Osteopontin: The Molecular Bridge between Fat and Cardiac–Renal Disorders


Abstract
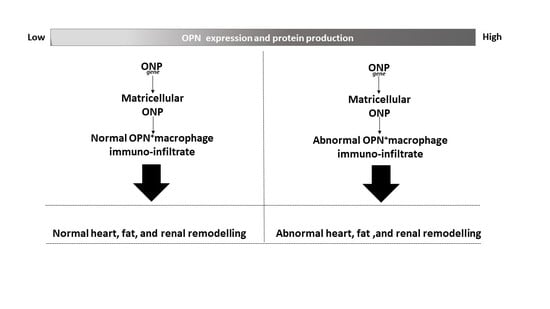
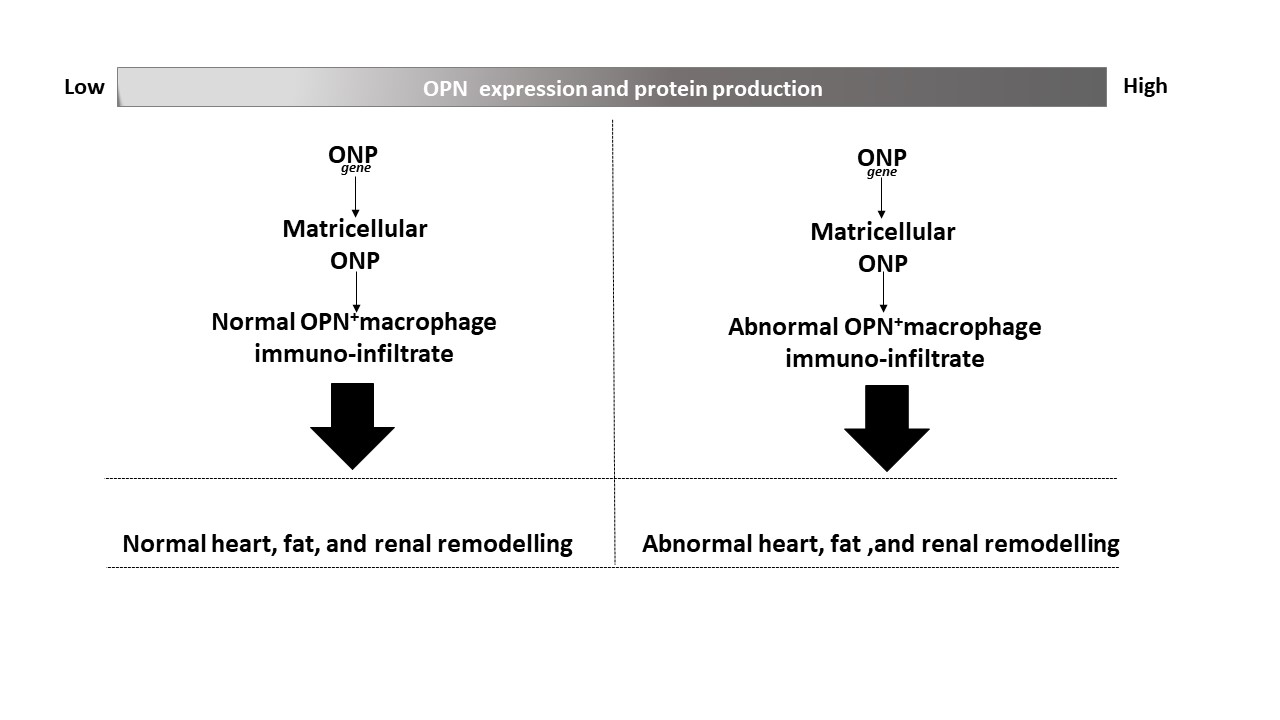
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Osteopontin (OPN) Structure and Physiological Functions
3. OPN as a Matricellular Protein in Fat, Cardiac and Renal Tissues
4. OPN in Obesity
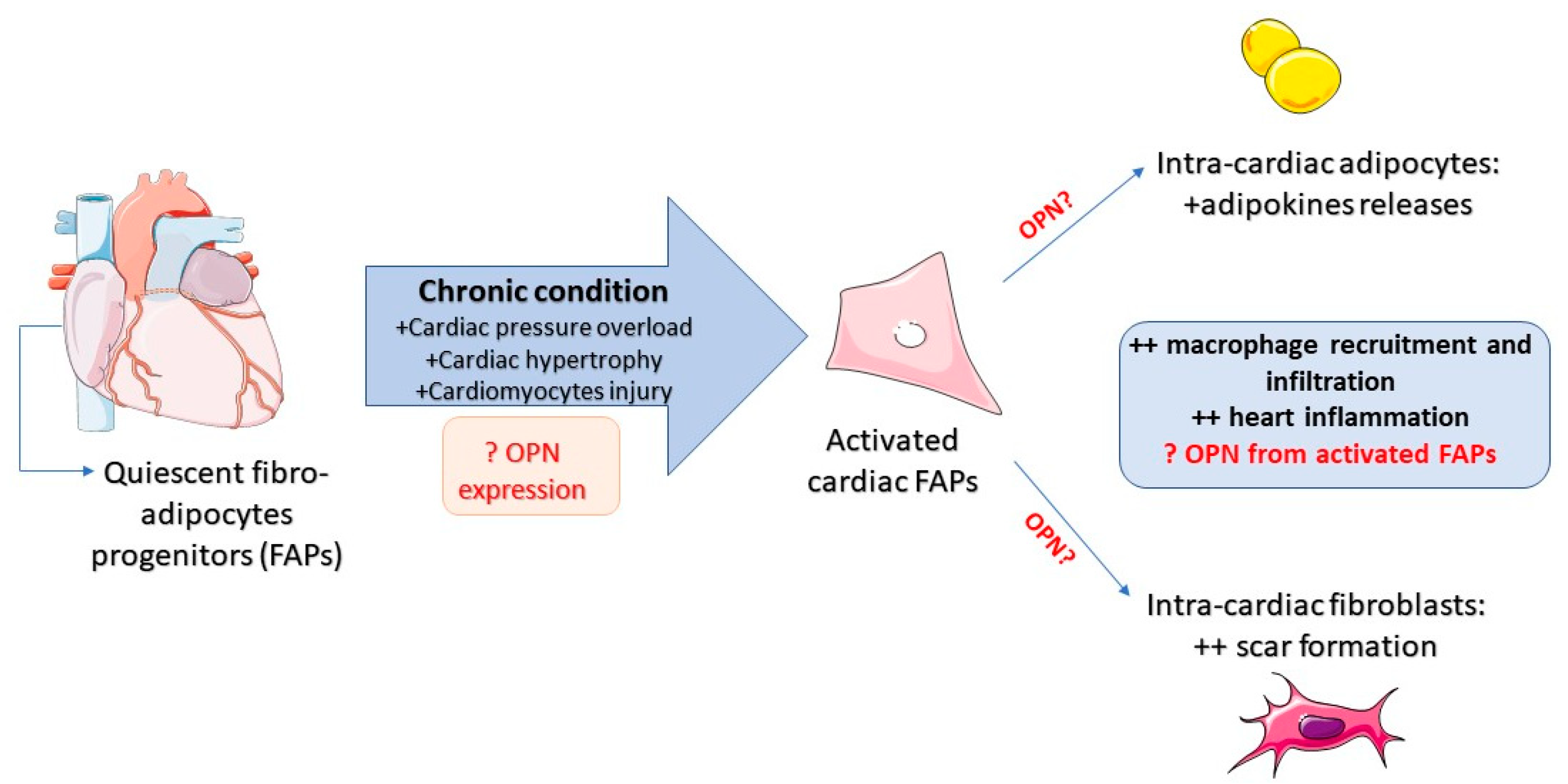
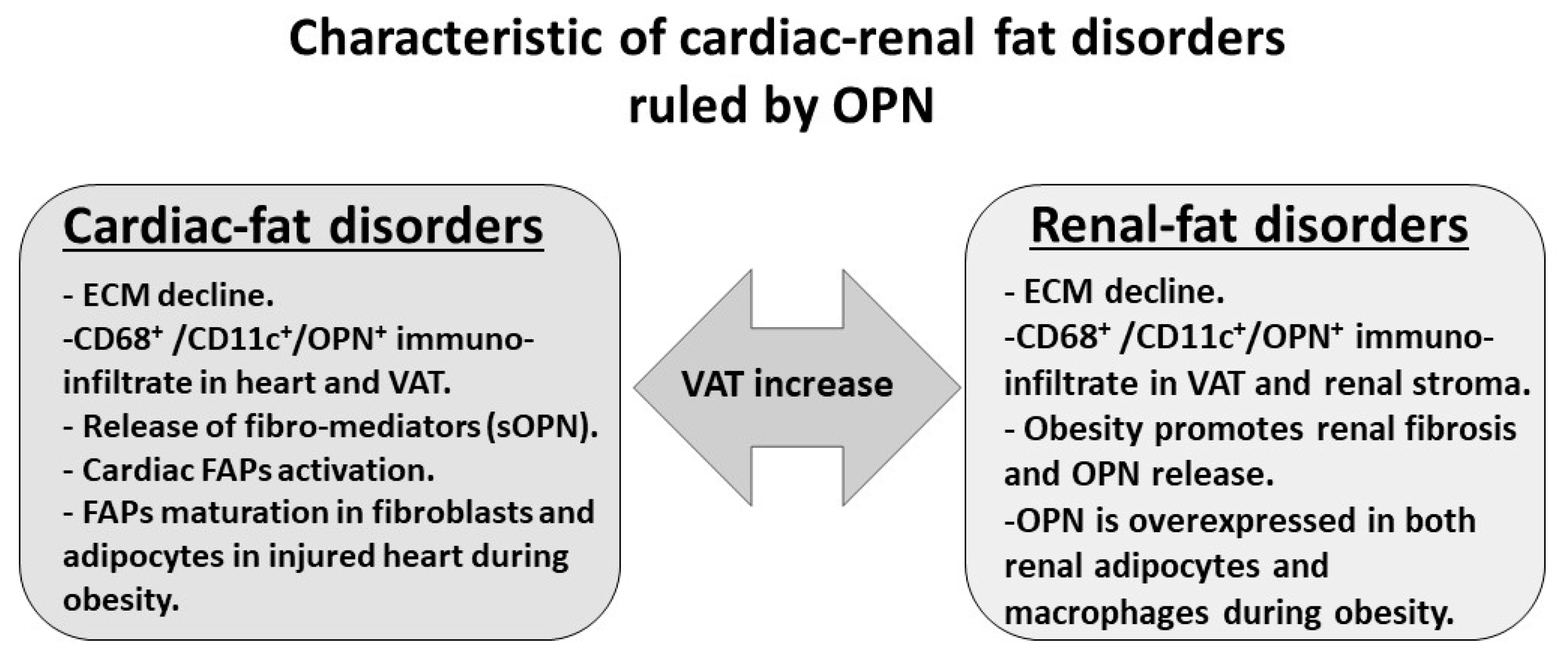
5. Cardiac–Fat Diseases Ruled by OPN
6. Renal–Fat Disease Ruled by OPN
7. Obesity Increase Renal OPN
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OPN | Osteopontin |
| ECM | Extracellular matrix |
| CVDs | Cardiovascular diseases |
| VAT | Visceral adipose tissue |
| HF | Heart failure |
| SPP1 | Secreted phosphoprotein 1 |
| Eta-1 | Early T lymphocyte activation 1 |
| SIBLING | Small integrin-binding ligand N-linked glycoprotein |
| NFkB | Nuclear factor kappa B |
| IL-10 | Interleukin-10 |
| IFNγ | Interferon-γ |
| CKD | Chronic kidney disease |
| TGF-β | Transforming growth factor-β |
| ATMs | Adipose tissue macrophages |
| CLs | Crown-like structures |
| TNFα | Tumor necrosis factor-α |
| IL-6 | Interleukin-6 |
| MCP-1 | Monocyte chemoattractant protein 1 |
| sOPN | Soluble OPN |
| FAPs | Fibro-adipocyte progenitors |
| BMI | Body mass index |
| AER | Albumin excretion rate |
| IL-6 | Interleukin-6 |
References
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Omar, B.; Banke, E.; Guirguis, E.; Akesson, L.; Manganiello, V.; Lyssenko, V.; Groop, L.; Gomez, M.F.; Degerman, E. Regulation of the pro-inflammatory cytokine osteopontin by GIP in adipocytes—A role for the transcription factor NFAT and phosphodiesterase 3B. Biochem. Biophys. Res. Commun. 2012, 425, 812–817. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 2012, 92, 635–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzali, M.; Kipari, T.; Ophascharoensuk, V.; Wesson, J.A.; Johnson, R.; Hughes, J. Osteopontin—A molecule for all seasons. QJM 2002, 95, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashkar, S.; Weber, G.F.; Panoutsakopoulou, V.; Sanchirico, M.E.; Jansson, M.; Zawaideh, S.; Rittling, S.R.; Denhardt, D.T.; Glimcher, M.J.; Cantor, H. Eta-1 (osteopontin): An early component of type-1 (cell-mediated) immunity. Science 2000, 287, 860–864. [Google Scholar] [CrossRef]
- Kazanecki, C.C.; Uzwiak, D.J.; Denhardt, D.T. Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J. Cell. Biochem. 2007, 102, 912–924. [Google Scholar] [CrossRef]
- Gimba, E.R.; Tilli, T.M. Human osteopontin splicing isoforms: Known roles, potential clinical applications and activated signaling pathways. Cancer Lett. 2013, 331, 11–17. [Google Scholar] [CrossRef]
