In Vitro Modeling of Non-Solid Tumors: How Far Can Tissue Engineering Go?


Abstract
:
1. Introduction
2. Modeling Solid Tumors in Vitro
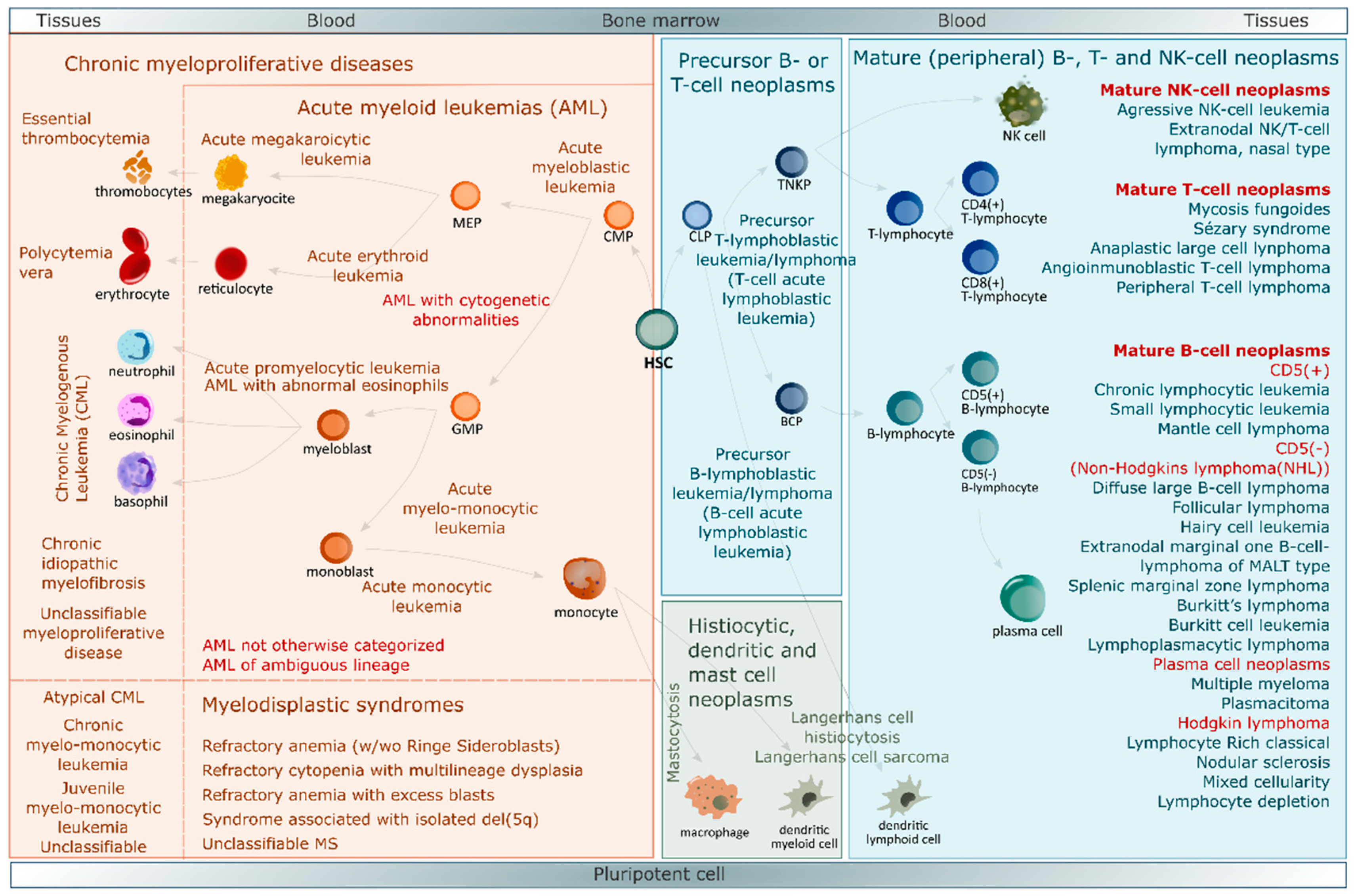
3. Blood Cancers
4. Bone Marrow Microenvironment, Home of Hematological Malignancies
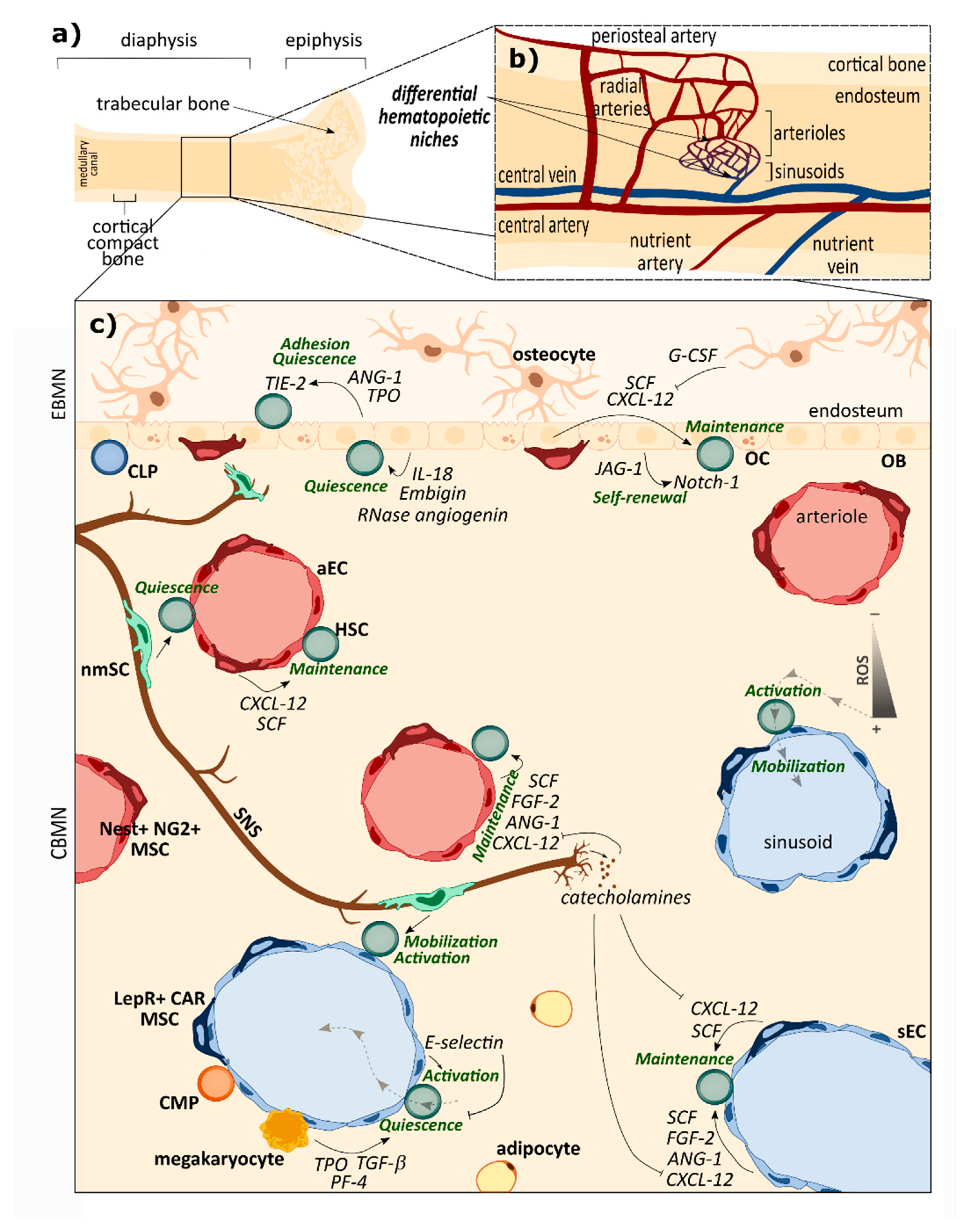
4.1. Healthy BM Niche: An Intricate and Precisely Organized Network Sustaining HSCs Homeostasis
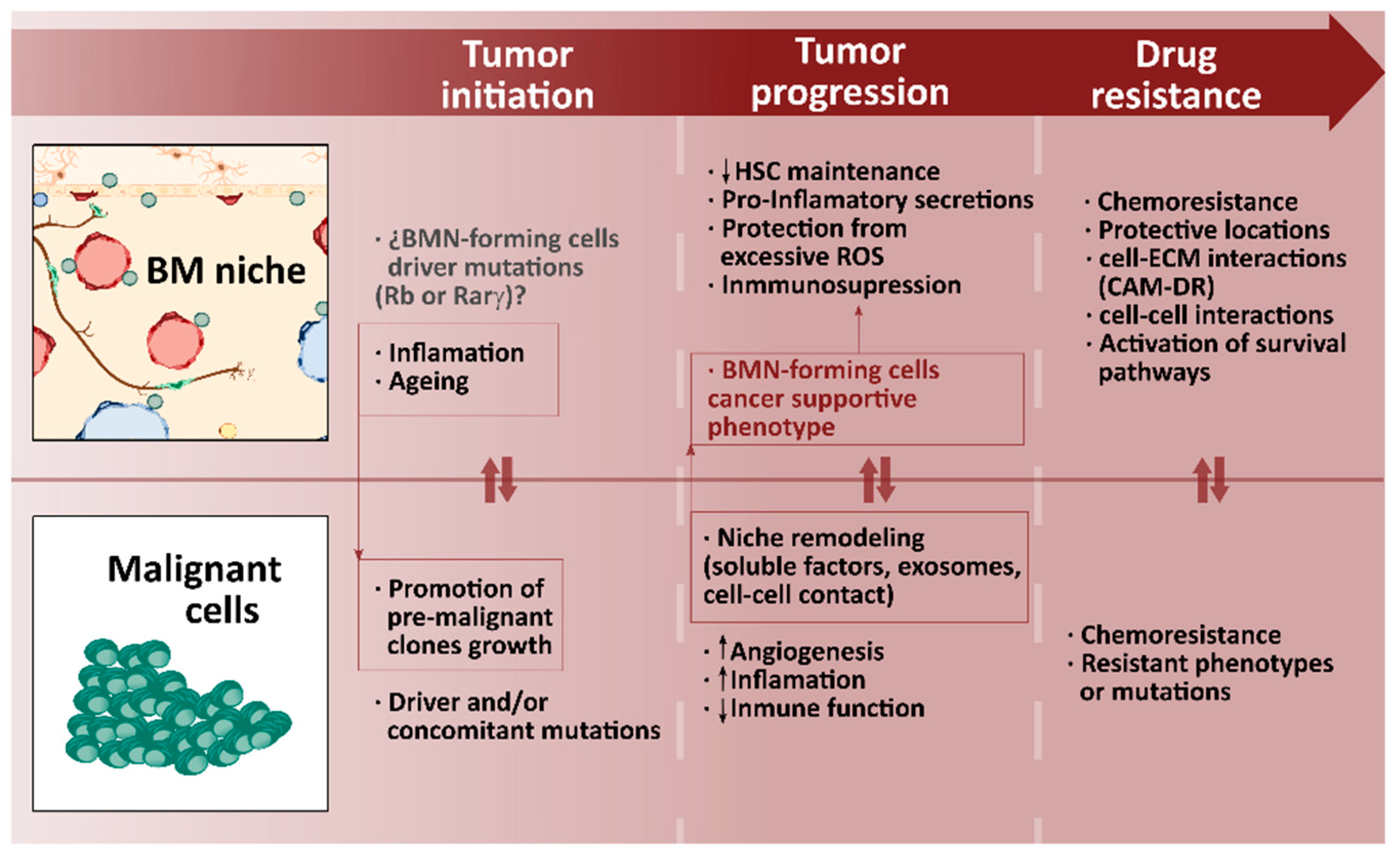
4.2. BM Niche in Hematological Malignancies: When the Regulatory Machinery Becomes the Perfect Tumor Partner
5. Advances in BM Models
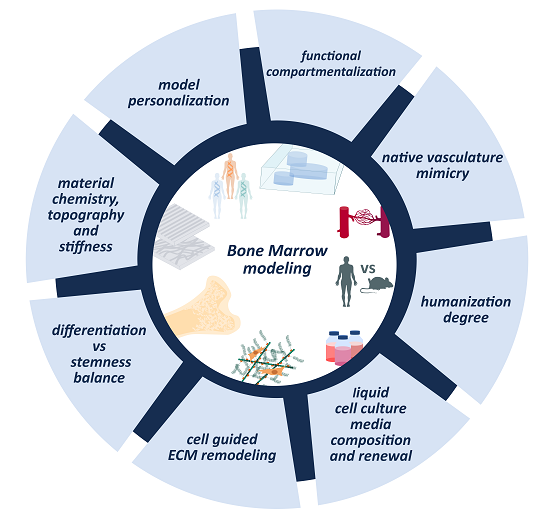
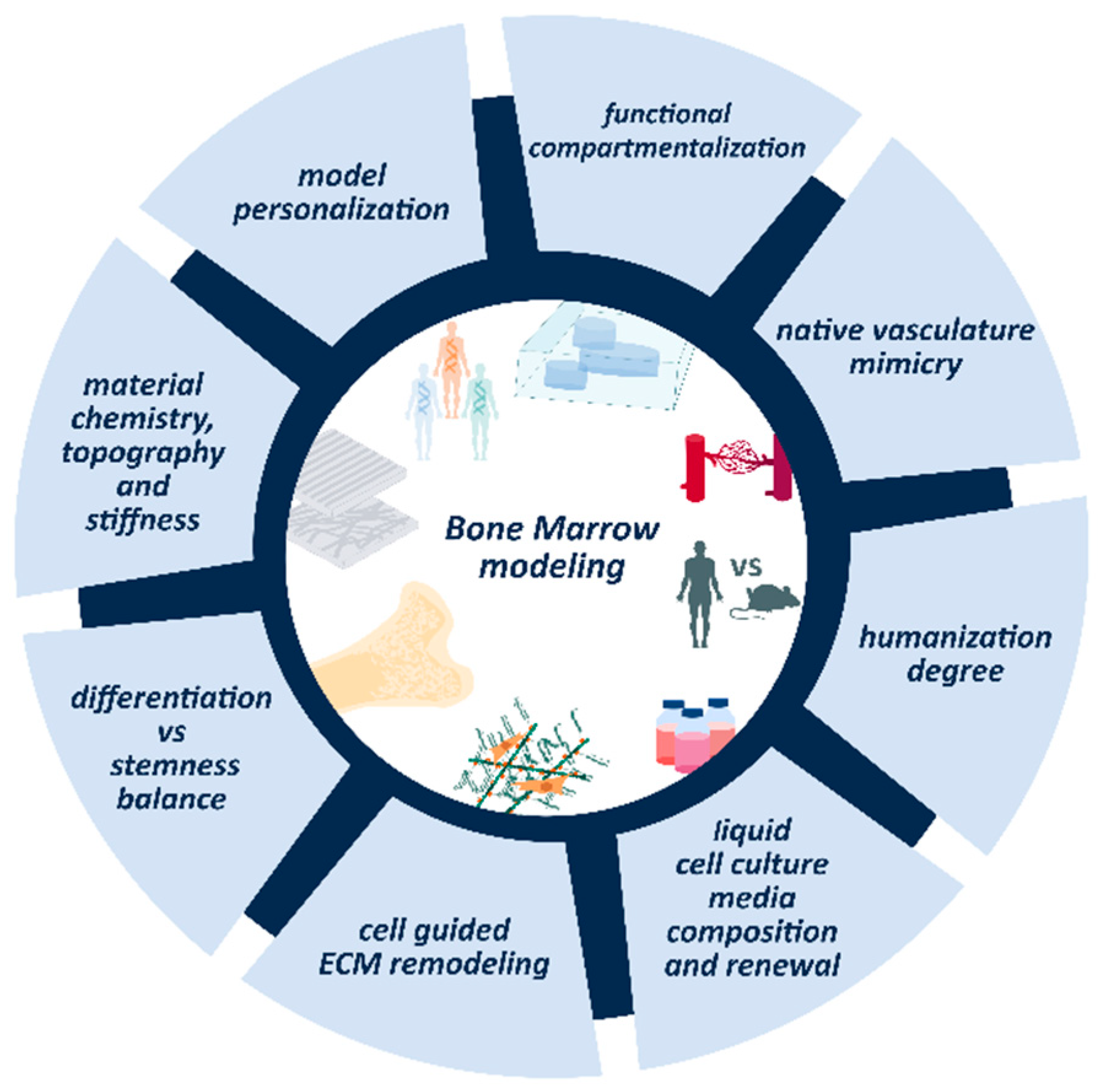
6. Unresolved Questions in Modeling BM and Non-Solid Tumors
6.1. ECM Dynamics and Remodeling
6.2. Vascularization of the Model
6.3. Compartmentalization
6.4. Stemness Maintenance Vs. Differentiation Balance
6.5. Cell Culture Media Renewal and Composition
6.6. Towards Personalized Medicine
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3DTEBM | 3D tissue engineered bone marrow |
| aBMN | Arteriolar bone marrow niche |
| ADSC | Adipose derived stem cells |
| aEC | Arteriolar endothelial cell |
| AML | Acute myeloid leukemia |
| ANG-1 | Angiopoietin |
| B-ALL | B cell acute lymphoblastic leukemia |
| BCP | B cell progenitor |
| BM | Bone marrow |
| BMN | Bone marrow niche |
| BMP | Bone morphogenetic protein |
| CAM-DR | Cell adhesion-mediated drug resistance |
| CBMN | Central bone marrow niche |
| CCL3 | CC motif ligand 3 |
| CLL | Chronic lymphocytic leukemia |
