Genome-Wide Analysis Reveals Changes in Long Noncoding RNAs in the Differentiation of Canine BMSCs into Insulin-Producing Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overview of lncRNA Sequencing
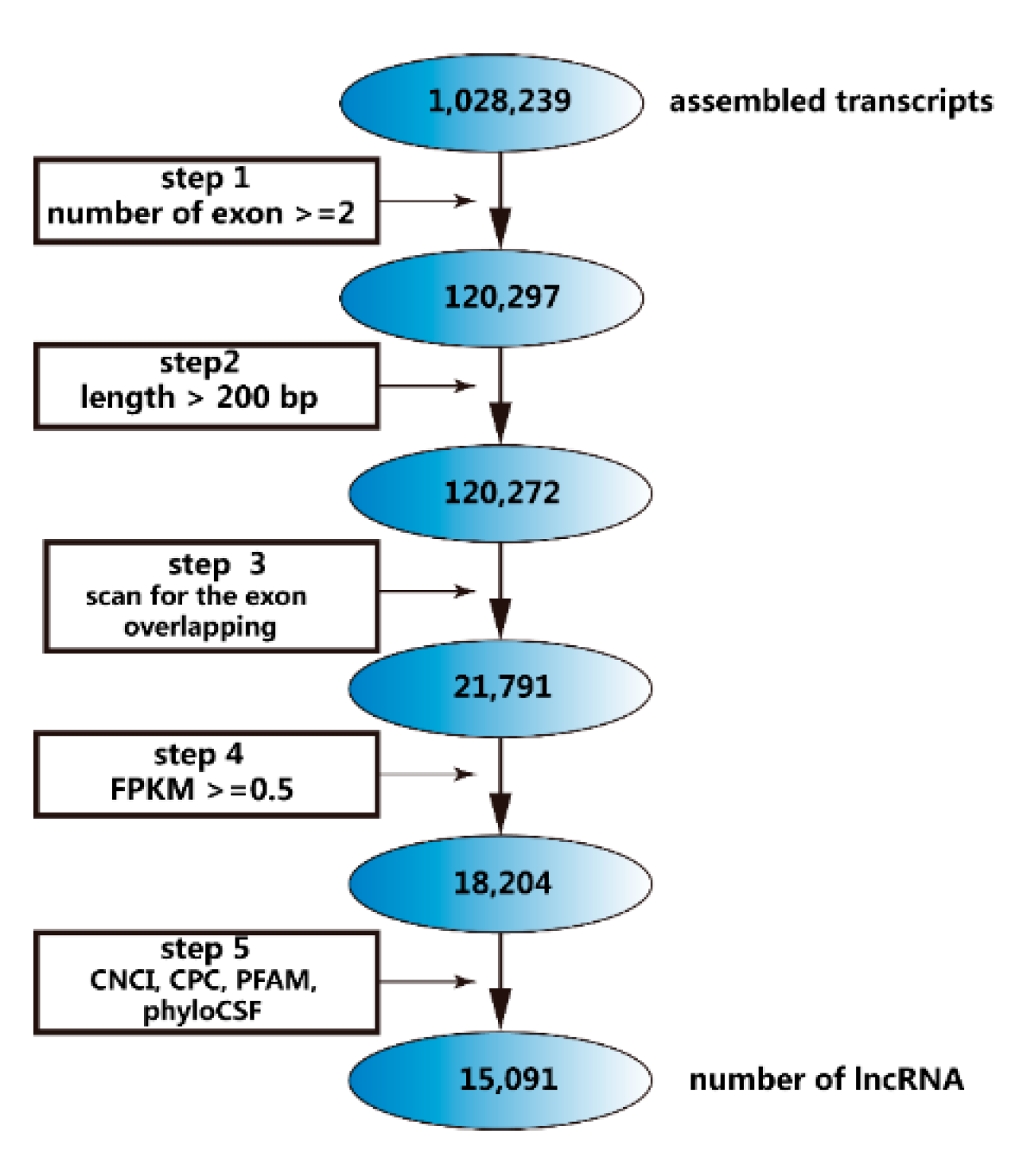
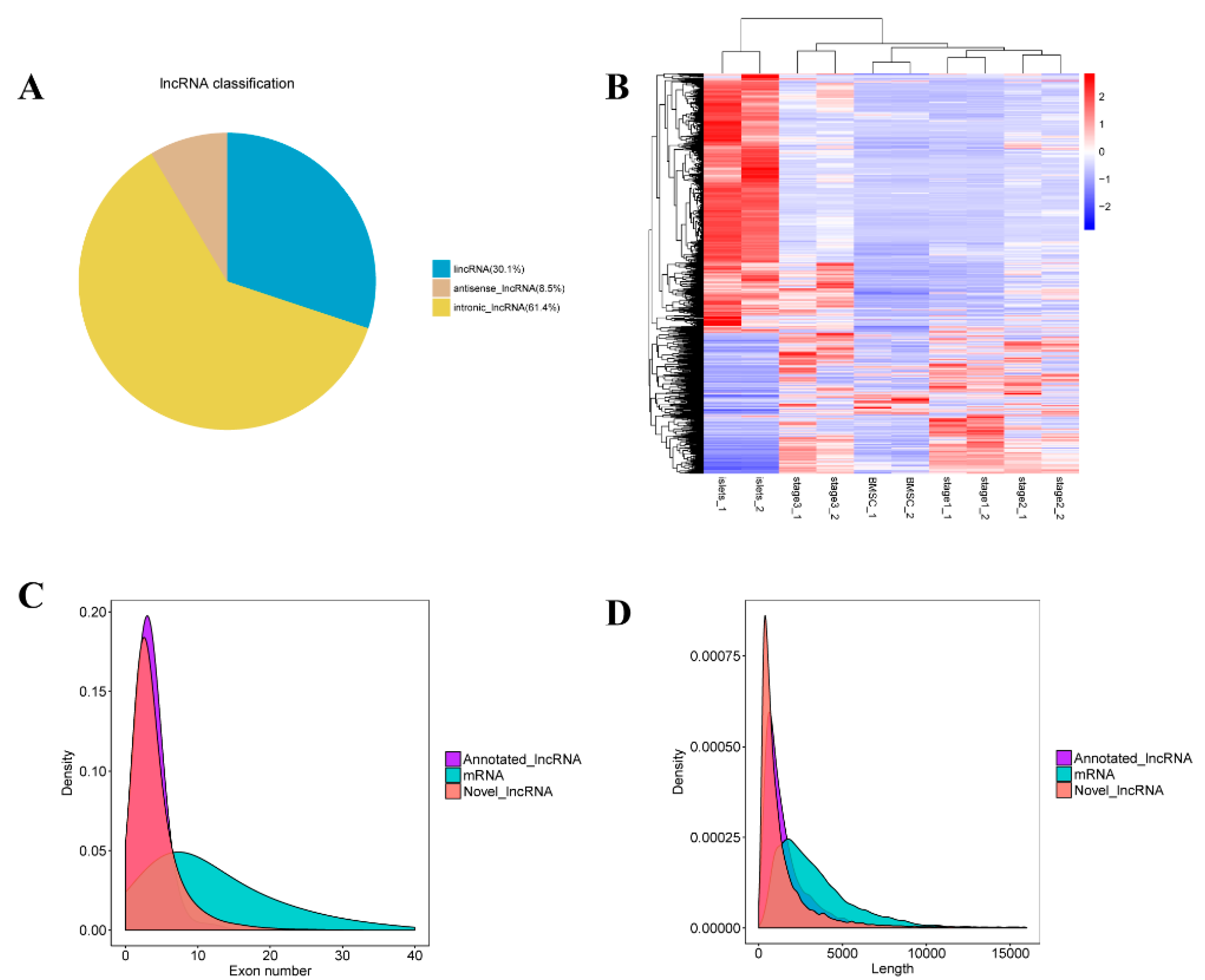
2.2. Identification of lncRNAs
2.3. Differentially Expressed lncRNAs
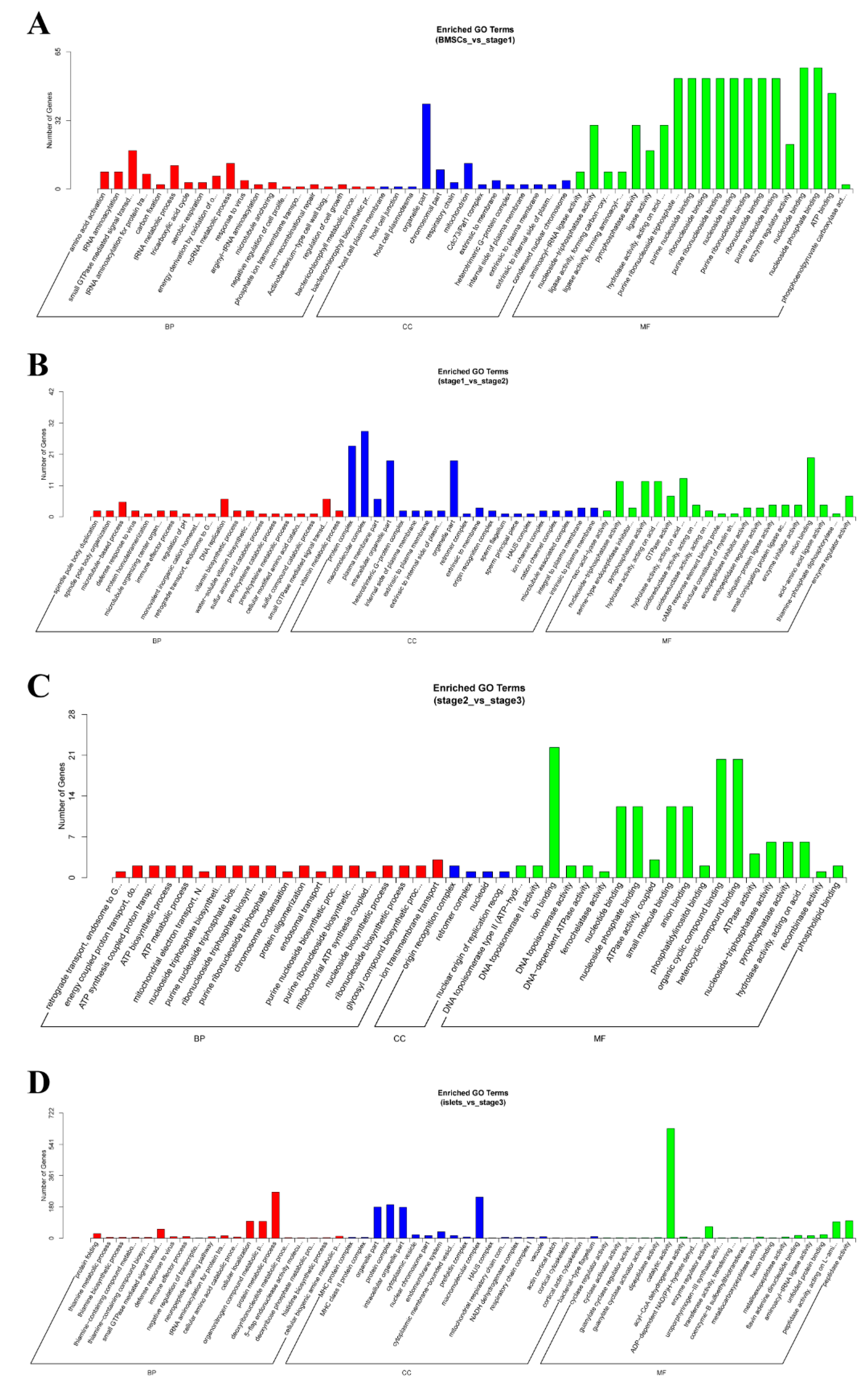
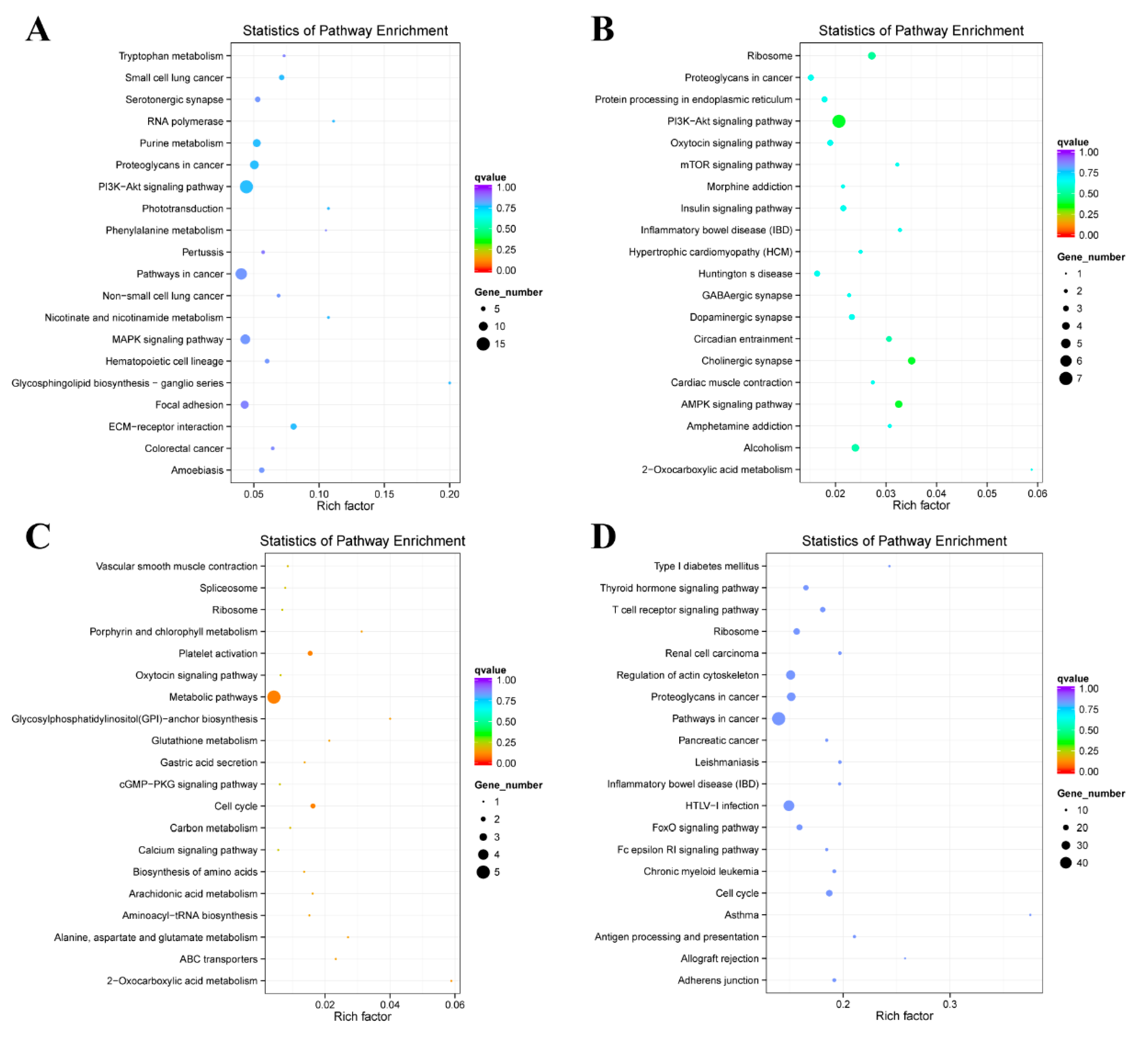
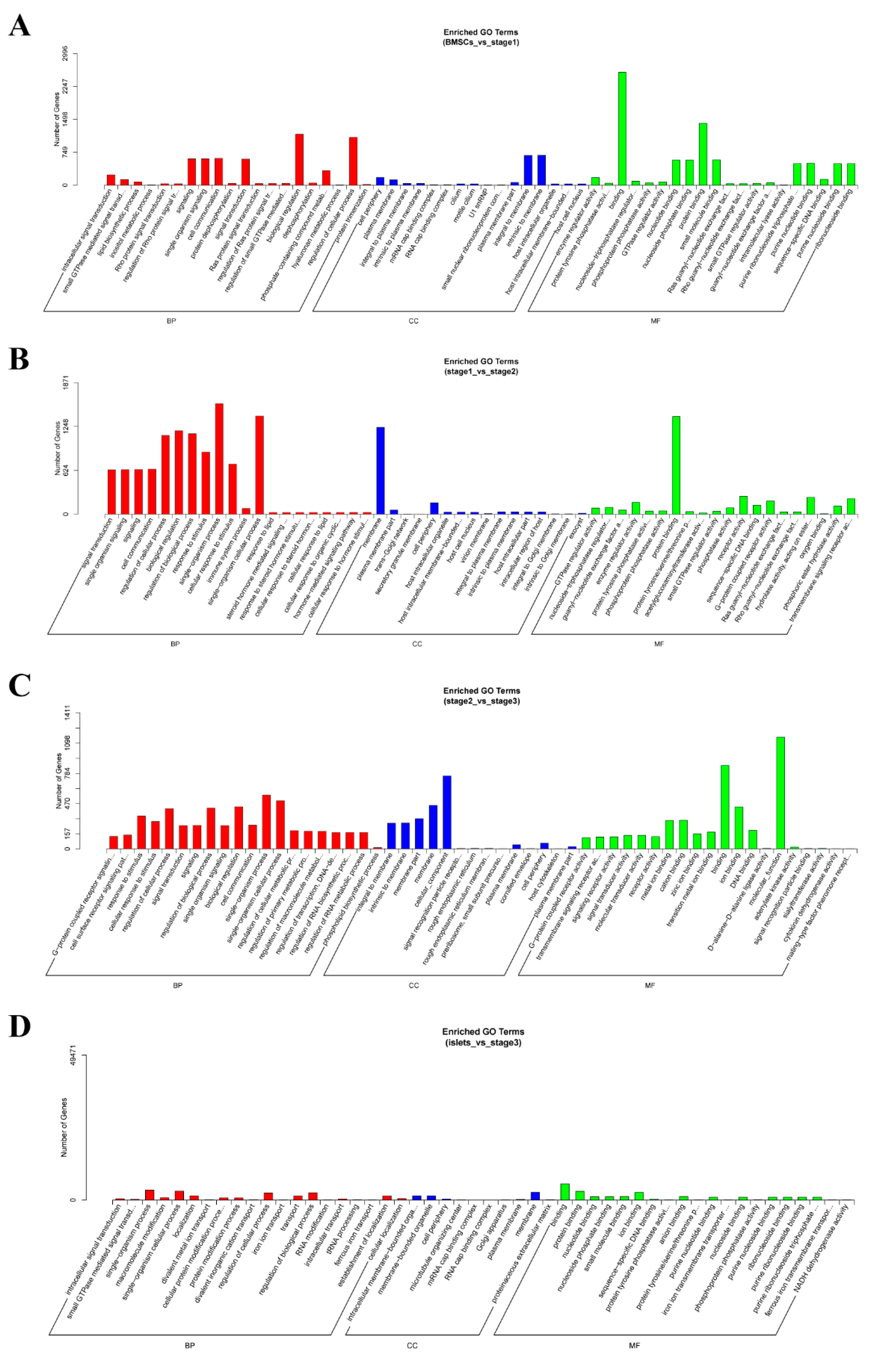
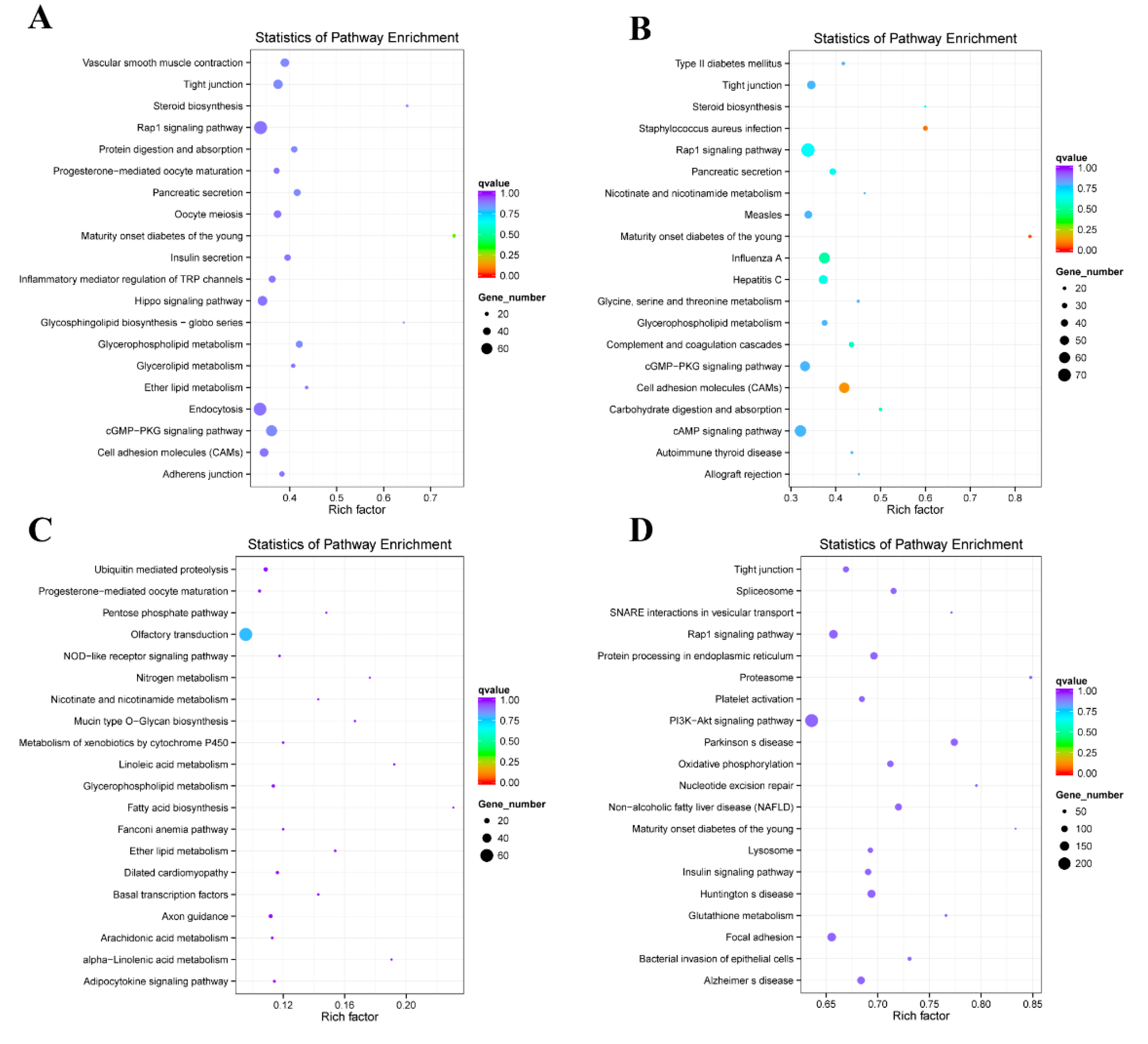
2.4. Enrichment Analysis of Colocalized Genes
2.5. Enrichment Analysis of Coexpressed Genes
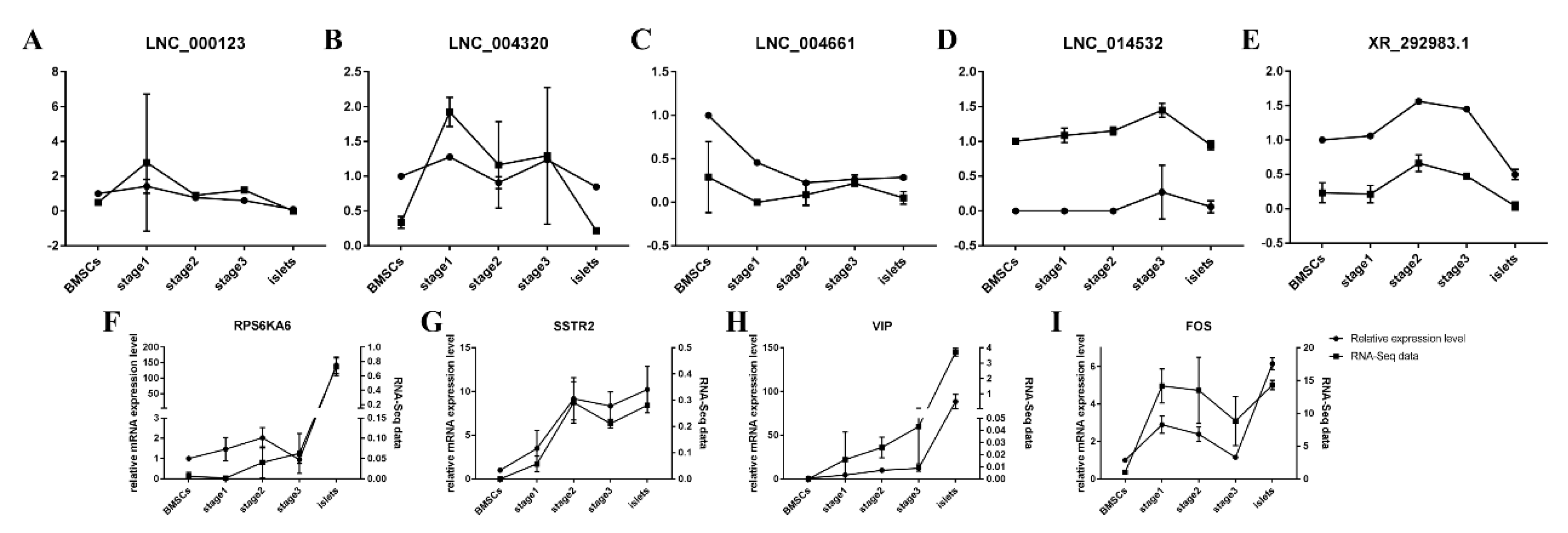
2.6. Verification of Target Genes and lncRNAs
3. Discussion
4. Materials and Methods
4.1. Separation, Cultivation, and Induction of cBMSCs
4.2. RNA Isolation and Qualification
4.3. Library Preparation
5. Sequencing
5.1. Transcriptome Assembly
5.2. Identification of lncRNAs
5.3. Differential Expression Analysis
5.4. GO and KEGG Enrichment Analyses
5.5. Target Gene Prediction
5.6. Gene Expression Validation by qRT-PCR