- Viloria, K.; Hill, N.J. Embracing the complexity of matricellular proteins: The functional and clinical significance of splice variation. Biomol. Concepts 2016, 7, 117–132. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14. [Google Scholar] [CrossRef]
- Morris, A.H.; Kyriakides, T.R. Matricellular proteins and biomaterials. Matrix Biol. 2014, 37, 183–191. [Google Scholar] [CrossRef]
- Denhardt, D.T.; Noda, M.; O’Regan, A.W.; Pavlin, D.; Berman, J.S. Osteopontin as a means to cope with environmental insults: Regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Investig. 2001, 107, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, K.W.; Lee, S.J.; Ye, B.H.; Kim, Y.W.; Bae, S.S.; Kim, C.D. Mechanical stretch enhances the expression and activity of osteopontin and MMP-2 via the Akt1/AP-1 pathways in VSMC. J. Mol. Cell. Cardiol. 2015, 85, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.M.; Desimine, V.L.; Wertz, S.L.; Perez, A.; Parker, B.M.; Maning, J.; McCrink, K.A.; Shehadeh, L.A.; Lymperopoulos, A. Deletion of Osteopontin Enhances beta(2)-Adrenergic Receptor-Dependent Anti-Fibrotic Signaling in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 1396. [Google Scholar] [CrossRef] [Green Version]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug. Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Bostan Gayret, O.; Tasdemir, M.; Erol, M.; Tekin Nacaroglu, H.; Zengi, O.; Yigit, O. Are there any new reliable markers to detect renal injury in obese children? Ren. Fail. 2018, 40, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Fitter, S.; Zannettino, A.C.W. Osteopontin in the pathophysiology of obesity: Is Opn a fat cell foe? Obes. Res. Clin. Pract. 2018, 12, 249–250. [Google Scholar] [CrossRef]
- Schinzari, F.; Tesauro, M.; Bertoli, A.; Valentini, A.; Veneziani, A.; Campia, U.; Cardillo, C. Calcification biomarkers and vascular dysfunction in obesity and type 2 diabetes: Influence of oral hypoglycemic agents. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E658–E666. [Google Scholar] [CrossRef]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Luczak, M.; Suszynska-Zajczyk, J.; Marczak, L.; Formanowicz, D.; Pawliczak, E.; Wanic-Kossowska, M.; Stobiecki, M. Label-Free Quantitative Proteomics Reveals Differences in Molecular Mechanism of Atherosclerosis Related and Non-Related to Chronic Kidney Disease. Int. J. Mol. Sci. 2016, 17, 631. [Google Scholar] [CrossRef]
- Brankovic, M.; Martijn Akkerhuis, K.; Mouthaan, H.; Constantinescu, A.; Caliskan, K.; van Ramshorst, J.; Germans, T.; Umans, V.; Kardys, I. Utility of temporal profiles of new cardio-renal and pulmonary candidate biomarkers in chronic heart failure. Int. J. Cardiol. 