| CLP | Common lynphoid progenitor |
| CML | Chronic myeloid leukemia |
| CMP | Common myeloid progenitor |
| COL | Collagen |
| CSC | Cancer stem cells |
| CXCL-12 | CXC motif chemokine ligand 12 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DBP | Demineralized bone powder |
| Del | deletion |
| DPN | Dip pen nanolithography |
| DWJM | decellularized Wharton’s Jelly matrix |
| EBMN | Endosteal bone marrow niche |
| ECM | Extracellular matrix |
| EPC | Endothelial progenitor cell |
| FGF-2 | Fibroblast growth factor 2 |
| FN | Fibronectin |
| G-CSF | Granulocyte-colony stimulating factor |
| GMP | Granulocyte-monocyte progenitor |
| HA | Hyaluronic acid |
| HCoMECs | Human colonic microvascular endothelial cells |
| HCT | Hematopoietic cell transplantation |
| HDMEC | Human dermal microvascular endothelial cell |
| HSC | Hematopoietic stem cell |
| HSPC | Hematopoietic stem and progenitor cell |
| HTS | High troughput technologies |
| HUVEC | Human umbilical vein endothelial cell |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IL | Interleukin |
| JAG-1 | Jagged-1 |
| LepR+ CAR MSCs | Leptin receptor expressing cells and abundant reticular mesenchymal stem cells |
| MCFS | Breast cancer derived tumor initiating cells |
| MEP | Megakaryocyte-erythrocyte progenitor |
| MM | Multiple myeloma |
| MOC | Multi-organ-chip |
| MPN | Myeloproliferative neoplasms |
| MSC | Mesenchymal stem cells |
| MVs | Mirovesicles |
| ND | Normal donnor |
| Nest+NG2+ | Nestin and neural glial antigen expressing mesenchymal stem cells |