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anderson, D.M.; Anderson, K.M.; Chang, C.-L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.J.; Hendrich, B.D.; Rupert, J.L.; Lafrenière, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar] [CrossRef]
- Marques, A.C.; Ponting, C.P. Intergenic lncRNAs and the evolution of gene expression. Curr. Opin. Genet. Dev. 2014, 27, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rearick, D.; Prakash, A.; McSweeny, A.; Shepard, S.S.; Fedorova, L.; Fedorov, A. Critical association of ncRNA with introns. Nucleic Acids Res. 2011, 39, 2357–2366. [Google Scholar] [CrossRef] [Green Version]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science 2005, 309, 1564–1566. [Google Scholar]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [Green Version]
- Ravasi, T.; Suzuki, H.; Pang, K.C.; Katayama, S.; Furuno, M.; Okunishi, R.; Fukuda, S.; Ru, K.; Frith, M.C.; Gongora, M.M.; et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006, 16, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Subtelny, A.O.; Koppstein, D.; Bell, G.W.; Sive, H.; Bartel, D.P. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012, 22, 2054–2066. [Google Scholar] [CrossRef] [Green Version]
- Tupy, J.L.; Bailey, A.M.; Dailey, G.; Evans-Holm, M.; Siebel, C.W.; Misra, S.; Celniker, S.E.; Rubin, G.M. Identification of putative noncoding polyadenylated transcripts in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2005, 102, 5495–5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomar, D.; Yadav, A.S.; Kumar, D.; Bhadauriya, G.; Kundu, G.C. Non-coding RNAs as potential therapeutic targets in breast cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194378. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, Q.; Piao, H.-Y.; Wang, B.; Zhu, G.-Q.; Chen, E.-B.; Xiao, K.; Zhou, Z.-J.; Shi, G.-M.; Shi, Y.-H.; et al. HNRNPAB-regulated lncRNA-ELF209 inhibits the malignancy of hepatocellular carcinoma. Int. J. Cancer 2020, 146, 169–180. [Google Scholar] [CrossRef]
- Lee, J.-S.; Mendell, J.T. Antisense-Mediated Transcript Knockdown Triggers Premature Transcription Termination. Mol. Cell 2020, 77, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Nair, L.; Chung, H.; Basu, U. Regulation of long non-coding RNAs and genome dynamics by the RNA surveillance machinery. Nat. Rev. Mol. Cell Biol. 2020, 21, 123–136. [Google Scholar] [CrossRef]