2019, 276, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y. The metabolic syndrome and adipocytokines. FEBS Lett. 2006, 580, 2917–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaki, D.; Czibik, G.; Pini, M.; Ternacle, J.; Suffee, N.; Mercedes, R.; Marcelin, G.; Surenaud, M.; Marcos, E.; Gual, P.; et al. Visceral Adipose Tissue Drives Cardiac Aging Through Modulation of Fibroblast Senescence by Osteopontin Production. Circulation 2018, 138, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Ananthula, S.; Milhorn, D.M.; Krishnaswamy, G.; Singh, K. Osteopontin: A novel inflammatory mediator of cardiovascular disease. Front. Biosci. 2007, 12, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Dalal, S.; Singh, K. Osteopontin: At the cross-roads of myocyte survival and myocardial function. Life Sci. 2014, 118, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamate, T.; Kohri, K.; Umekawa, T.; Konya, E.; Ishikawa, Y.; Iguchi, M.; Kurita, T. Interaction between osteopontin on madin darby canine kidney cell membrane and calcium oxalate crystal. Urol. Int. 1999, 62, 81–86. [Google Scholar] [CrossRef]
- Kaleta, B. The role of osteopontin in kidney diseases. Inflamm. Res. 2019, 68, 93–102. [Google Scholar] [CrossRef]
- Clemente, N.; Raineri, D.; Cappellano, G.; Boggio, E.; Favero, F.; Soluri, M.F.; Dianzani, C.; Comi, C.; Dianzani, U.; Chiocchetti, A. Osteopontin bridging innate and adaptive immunity in autoimmune diseases. J. Immunol. Res. 2016, 2016, 7675437. [Google Scholar] [CrossRef]
- Fisher, L.W.; Torchia, D.A.; Fohr, B.; Young, M.F.; Fedarko, N.S. Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochem. Biophys. Res. Commun. 2001, 280, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, F.W.; Zeyda, M.; Todoric, J.; Huber, J.; Geyeregger, R.; Weichhart, T.; Aszmann, O.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; et al. Osteopontin expression in human and murine obesity: Extensive local up-regulation in adipose tissue but minimal systemic alterations. Endocrinology 2008, 149, 1350–1357. [Google Scholar] [CrossRef] [Green Version]
- Herum, K.M.; Romaine, A.; Wang, A.; Melleby, A.O.; Strand, M.E.; Pacheco, J.; Braathen, B.; Duner, P.; Tonnessen, T.; Lunde, I.G.; et al. Syndecan-4 protects the heart from the profibrotic effects of thrombin-cleaved osteopontin. J. Am. Heart. Assoc. 2020, 9, e013518. [Google Scholar] [CrossRef]
- Gomez-Ambrosi, J.; Catalan, V.; Ramirez, B.; Rodriguez, A.; Colina, I.; Silva, C.; Rotellar, F.; Mugueta, C.; Gil, M.J.; Cienfuegos, J.A.; et al. Plasma osteopontin levels and expression in adipose tissue are increased in obesity. J. Clin. Endocrinol. Metab. 2007, 92, 3719–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraju, C.K.; Robinson, E.L.; Abdesselem, M.; Trenson, S.; Dries, E.; Gilbert, G.; Janssens, S.; Van Cleemput, J.; Rega, F.; Meyns, B.; et al. Myofibroblast phenotype and reversibility of fibrosis in patients with end-stage heart failure. J. Am. Coll. Cardiol. 2019, 73, 2267–2282. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.; Jones, E.; Chipitsyna, G.; Al-Zoubi, M.; Kang, C.; Saxena, S.; Gandhi, A.V.; Sendiky, J.; Yeo, C.J.; Arafat, H.A. Osteopontin (OPN) isoforms, diabetes, obesity, and cancer; what is one got to do with the other? A new role for OPN. J. Gastrointest. Surg. 2015, 19, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Podzimkova, J.; Palecek, T.; Kuchynka, P.; Marek, J.; Danek, B.A.; Jachymova, M.; Kalousova, M.; Zima, T.; Linhart, A. Plasma osteopontin levels in patients with dilated and hypertrophic cardiomyopathy. Herz 2019, 44, 347–353. [Google Scholar] [CrossRef]
- Pan, W.; Liang, J.; Tang, H.; Fang, X.; Wang, F.; Ding, Y.; Huang, H.; Zhang, H. Differentially expressed microRNA profiles in exosomes from vascular smooth muscle cells associated with coronary artery calcification. Int. J. Biochem. Cell Biol. 2020, 118, 105645. [Google Scholar] [CrossRef]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Chun, T.H.; Kang, L. Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochem. Pharmacol. 2016, 119, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Lancha, A.; Rodriguez, A.; Catalan, V.; Becerril, S.; Sainz, N.; Ramirez, B.; Burrell, M.A.; Salvador, J.; Fruhbeck, G.; Gomez-Ambrosi, J. Osteopontin deletion prevents the development of obesity and hepatic steatosis via impaired adipose tissue matrix remodeling and reduced inflammation and fibrosis in adipose tissue and liver in mice. PLoS ONE 2014, 9, e98398. [Google Scholar] [CrossRef]
- Schuch, K.; Wanko, B.; Ambroz, K.; Castelo-Rosa, A.; Moreno-Viedma, V.; Grun, N.G.; Leitner, L.; Staffler, G.; Zeyda, M.; Stulnig, T.M. Osteopontin affects macrophage polarization promoting endocytic but not inflammatory properties. Obesity 2016, 24, 1489–1498. [Google Scholar] [CrossRef]
- Leitner, L.; Schuch, K.; Jurets, A.; Itariu, B.K.; Keck, M.; Grablowitz, V.; Aszmann, O.C.; Prager, G.; Staffler, G.; Zeyda, M.; et al. Immunological blockade of adipocyte inflammation caused by increased matrix metalloproteinase-cleaved osteopontin in obesity. Obesity 2015, 23, 779–785. [Google Scholar] [CrossRef]
- Chen, C.; Li, R.; Ross, R.S.; Manso, A.M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell. Cardiol. 2016, 93, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey, M.L.; Iyer, R.P.; Jung, M.; DeLeon-Pennell, K.Y.; Ma, Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J. Mol. Cell. Cardiol. 2016, 91, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Guo, J.; Hua, Y.; Huang, K.; Magaye, R.; Cornell, J.; Kelly, D.J.; Reid, C.; Liew, D.; Zhou, Y.; et al. Cardiac fibrosis in the ageing heart: Contributors and mechanisms. Clin. Exp. Pharmacol. Physiol. 2017, 44, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, R.P.; Jung, M.; Lindsey, M.L. Using the laws of thermodynamics to understand how matrix metalloproteinases coordinate the myocardial response to injury. Met. Med. 2015, 2, 75–82. [Google Scholar]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [Green Version]
