All d-Lysine Analogues of the Antimicrobial Peptide HPA3NT3-A2 Increased Serum Stability and without Drug Resistance

Abstract
:1. Introduction
2. Results
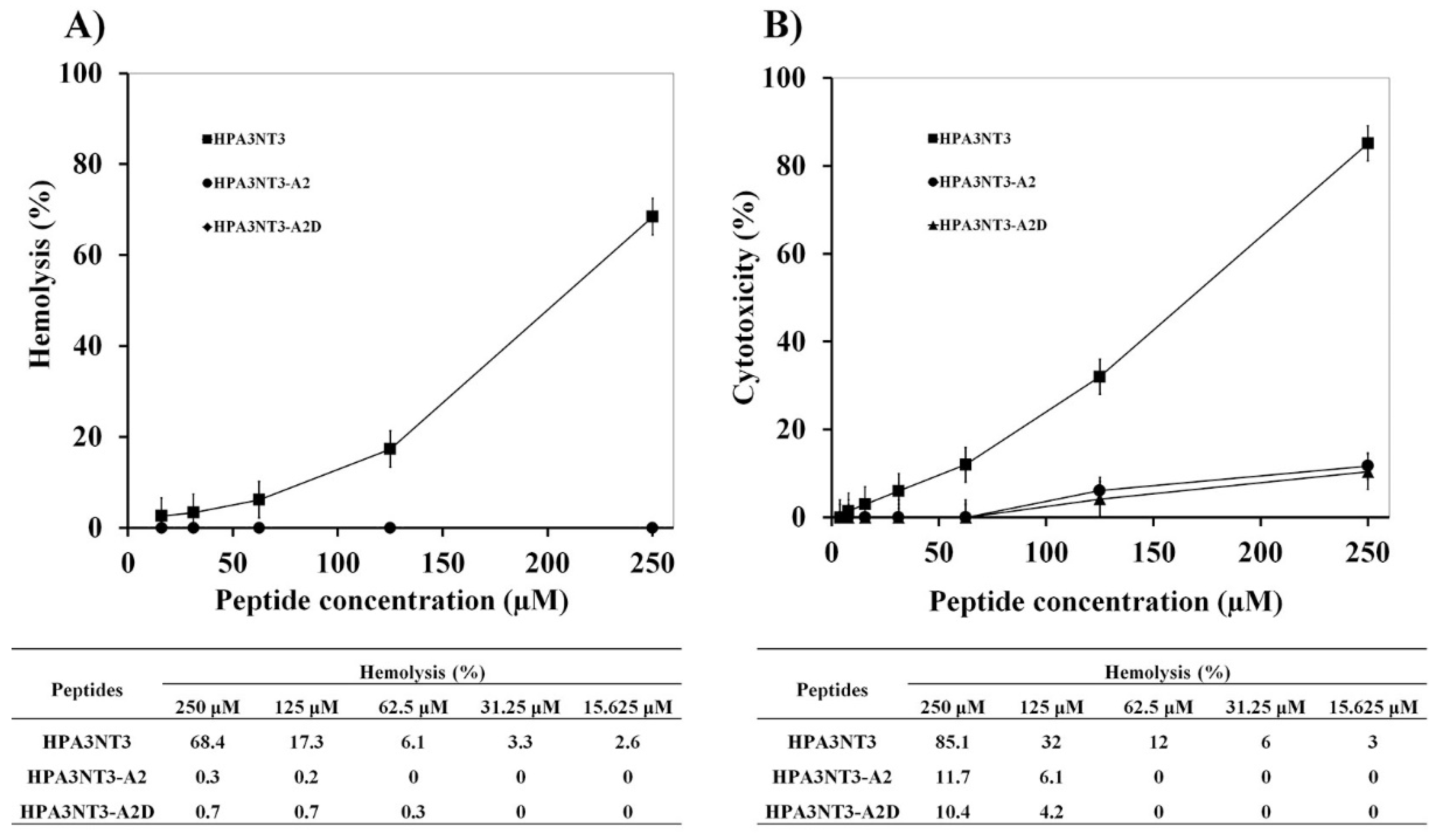
2.1. Peptide Design, Antimicrobial Activity, and Cytotoxic Activities of HPA3NT3-A2 and HPA3NT3-A2D
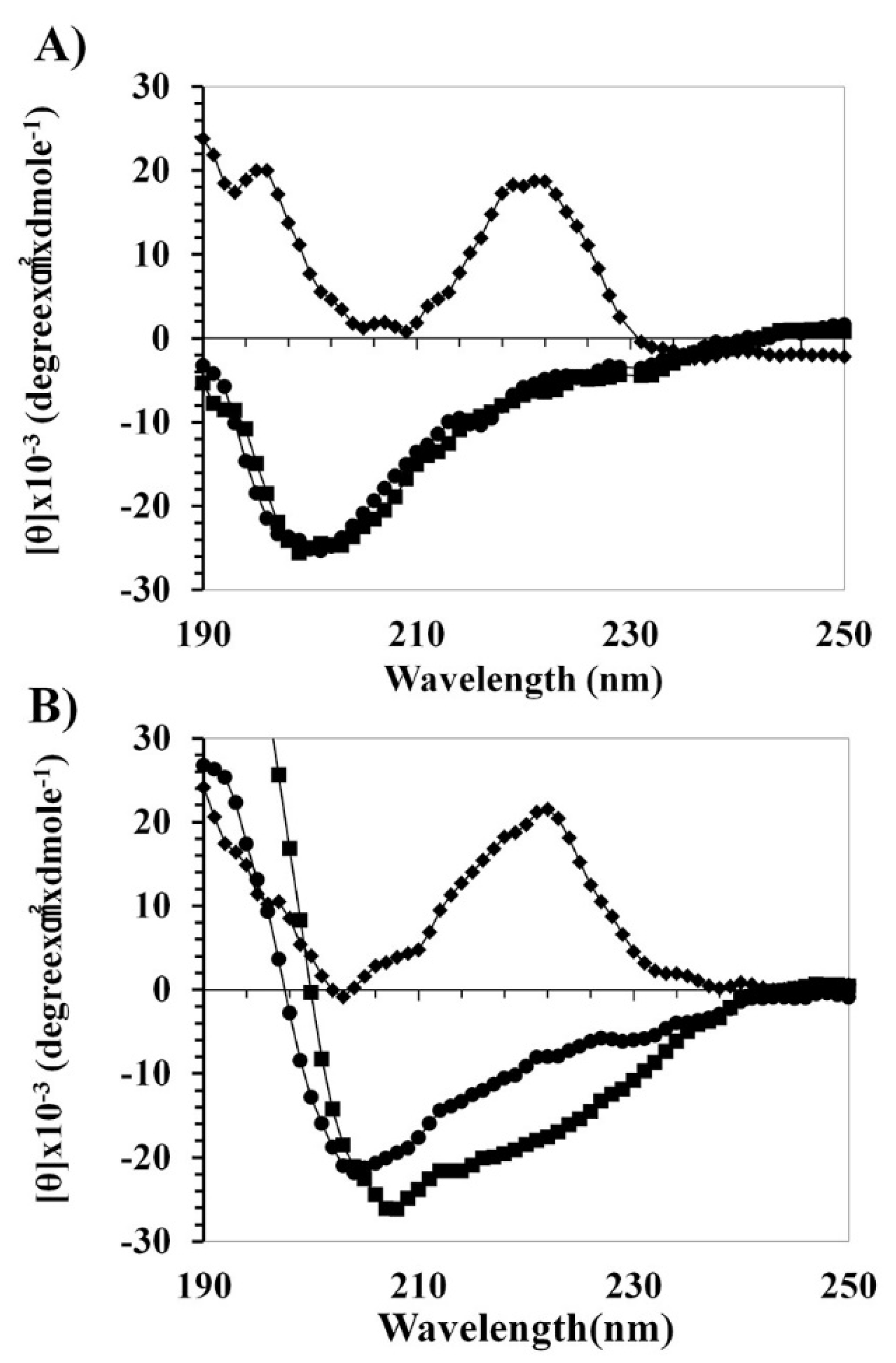
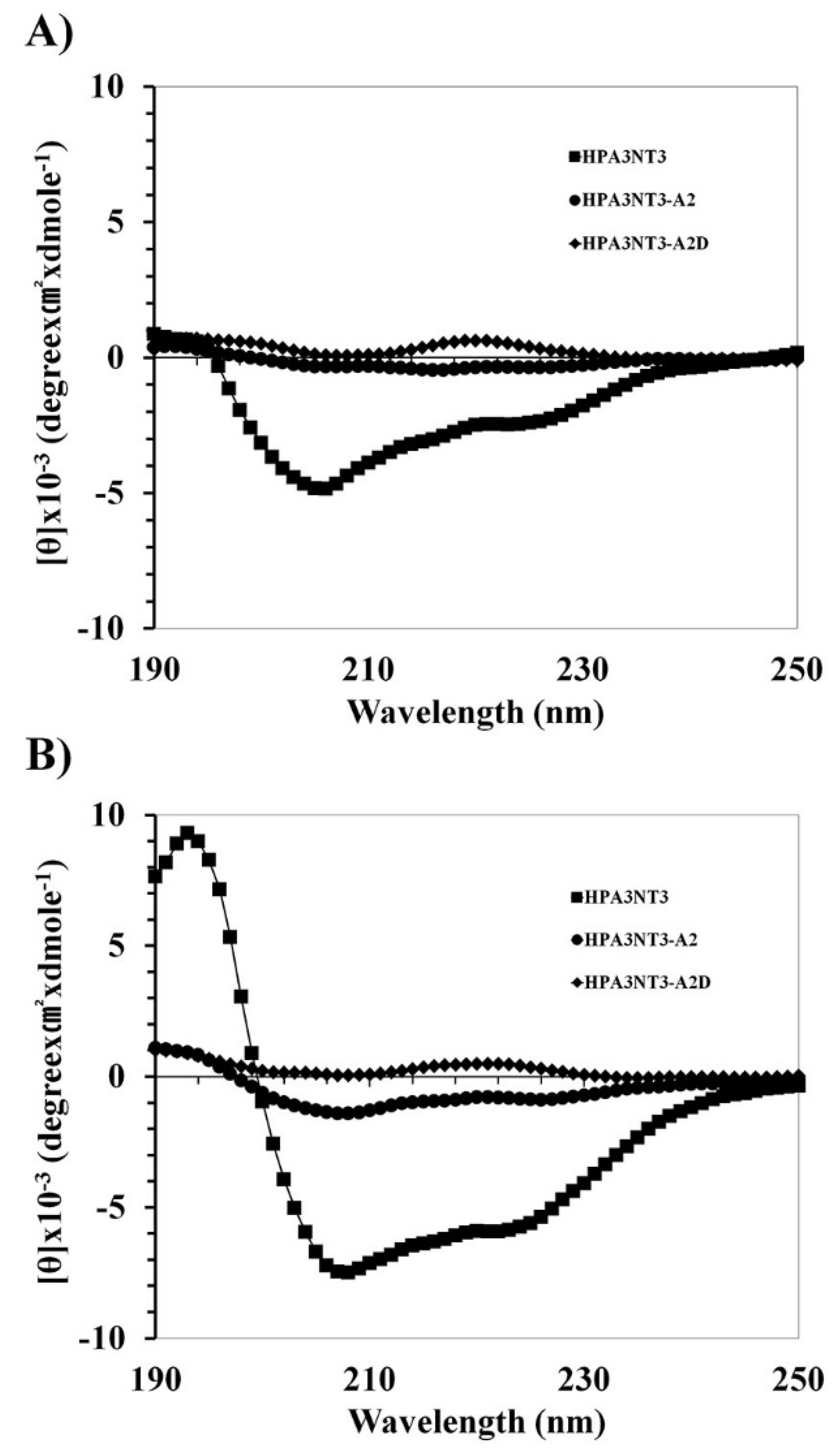
2.2. Peptide Mechanism by Using CD Spectrometer
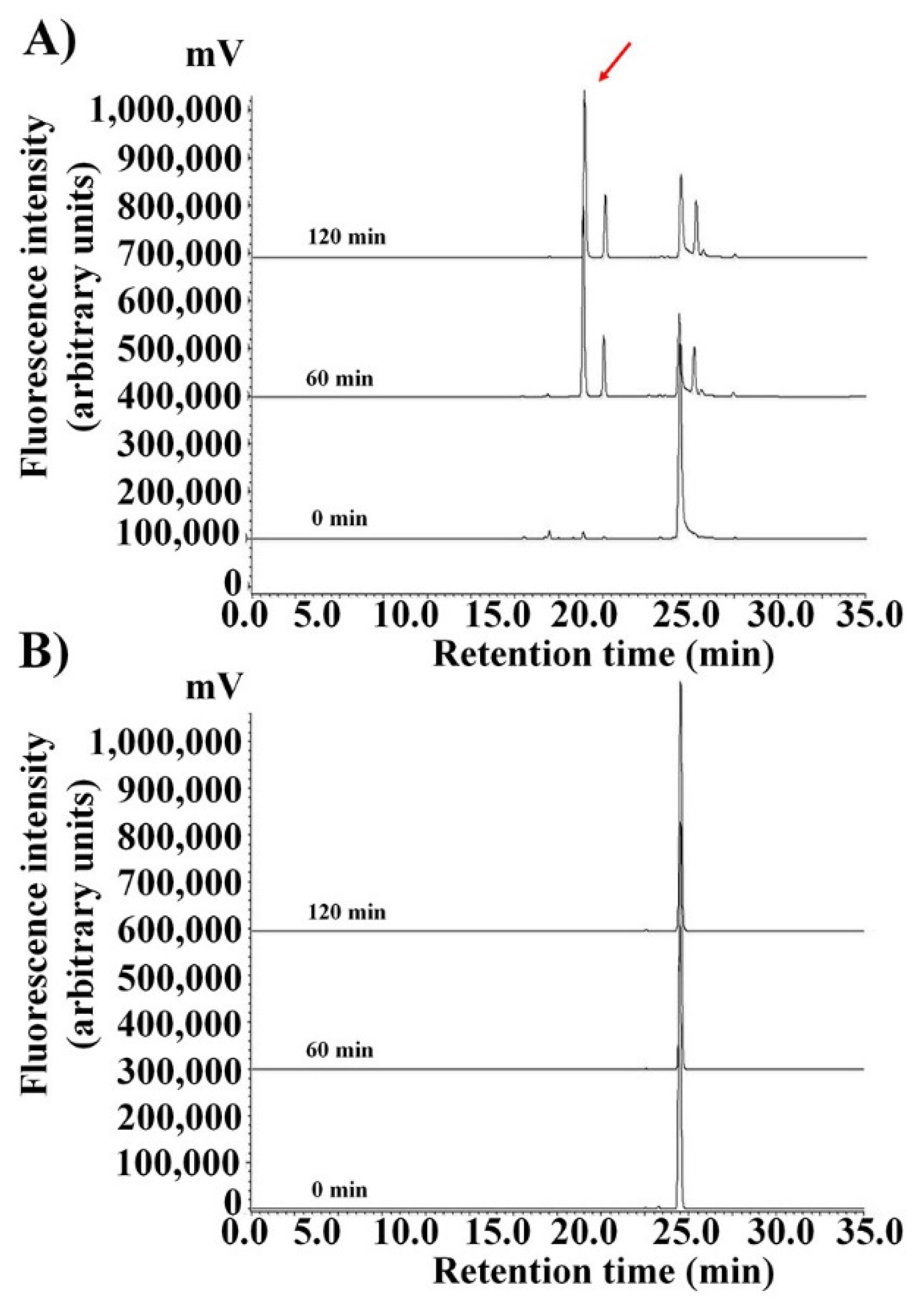
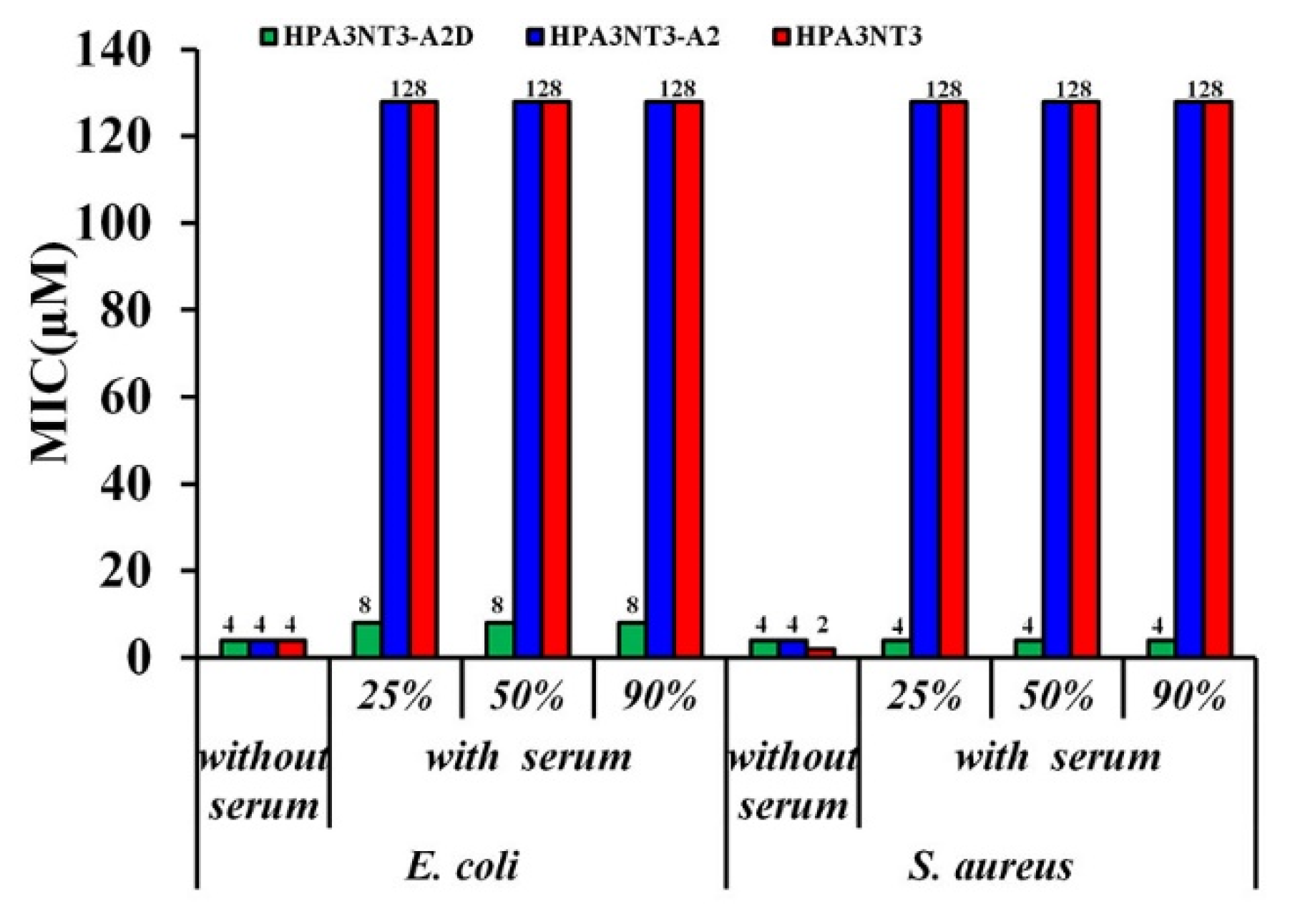
2.3. Maintenance of HPA3NT3-A2 Peptide in Serum by d-Enantiomer Substitution of Lysine
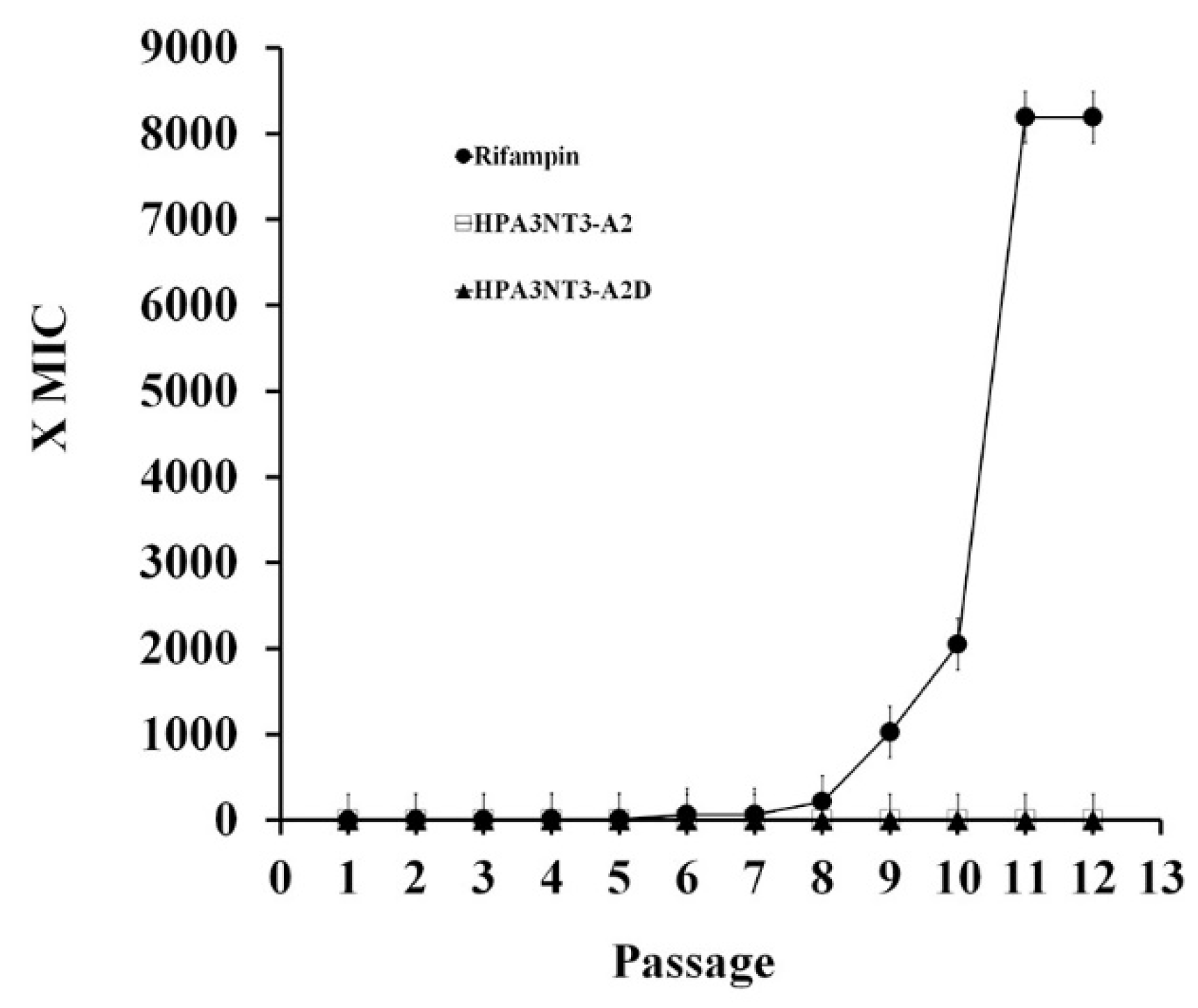
2.4. Non-Inducing Resistant and Drug-Resistant Bacteria Activity of HPA3NT3 and Its Analogue Peptides
3. Discussion
4. Materials and Methods
4.1. Microbial Strains
4.1.1. Peptide Synthesis, Rhodamine Labeling, and Purification
4.1.2. Antimicrobial Activity
4.1.3. Hemolysis
4.1.4. Cytotoxicity
4.1.5. Circular Dichroism (CD) Analysis
4.1.6. Binding Assay
4.1.7. Metabolic Stability of HPA3NT3-A2 in Serum
4.1.8. Stability of Peptides in Serum
4.1.9. Resistance Development Assay
