Development of Androgen-Antagonistic Coumarinamides with a Unique Aromatic Folded Pharmacophore

 and
and
Abstract
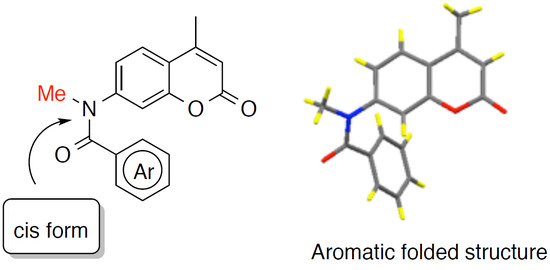
:
1. Introduction
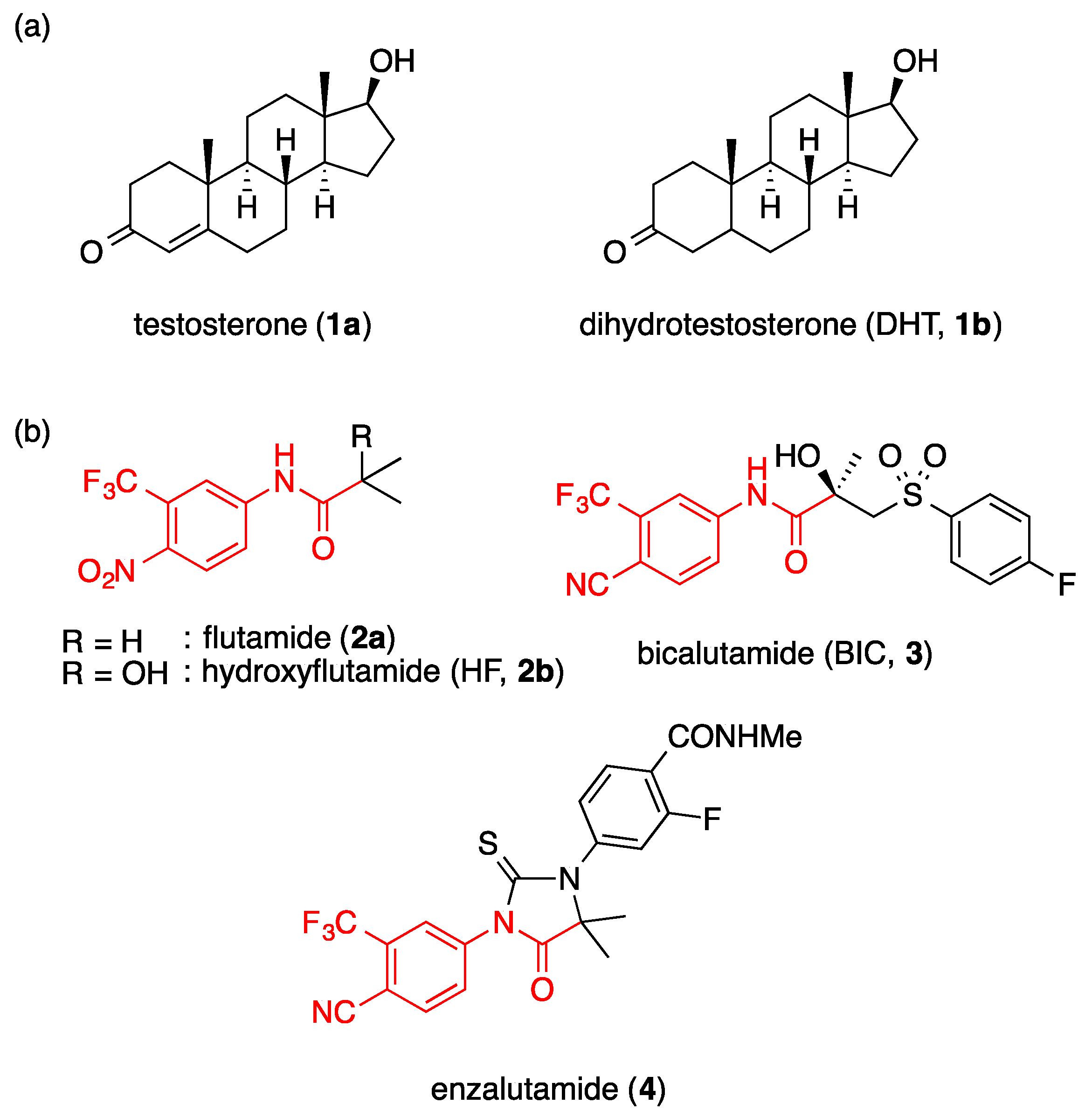
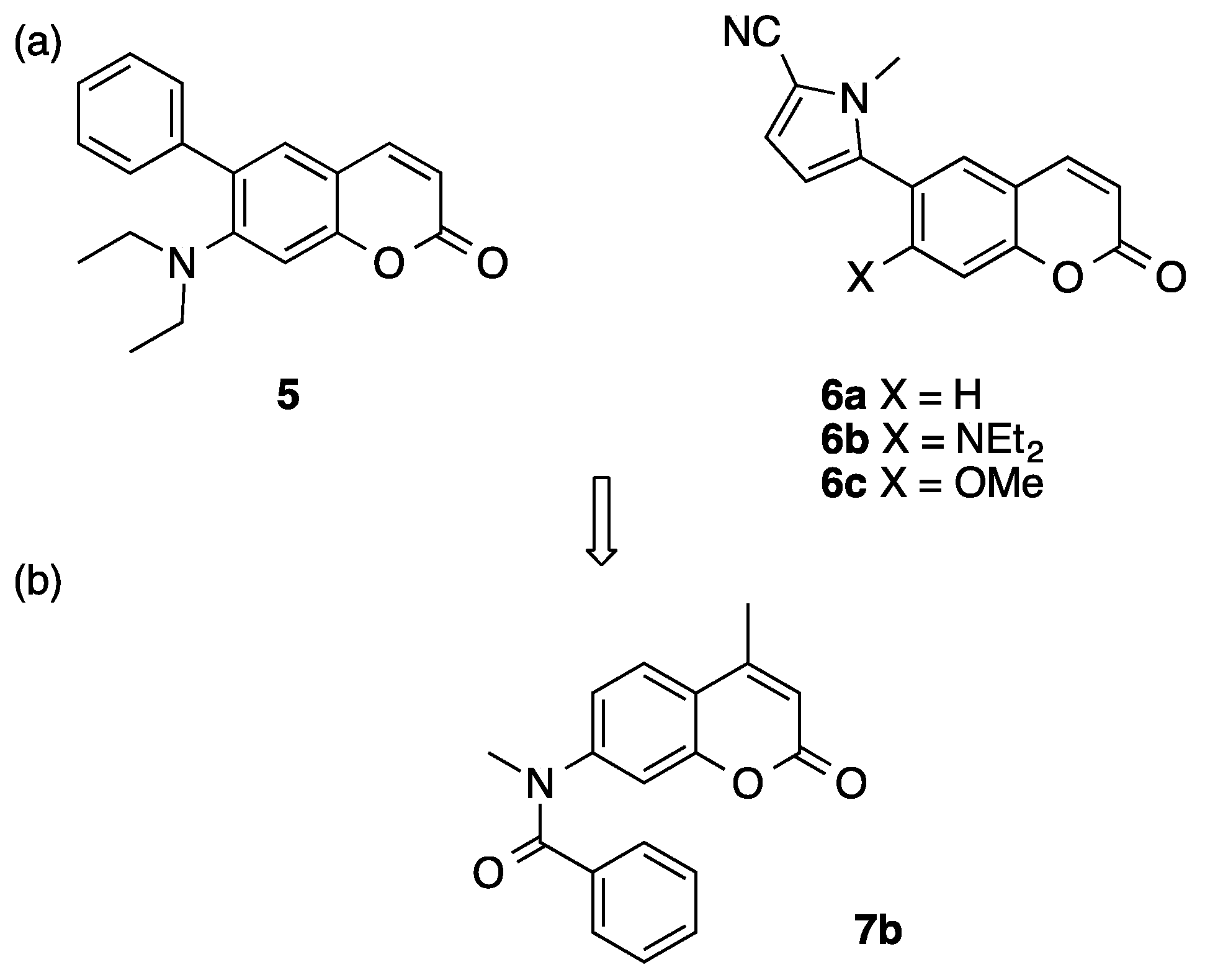
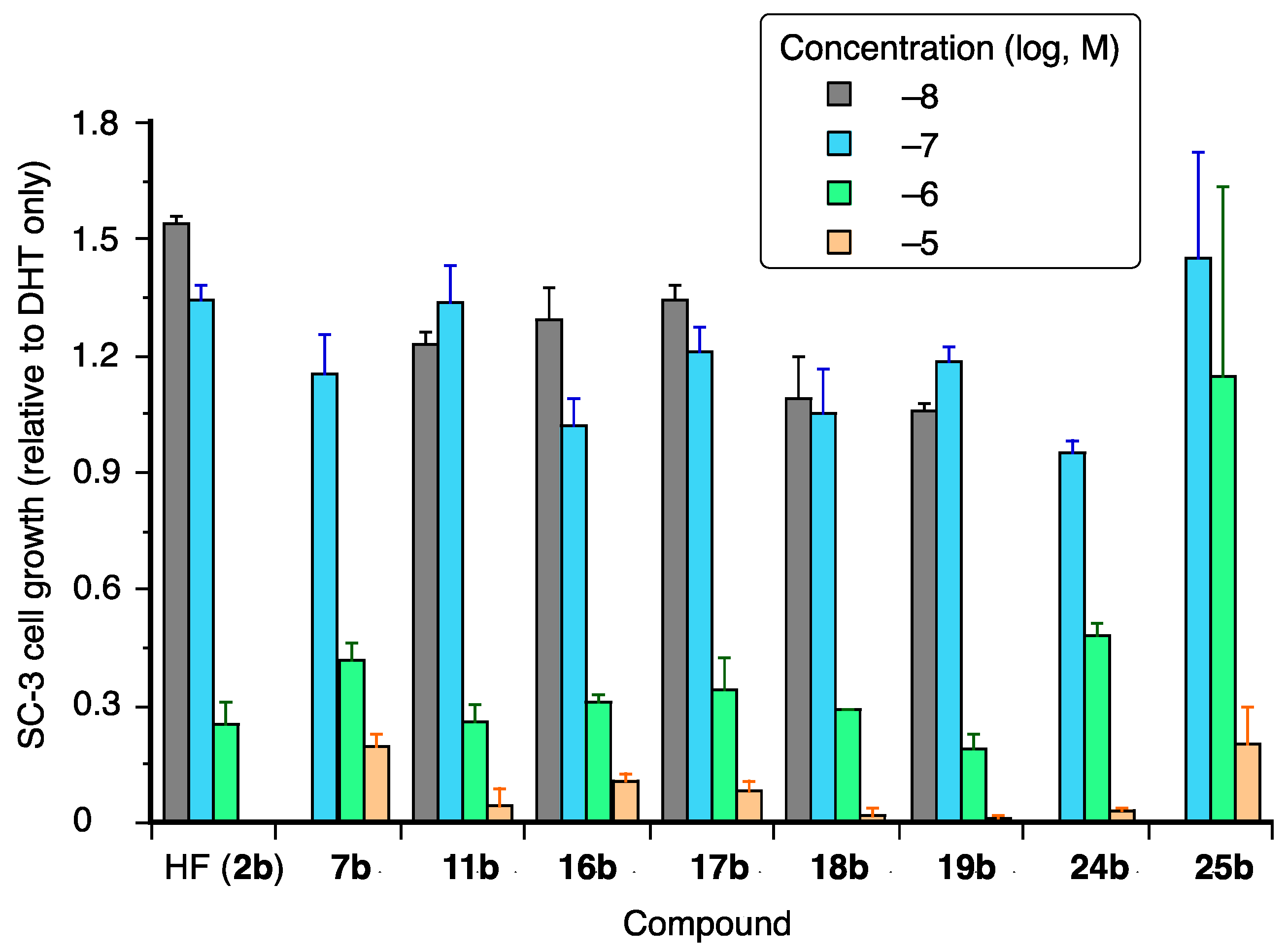
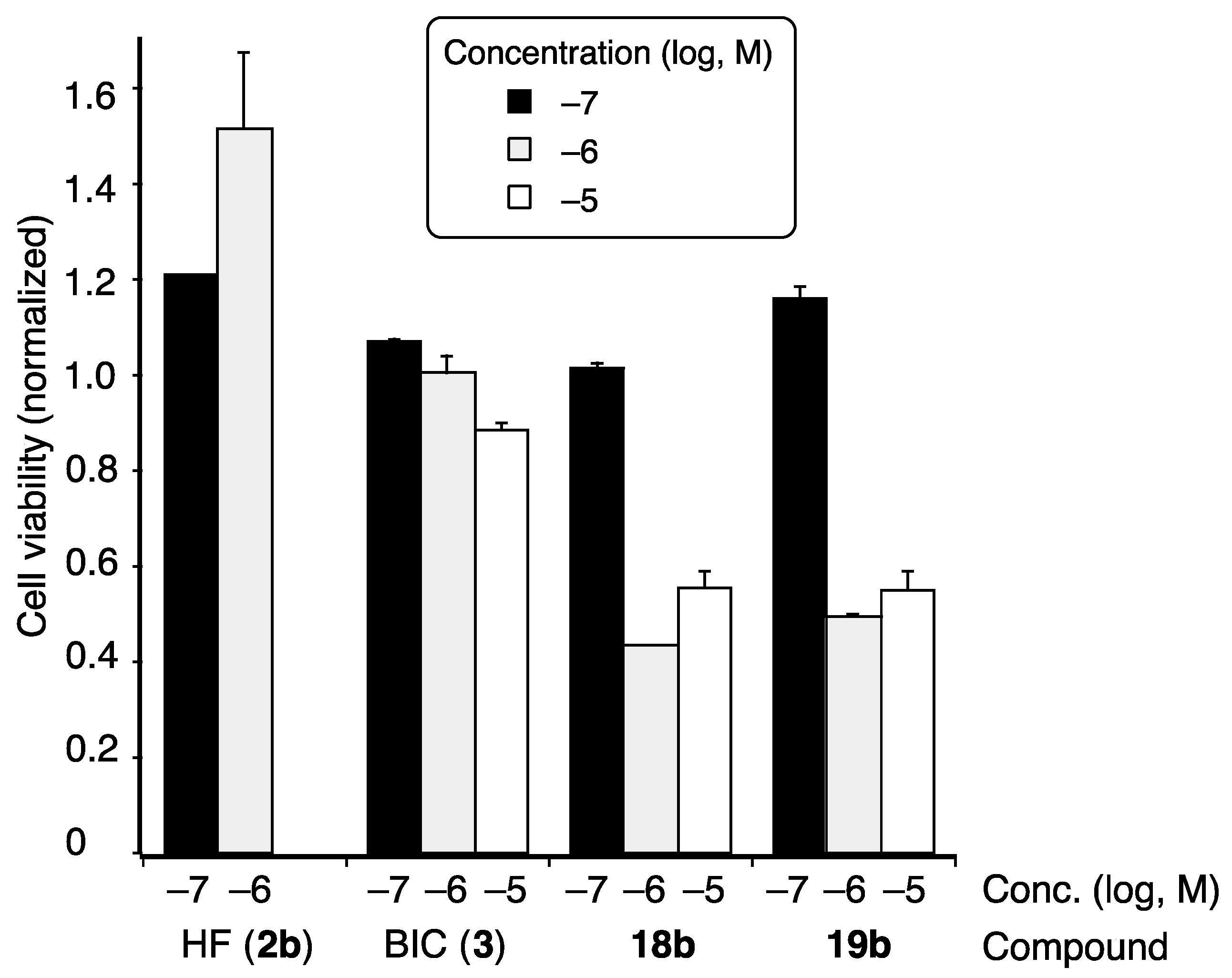
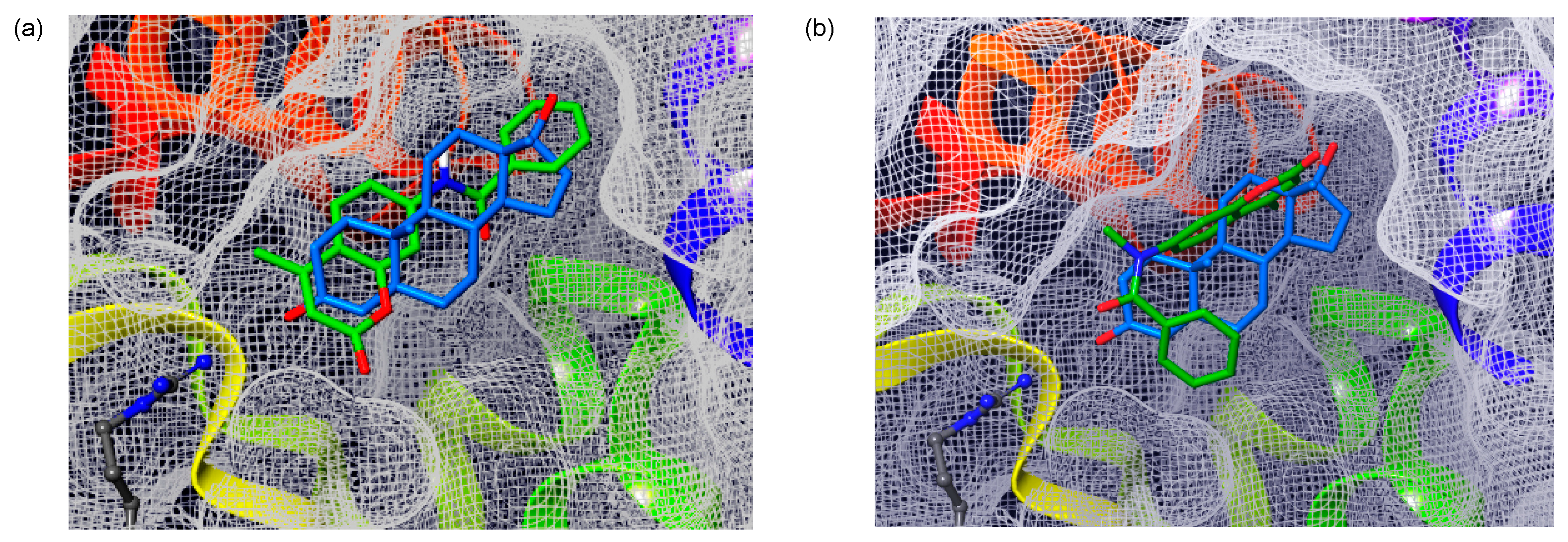
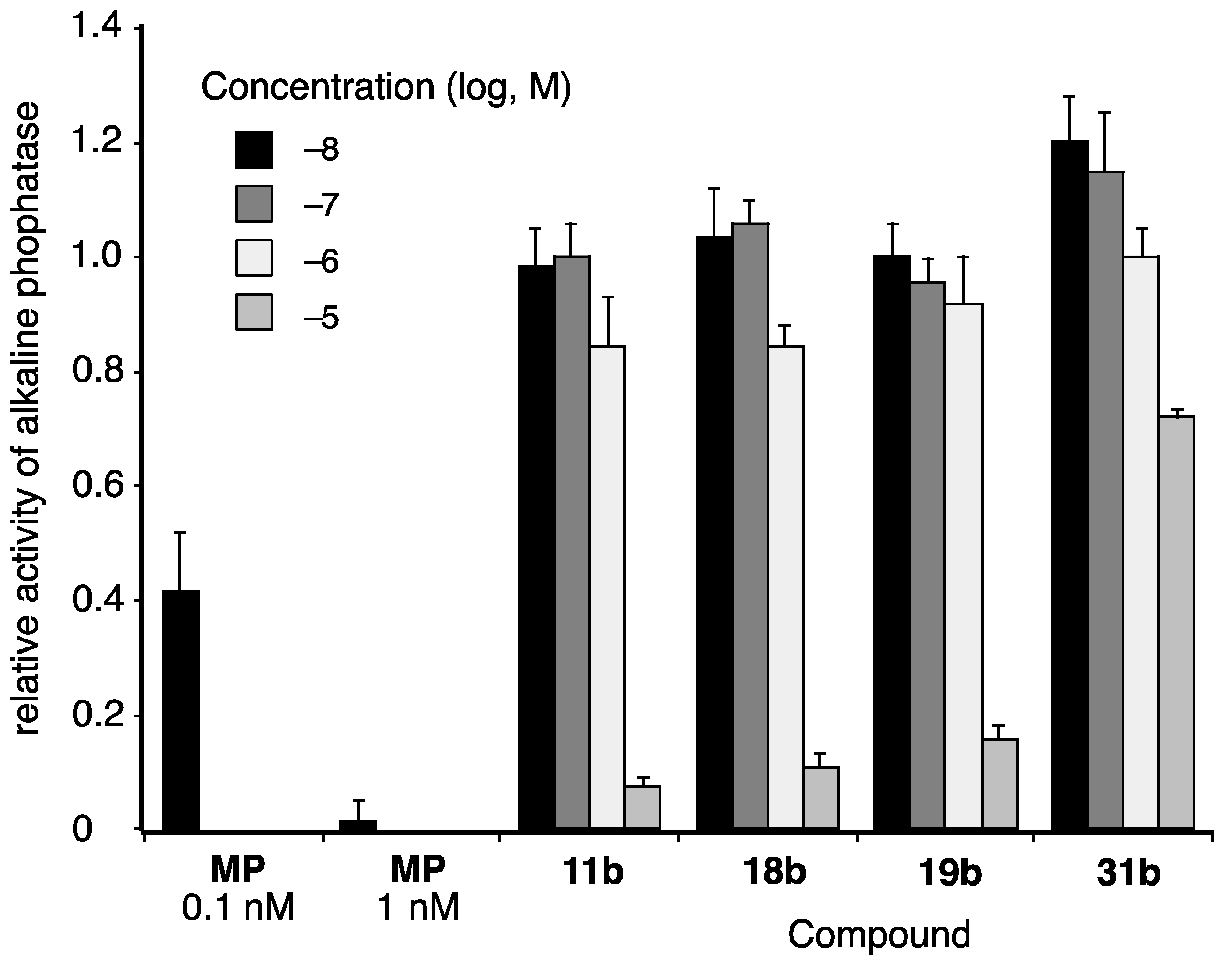
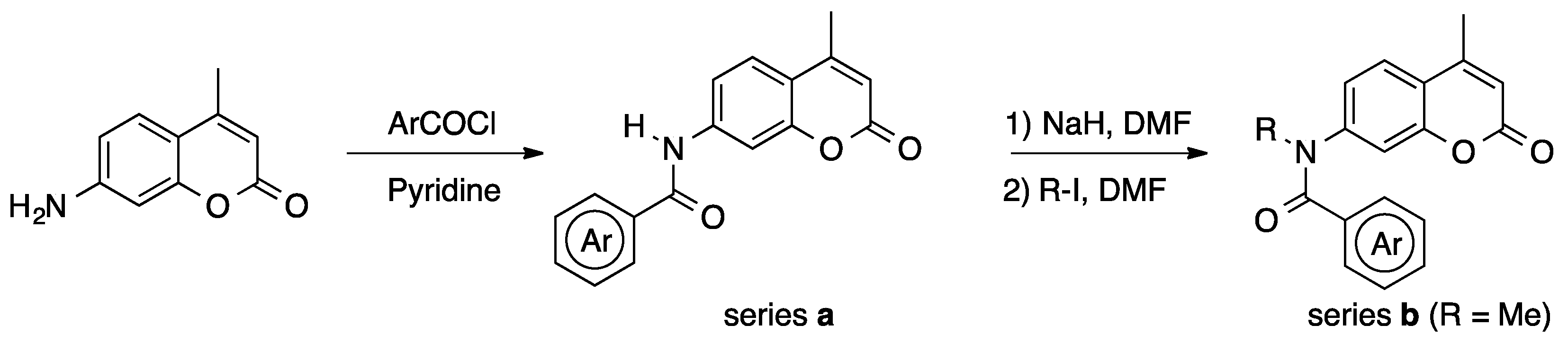
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis of 23a (General Procedure for Coumarinamides with Secondary Amide Bond)
3.1.3. Synthesis of 23b (General Procedure for N-Alkylated Coumarinamides)
3.2. SC-3 Growth Inhibition Assay
3.3. LNCaP Cell Proliferation Assay
3.4. Alkaline Phosphatase Assay Using T-47D Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AR | androgen receptor |
| DHT | dihydrotestosterone |
| BIC | bicalutamide |
| CRPC | castration-resistant prostate cancer |
| PR | progesterone receptor |
| NMR | ligand-binding domain |
| LBD | keratinocyte growth factor |
References
- Gao, W.; Bohl, C.E.; Dalton, J.T. Chemistry and structural biology of androgen receptor. Chem. Rev 2005, 105, 3352–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, N.Z.; Wardell, S.E.; Burnstein, K.L.; DeFranco, D.; Fuller, P.J.; Giguère, V.; Hochberg, R.B.; McKay, L.; Renoir, J.