- Sorop, O.; Heinonen, I.; van Kranenburg, M.; van de Wouw, J.; de Beer, V.J.; Nguyen, I.T.N.; Octavia, Y.; van Duin, R.W.B.; Stam, K.; van Geuns, R.J.; et al. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc. Res. 2018, 114, 954–964. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix. Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Passmore, M.; Nataatmadja, M.; Fung, Y.L.; Pearse, B.; Gabriel, S.; Tesar, P.; Fraser, J.F. Osteopontin alters endothelial and valvular interstitial cell behaviour in calcific aortic valve stenosis through HMGB1 regulation. Eur. J. Cardiothorac. Surg. 2015, 48, e20–e29. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Qiao, W.; Zhang, W.; Li, F.; Shi, J.; Dong, N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J. Thorac. Cardiovasc. Surg. 2017, 153, 1318–1327. [Google Scholar] [CrossRef] [Green Version]
- Trostel, J.; Truong, L.D.; Roncal-Jimenez, C.; Miyazaki, M.; Miyazaki-Anzai, S.; Kuwabara, M.; McMahan, R.; Andres-Hernando, A.; Sato, Y.; Jensen, T.; et al. Different effects of global osteopontin and macrophage osteopontin in glomerular injury. Am. J. Physiol. Renal. Physiol. 2018, 315, F759–F768. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Toba, H.; Lindsey, M.L. Extracellular matrix roles in cardiorenal fibrosis: Potential therapeutic targets for CVD and CKD in the elderly. Pharm. Ther. 2019, 193, 99–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reif, G.; Wallace, D.P. Extracellular matrix, integrins, and focal adhesion signaling in polycystic kidney disease. Cell. Signal. 2020, 72, 109646. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Ngov, C.; Henley, N.; Boufaied, N.; Gerarduzzi, C. Characterization of matricellular protein expression signatures in mechanistically diverse mouse models of kidney injury. Sci. Rep. 2019, 9, 16736. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological mechanisms of renal fibrosis: A review of animal models and therapeutic strategies. Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeyda, M.; Gollinger, K.; Todoric, J.; Kiefer, F.W.; Keck, M.; Aszmann, O.; Prager, G.; Zlabinger, G.J.; Petzelbauer, P.; Stulnig, T.M. Osteopontin is an activator of human adipose tissue macrophages and directly affects adipocyte function. Endocrinology 2011, 152, 2219–2227. [Google Scholar] [CrossRef] [Green Version]
- Nomiyama, T.; Perez-Tilve, D.; Ogawa, D.; Gizard, F.; Zhao, Y.; Heywood, E.B.; Jones, K.L.; Kawamori, R.; Cassis, L.A.; Tschop, M.H.; et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J. Clin. Investig. 2007, 117, 2877–2888. [Google Scholar] [CrossRef]