| NF-κB | nuclear factor κB |
| NK | Natural killer |
| nmSC | Non myelinating Schwann cell |
| OB | Osteoblast |
| OC | Osteoclast |
| PDMS | Poly(dimethyl siloxane) |
| PDTX | Patient derived tumor xenograft |
| PEGDA | Poly (ethylene glycol) diacrylate |
| PF-4 | Platelet factor 4 |
| PGE2 | Prostaglandin E2 |
| PKCβ | Protein kinase C beta |
| PU | Polyurethane |
| PVA | Polyvinyl alcohol |
| Rarγ | Retinoic acid receptor γ |
| Rb | Retinoblastoma |
| ROS | Reactive oxygen species |
| sBMN | Sinusoidal bone marrow niche |
| SCF | Stem cell factor |
| sEC | Sinusoidal endothelial cell |
| SEM | Scanning electron microscopy |
| SNS | Sympathetic nerve fiber |
| T-ALL | T cell acute lymphoblastic leukemia |
| TERM | Tissue Engineering and Regenerative Medicine |
| TGF-β | Transforming growth factor beta |
| TIE-2 | Angiopoietin receptor |
| TNF | Tumor necrosis factor |
| TNKP | T-natural killer cell progenitor |
| TPO | Thrombopoietin |
| UC | Umbilical cord |
| VCAM-1 | Vascular adhesion molecule 1 |
| VEGF-A | Vascular endothelial growth factor A |
| WHO | World Health Organization |
References
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelm, J.M.; Lal-Nag, M.; Sittampalam, G.S.; Ferrer, M. Translational in vitro research: Integrating 3D drug discovery and development processes into the drug development pipeline. Drug Discov. Today. 