-M.; Weigel, N.L.; et al. International Union of Pharmacology. LXV. The Pharmacology and Classification of the Nuclear Receptor Superfamily: Glucocorticoid, Mineralocorticoid, Progesterone, and Androgen Receptors. Pharmacol. Rev. 2006, 58, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Morley, J.E.; Korenman, S.G. Biological Actions of Androgens. Endocr. Rev. 1987, 8, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, C.J.; Bremner, W.J. Androgens in Men—Uses and Abuses. N. Engl. J. Med. 1996, 334, 707–715. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sawyers, C.L.; I Scher, H. Targeting the androgen receptor pathway in prostate cancer. Curr. Opin. Pharmacol. 2008, 8, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Taplin, M.-E. Androgen receptor: Role and novel therapeutic prospects in prostate cancer. Expert Rev. Anticancer. Ther. 2008, 8, 1495–1508. [Google Scholar] [CrossRef]
- Neri, R.; Florance, K.; Koziol, P.; Van Cleave, S. A Biological Profile of a Nonsteroidal Antiandrogen, SCH 13521 (4′–Nitro–3′–Trifluoromethylisobutyranilide). Endocrinology 1972, 91, 427–437. [Google Scholar] [CrossRef]
- Schellhammer, P. An update on bicalutamide in the treatment of prostate cancer. Expert Opin. Investig. Drugs 1999, 8, 849–860. [Google Scholar] [CrossRef]