- West, M. Dead adipocytes and metabolic dysfunction: Recent progress. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 178–182. [Google Scholar] [CrossRef]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem. Cells. Int. 2019, 2019, 4761427. [Google Scholar] [CrossRef]
- Zuo, L.; Tozawa, K.; Okada, A.; Yasui, T.; Taguchi, K.; Ito, Y.; Hirose, Y.; Fujii, Y.; Niimi, K.; Hamamoto, S.; et al. A paracrine mechanism involving renal tubular cells, adipocytes and macrophages promotes kidney stone formation in a simulated metabolic syndrome environment. J. Urol. 2014, 191, 1906–1912. [Google Scholar] [CrossRef]
- Aouadi, M.; Tencerova, M.; Vangala, P.; Yawe, J.C.; Nicoloro, S.M.; Amano, S.U.; Cohen, J.L.; Czech, M.P. Gene silencing in adipose tissue macrophages regulates whole-body metabolism in obese mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8278–8283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieur, X.; Mok, C.Y.; Velagapudi, V.R.; Nunez, V.; Fuentes, L.; Montaner, D.; Ishikawa, K.; Camacho, A.; Barbarroja, N.; O’Rahilly, S.; et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes 2011, 60, 797–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna-Luna, M.; Medina-Urrutia, A.; Vargas-Alarcon, G.; Coss-Rovirosa, F.; Vargas-Barron, J.; Perez-Mendez, O. Adipose tissue in metabolic syndrome: Onset and progression of atherosclerosis. Arch. Med. Res. 2015, 46, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Luna-Luna, M.; Cruz-Robles, D.; Avila-Vanzzini, N.; Herrera-Alarcon, V.; Martinez-Reding, J.; Criales-Vera, S.; Sandoval-Zarate, J.; Vargas-Barron, J.; Martinez-Sanchez, C.; Tovar-Palacio, A.R.; et al. Differential expression of osteopontin, and osteoprotegerin mRNA in epicardial adipose tissue between patients with severe coronary artery disease and aortic valvular stenosis: Association with HDL subclasses. Lipids Health Dis. 2017, 16, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.; Miles, P.D.; Ofrecio, J.M.; Neels, J.G.; Yu, J.G.; Resnik, J.L.; Wilkes, J.; Talukdar, S.; Thapar, D.; Johnson, K.; et al. Osteopontin is required for the early onset of high fat diet-induced insulin resistance in mice. PLoS ONE 2010, 5, e13959. [Google Scholar] [CrossRef] [Green Version]
- Kahles, F.; Findeisen, H.M. Does osteopontin induce adipose tissue inflammation by local macrophage proliferation? Mol. Metab. 2016, 5, 1147–1148. [Google Scholar] [CrossRef]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef]
- Mohamed, I.A.; Mraiche, F. Targeting osteopontin, the silent partner of Na+/H+ exchanger isoform 1 in cardiac remodeling. J. Cell. Physiol. 2015, 230, 2006–2018. [Google Scholar] [CrossRef]
- Landecho, M.F.; Tuero, C.; Valenti, V.; Bilbao, I.; de la Higuera, M.; Fruhbeck, G. Relevance of leptin and other adipokines in obesity-associated cardiovascular risk. Nutrients 2019, 11, 2664. [Google Scholar] [CrossRef] [Green Version]