4.2. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Wu, M.; Tong, X.; Liu, S.; Wang, D.; Wang, L.; Fan, H. Prevalence of methicillin-resistant Staphylococcus aureus in healthy Chinese population: A system review and meta-analysis. PLoS ONE 2019, 14, e0223599. [Google Scholar] [CrossRef] [PubMed]
- Satlin, M.J.; Walsh, T.J. Multidrug-resistant Enterobacteriaceae, Pseudomonas aeruginosa, and vancomycin-resistant Enterococcus: Three major threats to hematopoietic stem cell transplant recipients. Transpl. Infect. Dis. Off. J. Transplant. Soc. 2017, 19. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayaz, M.; Ullah, F.; Sadiq, A.; Ullah, F.; Ovais, M.; Ahmed, J.; Devkota, H.P. Synergistic interactions of phytochemicals with antimicrobial agents: Potential strategy to counteract drug resistance. Chem.-Biol. Interact. 2019, 308, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Chowanski, S.; Adamski, Z.; Lubawy, J.; Marciniak, P.; Pacholska-Bogalska, J.; Slocinska, M.; Spochacz, M.; Szymczak, M.; Urbanski, A.; Walkowiak-Nowicka, K.; et al. Insect Peptides—Perspectives in Human Diseases Treatment. Curr. Med. Chem. 2017, 24, 3116–3152. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jung, J.H.; Liu, Y. Antimicrobial Compounds from Marine Invertebrates-Derived Microorganisms. Curr. Med. Chem. 2016, 23, 2892–2905. [Google Scholar] [CrossRef]