- Fradet, Y. Bicalutamide (Casodex®) in the treatment of prostate cancer. Expert Rev. Anticancer. Ther. 2004, 4, 37–48. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Smith, A.D.; Ferraldeschi, R.; Al-Lazikani, B.; Workman, P.; De Bono, J.S. Drug discovery in advanced prostate cancer: Translating biology into therapy. Nat. Rev. Drug Discov. 2016, 15, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.M.; Gao, A.C. Drug resistance in castration resistant prostate cancer: Resistance mechanisms and emerging treatment strategies. Am. J. Clin. Exp. Urol. 2015, 3, 64–76. [Google Scholar] [PubMed]
- Steketee, K.; Timmerman, L.; Der Made, A.C.Z.-V.; Doesburg, P.; Brinkmann, A.O.; Trapman, J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int. J. Cancer 2002, 100, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Bohl, C.E.; Gao, W.; Miller, D.D.; Bell, C.E.; Dalton, J.T. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 6201–6206. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, N.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mülders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Fizazi, K.; Albigès, L.; Loriot, Y.; Massard, C. ODM-201: A new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev. Anticancer. Ther. 2015, 15, 1007–1017. [Google Scholar] [CrossRef]
- Fujii, S.; Kagechika, H. Androgen receptor modulators: A review of recent patents and reports (2012-2018). Expert Opin. Ther. Patents 2019, 29, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Hirano, T.; Mori, S.; Fujii, S.; Masuno, H.; Kinoshita, M.; Kagechika, H.; Tanatani, A. 6-Arylcoumarins as Novel Nonsteroidal Type Progesterone Antagonists: An Example with Receptor-Binding-Dependent Fluorescence. J. Med. Chem. 2011, 54, 7055–7065. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Negishi, M.; Sakai, H.; Hirano, T.; Mori, S.; Fujii, S.; Kagechika, H.; Tanatani, A. Development of 6-arylcoumarins as nonsteroidal progesterone antagonists. Structure–activity relationships and fluorescence properties. Bioorganic Med. Chem. 2016, 24, 5602–5610. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, D.; Nakamura, N.; Nishizawa, Y.; Uchida, N.; Noguchi, S.; Matsumoto, K.; Sato, B. Inhibitory and stimulatory effects of glucocorticoid on androgen- induced growth of murine shionogi carcinoma 115 in vivo and in cell culture. Cancer Res. 1987, 47, 6560–6564. [Google Scholar]