- Rubis, P.; Wisniowska-Smialek, S.; Dziewiecka, E.; Rudnicka-Sosin, L.; Kozanecki, A.; Podolec, P. Prognostic value of fibrosis-related markers in dilated cardiomyopathy: A link between osteopontin and cardiovascular events. Adv. Med. Sci. 2018, 63, 160–166. [Google Scholar] [CrossRef]
- Yousefi, K.; Irion, C.I.; Takeuchi, L.M.; Ding, W.; Lambert, G.; Eisenberg, T.; Sukkar, S.; Granzier, H.L.; Methawasin, M.; Lee, D.I.; et al. Osteopontin promotes left ventricular diastolic dysfunction through a mitochondrial pathway. J. Am. Coll. Cardiol. 2019, 73, 2705–2718. [Google Scholar] [CrossRef] [PubMed]
- Coculescu, B.I.; Manole, G.; Dinca, G.V.; Coculescu, E.C.; Berteanu, C.; Stocheci, C.M. Osteopontin-a biomarker of disease, but also of stage stratification of the functional myocardial contractile deficit by chronic ischaemic heart disease. J. Enzyme. Inhib. Med. Chem. 2019, 34, 783–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matloch, Z.; Cinkajzlova, A.; Mraz, M.; Haluzik, M. The role of inflammation in epicardial adipose tissue in heart diseases. Curr. Pharm. Des. 2018, 24, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Lonn, E.; Lamy, A.; Singh, N.; Sharma, A.M. Epicardial fat thickness and coronary artery disease correlate independently of obesity. Int. J. Cardiol. 2011, 146, 452–454. [Google Scholar] [CrossRef]
- Iacobellis, G.; Bianco, A.C. Epicardial adipose tissue: Emerging physiological, pathophysiological and clinical features. Trends Endocrinol. Metab. 2011, 22, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Vianello, E.; Dozio, E.; Arnaboldi, F.; Marazzi, M.G.; Martinelli, C.; Lamont, J.; Tacchini, L.; Sigruner, A.; Schmitz, G.; Corsi Romanelli, M.M. Epicardial adipocyte hypertrophy: Association with M1-polarization and toll-like receptor pathways in coronary artery disease patients. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 246–253. [Google Scholar] [CrossRef]
- Villasante Fricke, A.C.; Iacobellis, G. Epicardial adipose tissue: Clinical biomarker of cardio-metabolic risk. Int. J. Mol. Sci. 2019, 20, 5989. [Google Scholar] [CrossRef] [Green Version]
- Tardelli, M.; Zeyda, K.; Moreno-Viedma, V.; Wanko, B.; Grun, N.G.; Staffler, G.; Zeyda, M.; Stulnig, T.M. Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol. Metab. 2016, 5, 1131–1137. [Google Scholar] [CrossRef]
- Pierzynova, A.; Sramek, J.; Cinkajzlova, A.; Kratochvilova, H.; Lindner, J.; Haluzik, M.; Kucera, T. The number and phenotype of myocardial and adipose tissue CD68+ cells is associated with cardiovascular and metabolic disease in heart surgery patients. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 946–955. [Google Scholar] [CrossRef]