- Elhag, O.; Zhou, D.; Song, Q.; Soomro, A.A.; Cai, M.; Zheng, L.; Yu, Z.; Zhang, J. Screening, Expression, Purification and Functional Characterization of Novel Antimicrobial Peptide Genes from Hermetia illucens (L.). PLoS ONE 2017, 12, e0169582. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility in Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Hahm, K.S. Effects of N- and C-terminal truncation of HP (2-20) from Helicobacter pylori ribosomal protein L1 (RPL1) on its anti-microbial activity. Biotechnol. Lett. 2005, 27, 193–199. [Google Scholar] [CrossRef]
- Li, J.; Koh, J.J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alghalayini, A.; Garcia, A.; Berry, T.; Cranfield, C.G. The Use of Tethered Bilayer Lipid Membranes to Identify the Mechanisms of Antimicrobial Peptide Interactions with Lipid Bilayers. Antibiotics 2019, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Nichols, M.; Kuljanin, M.; Nategholeslam, M.; Hoang, T.; Vafaei, S.; Tomberli, B.; Gray, C.G.; DeBruin, L.; Jelokhani-Niaraki, M. Dynamic turn conformation of a short tryptophan-rich cationic antimicrobial peptide and its interaction with phospholipid membranes. J. Phys. Chem. B 2013, 117, 14697–14708. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial Peptides: Interaction with Model and Biological Membranes and Synergism with Chemical Antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Park, S.C.; Hahm, K.S.; Park, Y. Antimicrobial HPA3NT3 peptide analogs: Placement of aromatic rings and positive charges are key determinants for cell selectivity and mechanism of action. Biochim. Biophys. Acta 2013, 1828, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Böttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef]
- Starr, C.G.; Wimley, W.C. Antimicrobial peptides are degraded by the cytosolic proteases of human erythrocytes. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2319–2326. [Google Scholar] [CrossRef]
- Manabe, T.; Kawasaki, K. d-form KLKLLLLLKLK-NH(2) peptide exerts higher antimicrobial properties than its L-form counterpart via an association with bacterial cell wall components. Sci. Rep. 2017, 7, 43384. [Google Scholar] [CrossRef] [Green Version]