- Inoue, K.; Urushibara, K.; Kanai, M.; Yura, K.; Fujii, S.; Ishigami-Yuasa, M.; Hashimoto, Y.; Mori, S.; Kawachi, E.; Matsumura, M.; et al. Design and synthesis of 4-benzyl-1-(2H)-phthalazinone derivatives as novel androgen receptor antagonists. Eur. J. Med. Chem. 2015, 102, 310–319. [Google Scholar] [CrossRef]
- Veldscholte, J.; Berrevoets, C.A.; Brinkmann, A.O.; Grootegoed, J.A.; Mulder, E. Anti-androgens and the mutated androgen receptor of LNCaP cells: Differential effects on binding affinity, heat-shock protein interaction, and transcription activation. Biochemistry 1992, 31, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, B.; Geng, G.; Wu, J.H. Study of the impact of the T877A mutation on ligand-induced helix-12 positioning of the androgen receptor resulted in design and synthesis of novel antiandrogens. Proteins Struct. Funct. Bioinform. 2009, 78, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Kagechika, H.; Himi, T.; Kawachi, E.; Shudo, K. Retinobenzoic acids. 4. Conformation of aromatic amides with retinoidal activity. Importance of trans-amide structure for the activity. J. Med. Chem. 1989, 32, 2292–2296. [Google Scholar] [CrossRef] [PubMed]
- Tanatani, A.; Yokoyama, A.; Azumaya, I.; Takakura, Y.; Mitsui, C.; Shiro, M.; Uchiyama, M.; Muranaka, A.; Kobayashi, N.; Yokozawa, T. Helical Structures ofN-Alkylated Poly(p-benzamide)s. J. Am. Chem. Soc. 2005, 127, 8553–8561. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, D.; Albertini, A.; Zava, D. Progestin regulation of alkaline phosphatase in the human breast cancer cell line T47D. Cancer Res. 1991, 51, 4470–4475. [Google Scholar]
- Yamada, A.; Fujii, S.; Mori, S.; Kagechika, H. Design and Synthesis of 4-(4-Benzoylaminophenoxy)phenol Derivatives As Androgen Receptor Antagonists. ACS Med. Chem. Lett. 2013, 4, 937–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, K.; Goto, T.; Fujii, S.; Suzuki, T.; Ohta, S.; Endo, Y. Design and synthesis of carborane-containing androgen receptor (AR) antagonist bearing a pyridine ring. Bioorg. Med. Chem. 2008, 16, 8022–8028. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | N-Substituent | X | IC50, µM | Compound | N-Substituent | X | IC50, µM |
|---|---|---|---|---|---|---|---|
| Hydroxyflutamide (2b) | 0.29 ± 0.03 | ||||||
| 7a | H | none | inactive c | 7b | Me | none | 1.34 ± 0.21 |
| 8a | H | p-CN | inactive c | 8b | Me | p-CN | inactive c |
| 9a | H | p-NO2 | inactive c | 9b | Me | p-NO2 | inactive c |
| 10a | H | p-CF3 | inactive c | 10b | Me | p-CF3 | inactive c |
| 11a | H | p-F | inactive c | 11b | Me | p-F | 0.58 ± 0.06 |
| 12a | H | p-Cl | inactive c | 12b | Me | p-Cl | inactive c |
| 13a | H | p-Me | inactive c | 13b | Me | p-Me | inactive c |