2019, 24, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Hassani, I.; Clary, J.M.; Lipke, E.A. Polymeric biomaterials for in vitro cancer tissue engineering and drug testing applications. Tissue Eng. Part B Rev. 2016, 22, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Bhatia, S.N. Engineering tissues for in vitro applications. Curr. Opin. Biotechnol. 2006, 17, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.E.; Rodrigues, M.T.; Domingues, R.M.A.; Reis, R.L. Tissue engineering and regenerative medicine: New trends and directions-A year in review. Tissue Eng. Part B Rev. 2017, 23, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lee, S.J.; Cheng, H.; Yoo, J.J.; Atala, A. 3D bioprinted functional and contractile cardiac tissue constructs. Acta Biomater. 2018, 70, 48–56. [Google Scholar] [CrossRef]
- Tsukamoto, Y.; Akagi, T.; Akashi, M. Vascularized cardiac tissue construction with orientation by layer-by-layer method and 3D printer. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Van Grunsven, L.A. 3D in vitro models of liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 133–146. [Google Scholar] [CrossRef]
- Griffith, L.G.; Swartz, M.A. Capturing complex 3D tissue physiology in vitro. Nat. Rev. Mol. Cell Biol. 2006, 7, 211–224. [Google Scholar] [CrossRef]
- Schenke-Layland, K.; Nerem, R.M. In vitro human tissue models-moving towards personalized regenerative medicine. Adv. Drug Deliv. Rev. 2011, 63, 195–196. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.Y.