savelli, D.; Di Palma, M.; Sartini, S.; Eusebi, S.; Borroto-Escuela, D.O.; Cuppini, R.; Fuxe, K.; Ambrogini, P. Electrophysiological approach to GPCR–RTK interaction study in hippocampus of adult rats. In Receptor-Receptor Interactions in the Central Nervous System; Springer: Berlin/Heidelberg, Germany, 2018; pp. 71–90. [Google Scholar]
- Sartini, S.; Lattanzi, D.; Di Palma, M.; Savelli, D.; Eusebi, S.; Sestili, P.; Cuppini, R.; Ambrogini, P. Maternal Creatine Supplementation Positively Affects Male Rat Hippocampal Synaptic Plasticity in Adult Offspring. Nutrients 2019, 11, 2014. [Google Scholar] [CrossRef] [Green Version]
- Lattanzi, D.; Di Palma, M.; Cuppini, R.; Ambrogini, P. GABAergic Input Affects Intracellular Calcium Levels in Developing Granule Cells of Adult Rat Hippocampus. Int. J. Mol. Sci. 2020, 21, 1715. [Google Scholar] [CrossRef] [Green Version]
- Maravall, M.; Mainen, Z.F.; Sabatini, B.L.; Svoboda, K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 2000, 78, 2655–2667. [Google Scholar] [CrossRef] [Green Version]
- NeuronJ: An ImageJ Plugin for Neurite Tracing and Quantification. Available online: https://imagescience.org/meijering/software/neuronj/ (accessed on 4 June 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTRL | (d)CASQ-Null | |
|---|---|---|
| RMP (mV) ** | −65.1 ± 2.3 | −55.6 ± 2.6 |
| IR MΩ | 210.4 ± 19.1 | 249.7± 21.4 |
| C pF | 104.5 ± 9.1 | 93.6 ± 8.9 |
| AP-T (mV) ** | −33.2 ± 2.2 | −41.0 ± 1.2 |
| AP-A (mV) | 101.7 ± 3.6 | 101.8 ± 4.2 |
| CTRL | (d)CASQ-Null | |
|---|---|---|
| [Ca2+]0 (nM) | 39 ± 6.5 | 47 ± 18.9 |
| Δ[Ca2+]AP (nM) | 12 ± 1.2 | 14 ± 2.8 |
| Unperturbed [Ca2+] (nM) | 40 ± 12.8 | 61 ± 21.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrogini, P.; Lattanzi, D.; Di Palma, M.; Ciacci, C.; Savelli, D.; Galati, C.; Gioacchini, A.M.; Pietrangelo, L.; Vallorani, L.; Protasi, F.; et al. Calsequestrin Deletion Facilitates Hippocampal Synaptic Plasticity and Spatial Learning in Post-Natal Development. Int. J. Mol. Sci. 2020, 21, 5473. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155473
Ambrogini P, Lattanzi D, Di Palma M, Ciacci C, Savelli D, Galati C, Gioacchini AM, Pietrangelo L, Vallorani L, Protasi F, et al. Calsequestrin Deletion Facilitates Hippocampal Synaptic Plasticity and Spatial Learning in Post-Natal Development. International Journal of Molecular Sciences. 2020; 21(15):5473. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155473
Chicago/Turabian StyleAmbrogini, Patrizia, Davide Lattanzi, Michael Di Palma, Caterina Ciacci, David Savelli, Claudia Galati, Anna Maria Gioacchini, Laura Pietrangelo, Luciana Vallorani, Feliciano Protasi, and et al. 2020. "Calsequestrin Deletion Facilitates Hippocampal Synaptic Plasticity and Spatial Learning in Post-Natal Development" International Journal of Molecular Sciences 21, no. 15: 5473. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155473