| 14a | H | p-OMe | inactive c | 14b | Me | p-OMe | inactive c |
| 15a | H | m-CN | inactive c | 15b | Me | m-CN | 2.40 ± 0.39 |
| 16a | H | m-CF3 | 7.66 ± 3.57 | 16b | Me | m-CF3 | 0.60 ± 0.04 |
| 17a | H | m-F | inactive c | 17b | Me | m-F | 0.72 ± 0.12 |
| 18a | H | m-Cl | inactive c | 18b | Me | m-Cl | 0.73 ± 0.11 |
| 19a | H | m-Me | >10 b | 19b | Me | m-Me | 0.49 ± 0.04 |
| 20a | H | m-OMe | inactive c | 20b | Me | m-OMe | 0.67 ± 0.05 |
| 21a | H | o-F | inactive c | 21b | Me | o-F | 7.70 ± 0.45 |
| 22a | H | o-Cl | inactive c | 22b | Me | o-Cl | inactive c |
| 23a | H | o-OMe | inactive c | 23b | Me | o-OMe | >10 b |
| 24a | H | 3,5-Cl2 | inactive c | 24b | Me | m,m’-Cl2 | 1.04 ± 0.07 |
| 25a | H | 3,5-Me2 | inactive c | 25b | Me | m,m’-Me2 | 0.83 ± 0.07 |

| Compound | X | N-Substituent | IC50, µM | Compound | X | N-Substituent | IC50, µM |
|---|---|---|---|---|---|---|---|
| 18b | Cl | Me | 0.73 ± 0.11 | 19b | Me | Me | 0.49 ± 0.04 |
| 18c | Et | 1.70 ± 0.20 | 19c | Et | 2.46 ± 0.35 | ||
| 18d | n-Pr | 2.88 ± 0.28 | 19d | n-Pr | 8.49 ± 0.18 | ||
| 18e | CH2Ph | 5.55 ± 0.40 | 19e | CH2Ph | 1.83 ± 0.12 | ||
| 18f | CH2-2-naphthyl | Inactive b | 19f | CH2-2-naphthyl | Inactive b |

| Compound | Ar | IC50, µM |
|---|---|---|
| 7b | Ph | 1.34 ± 0.21 |