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Wright, W.E.; Shay, J.W. Telomere dynamics in cancer progression and prevention: Fundamental differences in human and mouse telomere biology. Nat. Med. 2000, 6, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Brancato, V.; Oliveira, J.M.; Correlo, V.M.; Reis, R.L.; Kundu, S.C. Could 3D models of cancer enhance drug screening? Biomaterials 2020, 232. [Google Scholar] [CrossRef]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2016, 387, 61–68. [Google Scholar] [CrossRef]
- Håkanson, M.; Cukierman, E.; Charnley, M. Miniaturized pre-clinical cancer models as research and diagnostic tools. Adv. Drug Deliv. Rev. 2013, 69–70, 52–66. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, J.; Varadaraj, S.; Dash, S.K.; Sharma, A.; Verma, R.S. Organotypic cancer tissue models for drug screening: 3D constructs, bioprinting and microfluidic chips. Drug Discov. Today 2020, 25. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Hayley, E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human Primary Liver Cancer-derived Organoid Cultures for disease modelling and drug screening. Nat. Med. 2018, 23, 1424–1435. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Angeloni, V.; Contessi, N.; De Marco, C.; Bertoldi, S.; Tanzi, M.C.; Daidone, M.G.; Farè, S. Polyurethane foam scaffold as in vitro model for breast cancer bone metastasis. Acta Biomater. 2017, 63, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Chi, B.H.; Yoo, J.J.; Ju, Y.M.; Whang, Y.M.; Chang, I.H. Structure establishment of three-dimensional (3D) cell culture printing model for bladder cancer. PLoS ONE 2019, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.R.; Barata, D.; Teixeira, L.M.; Giselbrecht, S.; Reis, R.L.; Oliveira, J.M.; Truckenmüller, R.; Habibovic, P. Colorectal