- Aboudehen, K. Regulation of mTOR signaling by long non-coding RNA. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194449. [Google Scholar] [CrossRef]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef]
- Ju, C.; Liu, R.; Zhang, Y.-W.; Zhang, Y.; Zhou, R.; Sun, J.; Lv, X.-B.; Zhang, Z. Mesenchymal stem cell-associated lncRNA in osteogenic differentiation. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 115, 108912. [Google Scholar] [CrossRef]
- Zhang, W.; Dong, R.; Diao, S.; Du, J.; Fan, Z.; Wang, F. Differential long noncoding RNA/mRNA expression profiling and functional network analysis during osteogenic differentiation of human bone marrow mesenchymal stem cells. Stem Cell Res. Ther. 2017, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, A.; Ismaily, M.; Bajaj, M. Diabetes and covid-19: A global health challenge. BMJ Open Diabetes Res. Care 2020, 8, e001450. [Google Scholar] [CrossRef]
- Jathar, S.; Kumar, V.; Srivastava, J.; Tripathi, V. Technological Developments in lncRNA Biology. Adv. Exp. Med. Biol. 2017, 1008, 283–323. [Google Scholar] [PubMed]
- Ferrè, F.; Colantoni, A.; Helmer-Citterich, M. Revealing protein-lncRNA interaction. Brief. Bioinform. 2016, 17, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, M.U.; Lam, M.T.Y.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Blanc, K.; Pittenger, M. Mesenchymal stem cells: Progress toward promise. Cytotherapy 2005, 7, 36–45. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [Green Version]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Bai, J.; Zhang, X.; Lv, Y.; Gong, Y.; Liu, L.; Zhao, H.; Yu, F.; Ping, Y.; Zhang, G.; et al. A comprehensive overview of lncRNA annotation resources. Brief. Bioinform. 2017, 18, 236–249. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-Z.; Chen, M.; Chen, D.; Gao, X.-C.; Zhu, S.; Huang, H.; Hu, M.; Zhu, H.; Yan, G.-R. A Peptide Encoded by a Putative lncRNA HOXB-AS3 Suppresses Colon Cancer Growth. Mol. Cell 2017, 68, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Chen, X.; Wang, Y.; Fan, B.; Guo, K.; Yang, C.; Yu, S.; Pang, Y.; Zhang, S. Chitooligosaccharide inhibits RANKL-induced osteoclastogenesis and ligation-induced periodontitis by suppressing MAPK/ c-fos/NFATC1 signaling. J. Cell. Physiol. 2020, 235, 3022–3032. [Google Scholar] [CrossRef]