- Melchionna, M.; Styan, K.E.; Marchesan, S. The Unexpected Advantages of Using D-Amino Acids for Peptide Self-Assembly into Nanostructured Hydrogels for Medicine. Curr. Top. Med. Chem. 2016, 16, 2009–2018. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Ding, J.; Li, H.; Li, L.; Zhao, R.; Shen, Z.; Fan, X.; Xi, T. Effects of cations and pH on antimicrobial activity of thanatin and s-thanatin against Escherichia coli ATCC25922 and B. subtilis ATCC 21332. Curr. Microbiol. 2008, 57, 552–557. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Shan, A.; Ma, Z.; Xu, W.; Wang, J.; Chou, S.; Cheng, B. Bactericidal efficiency and modes of action of the novel antimicrobial peptide T9W against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2015, 59, 3008–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cera, N. Serine Proteases. IUBMB Life 2009, 61, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Cataldo, M.A.; Dancer, S.J.; De Angelis, G.; Falcone, M.; Frank, U.; Kahlmeter, G.; Pan, A.; Petrosillo, N.; Rodríguez-Baño, J.; et al. ESCMID guidelines for the management of the infection control measures to reduce transmission of multidrug-resistant Gram-negative bacteria in hospitalized patients. Clin. Microbiol. Infect. 