- Madaro, L.; Passafaro, M.; Sala, D.; Etxaniz, U.; Lugarini, F.; Proietti, D.; Alfonsi, M.V.; Nicoletti, C.; Gatto, S.; De Bardi, M.; et al. Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell. Biol. 2018, 20, 917–927. [Google Scholar] [CrossRef]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Capote, J.; Kramerova, I.; Martinez, L.; Vetrone, S.; Barton, E.R.; Sweeney, H.L.; Miceli, M.C.; Spencer, M.J. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J. Cell. Biol. 2016, 213, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Foster, C.R.; Dalal, S.; Singh, K. Role of osteopontin in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell. Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef] [Green Version]
- Berezin, A.E.; Kremzer, A.A. Circulating osteopontin as a marker of early coronary vascular calcification in type two diabetes mellitus patients with known asymptomatic coronary artery disease. Atherosclerosis 2013, 229, 475–481. [Google Scholar] [CrossRef]
- Iglesias, P.; Diez, J.J. Adipose tissue in renal disease: Clinical significance and prognostic implications. Nephrol. Dial. Transplant. 2010, 25, 2066–2077. [Google Scholar] [CrossRef]
- Xie, Z.; Singh, M.; Singh, K. Osteopontin modulates myocardial hypertrophy in response to chronic pressure overload in mice. Hypertension 2004, 44, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Wajchenberg, B.L.; Giannella-Neto, D.; da Silva, M.E.; Santos, R.F. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Horm. Metab. Res. 2002, 34, 616–621. [Google Scholar] [CrossRef]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef]
- Mulyadi, L.; Stevens, C.; Munro, S.; Lingard, J.; Bermingham, M. Body fat distribution and total body fat as risk factors for microalbuminuria in the obese. Ann. Nutr. Metab. 2001, 45, 67–71. [Google Scholar] [CrossRef]
- Toita, R.; Kawano, T.; Murata, M.; Kang, J.H. Anti-obesity and anti-inflammatory effects of macrophage-targeted interleukin-10-conjugated liposomes in obese mice. Biomaterials 2016, 110, 81–88. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vianello, E.; Kalousová, M.; Dozio, E.; Tacchini, L.; Zima, T.; Corsi Romanelli, M.M. Osteopontin: The Molecular Bridge between Fat and Cardiac–Renal Disorders. Int. J. Mol. Sci. 2020, 21, 5568. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155568
Vianello E, Kalousová M, Dozio E, Tacchini L, Zima T, Corsi Romanelli MM. Osteopontin: The Molecular Bridge between Fat and Cardiac–Renal Disorders. International Journal of Molecular Sciences. 2020; 21(15):5568. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155568
Chicago/Turabian StyleVianello, Elena, Marta Kalousová, Elena Dozio, Lorenza Tacchini, Tomáš Zima, and Massimiliano Marco Corsi Romanelli. 2020. "Osteopontin: The Molecular Bridge between Fat and Cardiac–Renal Disorders" International Journal of Molecular Sciences 21, no. 15: 5568. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155568