| 26b | c-C6H11 | Inactive b |
| 27b | 1-naphthyl | 0.66 ± 0.04 |
| 28b | 2-naphthyl | 7.81 ± 1.91 |
| 29b | 2-pyridyl | Inactive b |
| 30b | 2-furyl | 0.92 ± 0.18 |
| 31b | 2-thiophenyl | 0.50 ± 0.07 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koga, H.; Negishi, M.; Kinoshita, M.; Fujii, S.; Mori, S.; Ishigami-Yuasa, M.; Kawachi, E.; Kagechika, H.; Tanatani, A. Development of Androgen-Antagonistic Coumarinamides with a Unique Aromatic Folded Pharmacophore. Int. J. Mol. Sci. 2020, 21, 5584. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155584
Koga H, Negishi M, Kinoshita M, Fujii S, Mori S, Ishigami-Yuasa M, Kawachi E, Kagechika H, Tanatani A. Development of Androgen-Antagonistic Coumarinamides with a Unique Aromatic Folded Pharmacophore. International Journal of Molecular Sciences. 2020; 21(15):5584. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155584
Chicago/Turabian StyleKoga, Hitomi, Mai Negishi, Marie Kinoshita, Shinya Fujii, Shuichi Mori, Mari Ishigami-Yuasa, Emiko Kawachi, Hiroyuki Kagechika, and Aya Tanatani. 2020. "Development of Androgen-Antagonistic Coumarinamides with a Unique Aromatic Folded Pharmacophore" International Journal of Molecular Sciences 21, no. 15: 5584. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155584