2014, 20 (Suppl. 1), 1–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araos, R.; Battaglia, T.; Ugalde, J.A.; Rojas-Herrera, M.; Blaser, M.J.; D’Agata, E.M.C. Fecal Microbiome Characteristics and the Resistome Associated with Acquisition of Multidrug-Resistant Organisms among Elderly Subjects. Front. Microbiol. 2019, 10, 2260. [Google Scholar] [CrossRef]
- Park, S.C.; Kim, M.H.; Hossain, M.A.; Shin, S.Y.; Kim, Y.; Stella, L.; Wade, J.D.; Park, Y.; Hahm, K.S. Amphipathic alpha-helical peptide, HP (2-20), and its analogues derived from Helicobacter pylori: Pore formation mechanism in various lipid compositions. Biochim. Biophys. Acta 2008, 1778, 229–241. [Google Scholar] [CrossRef]
- Lee, J.K.; Luchian, T.; Park, Y. New antimicrobial peptide kills drug-resistant pathogens without detectable resistance. Oncotarget 2018, 9, 15616–15634. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Seo, C.H.; Luchian, T.; Park, Y. Antimicrobial Peptide CMA3 Derived from the CA-MA Hybrid Peptide: Antibacterial and Anti-inflammatory Activities with Low Cytotoxicity and Mechanism of Action in Escherichia coli. Antimicrob. Agents Chemother. 2016, 60, 495–506. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Retention Time a | Net Charge |