- López-Vicente, L.; Pons, B.; Coch, L.; Teixidó, C.; Hernández-Losa, J.; Armengol, G.; Ramon YCajal, S. RSK4 inhibition results in bypass of stress-induced and oncogene-induced senescence. Carcinogenesis 2011, 32, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.; Boland, B.B.; Jensen, P.; Alarcon, C.; Nawrocki, A.; Grimsby, J.S.; Rhodes, C.J.; Larsen, M.R. Characterization of Signaling Pathways Associated with Pancreatic β-cell Adaptive Flexibility in Compensation of Obesity-linked Diabetes in Mice. Mol. Cell. Proteom. MCP 2020, 19, 971–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva Xavier, G.; Rutter, G.A. Metabolic and Functional Heterogeneity in Pancreatic β Cells. J. Mol. Biol. 2020, 432, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-X.; Qiu, W.-L.; Yang, L.; Zhang, Y.; He, M.-Y.; Li, L.-C.; Xu, C.-R. Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J. 2019, 38, e100164. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, E.; Kulkarni, R.N.; Scott, D.K.; Mauvais-Jarvis, F.; Stewart, A.F.; Garcia-Ocaña, A. Human β-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014, 63, 819–831. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Shibasaki, T.; Park, J.-H.; Hidaka, S.; Takahashi, T.; Ono, A.; Song, D.-K.; Seino, S. Role of Epac2A/Rap1 signaling in interplay between incretin and sulfonylurea in insulin secretion. Diabetes 2015, 64, 1262–1272. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zheng, S.; Zhang, X.; Dai, P.; Gao, Y.; Nan, L.; Zhang, Y. Transplantation of Amniotic Scaffold-Seeded Mesenchymal Stem Cells and/or Endothelial Progenitor Cells From Bone Marrow to Efficiently Repair 3-cm Circumferential Urethral Defect in Model Dogs. Tissue Eng. Part A 2018, 24, 47–56. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, W.; Hua, J.; Lei, A.; Lv, C.; Wang, H.; Yang, C.; Gao, Z.; Dou, Z. Pancreatic islet-like clusters from bone marrow mesenchymal stem cells of human first-trimester abortus can cure streptozocin-induced mouse diabetes. Rejuvenation Res. 2010, 13, 695–706. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef]
- Lin, M.F.; Jungreis, I.; Kellis, M. PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Dai, P.; Gao, D.; Zhang, X.; Ruan, C.; Li, J.; Chen, Y.; Zhang, L.; Zhang, Y. Genome-Wide Analysis Reveals Changes in Long Noncoding RNAs in the Differentiation of Canine BMSCs into Insulin-Producing Cells. Int. J. Mol. Sci. 2020, 21, 5549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155549
Wang J, Dai P, Gao D, Zhang X, Ruan C, Li J, Chen Y, Zhang L, Zhang Y. Genome-Wide Analysis Reveals Changes in Long Noncoding RNAs in the Differentiation of Canine BMSCs into Insulin-Producing Cells. International Journal of Molecular Sciences. 2020; 21(15):5549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155549
Chicago/Turabian StyleWang, Jinglu, Pengxiu Dai, Dengke Gao, Xia Zhang, Chenmei Ruan, Jiakai Li, Yijing Chen, Luwen Zhang, and Yihua Zhang. 2020. "Genome-Wide Analysis Reveals Changes in Long Noncoding RNAs in the Differentiation of Canine BMSCs into Insulin-Producing Cells" International Journal of Molecular Sciences 21, no. 15: 5549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155549