|---|---|---|---|
| HPA3NT3 | FKRLKKLFKKIWNWK-NH2 | 20.94 | +8 |
| HPA3NT3-A2 | AKRLKKLAKKIWKWK-NH2 | 15.20 | +9 |
| HPA3NT3-A2D b | AKRLKKLAKKIWKWK-NH2 | 11.97 | +9 |
| MIC (μM) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| E. coli | NT3a | 4 | 4 | 1 | 1 | 1 | 1 | 1 | 2 | >64 | >64 | 32 | 1 |
| -A2b | 4 | 4 | 1 | 1 | 1 | 1 | 1 | 2 | >64 | >64 | >64 | 2 | |
| -A2Dc | 4 | 16 | 0.5 | 1 | 2 | 1 | 2 | 4 | >64 | >64 | 32 | 2 | |
| S. aureus | NT3 | 1 | 32 | 1 | 1 | 1 | 1 | 1 | 1 | >64 | >64 | 4 | 0.5 |
| -A2 | 1 | 32 | 1 | 1 | 2 | 2 | 2 | 2 | >64 | >64 | >64 | 1 | |
| -A2D | 1 | 32 | 1 | 2 | 4 | 4 | 4 | 4 | >64 | >64 | 32 | 2 | |
| Strains | MIC (μM) | ||||
|---|---|---|---|---|---|
| -A2 | -A2D | Amp | Ery | Cip | |
| E. colia | 4 | 4 | - | - | - |
| S. aureusa | 2 | 2 | - | - | - |
| E. coli CCARM 1229 b | 2 | 1 | >512 | 256 | - |
| E. coli CCARM 1238 b | 2 | 2 | >512 | 256 | - |
| P. aeruginosa 3547 c | 4 | 2 | >512 | >512 | >512 |
| P. aeruginosa 4007 c | 1 | 4 | >512 | >512 | >512 |
| S. aureus CCARM 3089 b | 2 | 4 | >512 | >512 | >512 |
| S. aureus CCARM 3114 b | 4 | 2 | >512 | >512 | >512 |
| S. aureus PBEL 1 d | 1 | 4 | >512 | >512 | >512 |
| S. aureus PBEL 2 d | 1 | 4 | >512 | >512 | >512 |
| S. typhimurium CCARM 8009 b | 4 | 8 | >512 | 256 | - |
| S. typhimurium CCARM 8013 b | 1 | 4 | >512 | 128 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-K.; Park, Y. All d-Lysine Analogues of the Antimicrobial Peptide HPA3NT3-A2 Increased Serum Stability and without Drug Resistance. Int. J. Mol. Sci. 2020, 21, 5632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165632
Lee J-K, Park Y. All d-Lysine Analogues of the Antimicrobial Peptide HPA3NT3-A2 Increased Serum Stability and without Drug Resistance. International Journal of Molecular Sciences. 2020; 21(16):5632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165632
Chicago/Turabian StyleLee, Jong-Kook, and Yoonkyung Park. 2020. "All d-Lysine Analogues of the Antimicrobial Peptide HPA3NT3-A2 Increased Serum Stability and without Drug Resistance" International Journal of Molecular Sciences 21, no. 16: 5632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165632