tumor-on-a-chip system: A 3D tool for precision onco-nanomedicine. Sci. Adv. 2019, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolillo, M.; Colombo, R.; Serra, M.; Belvisi, L.; Papetti, A.; Ciusani, E.; Comincini, S.; Schinelli, S. Stem-like cancer cells in a dynamic 3d culture system: A model to study metastatic cell adhesion and anti-cancer drugs. Cells 2019, 8, 1434. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, M.A. Battling the hematological malignancies: The 200 Years’ War. Oncologist 2008, 13, 126–138. [Google Scholar] [CrossRef] [Green Version]
- Jagannathan-Bogdan, M.; Zon, L.I. Hematopoiesis. Development 2013, 140, 2463–2467. [Google Scholar] [CrossRef] [Green Version]
- Rieger, M.A.; Schroeder, T. Hematopoiesis. Cold Spring Harb. Perspect. Biol. 2012, 52, 173–180. [Google Scholar] [CrossRef]
- Harris, N.L.; Jaffe, E.S.; Diebold, J.; Flandrin, G.; Muller-Hermelink, H.K.; Vardiman, J.; Lister, T.A.; Bloomfield, C.D. The World Health Organization Classification of Neoplasms of the Hematopoietic and Lymphoid Tissues: Report of the Clinical Advisory Committee Meeting-Airlie House, Virginia, November, 1997. Hematol. J. 2000, 1, 53–66. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserijan, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Godavarthy, P.S.; Krause, D.S. The bone marrow microenvironment in health and disease at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galán-Díez, M.; Cuesta-Domínguez, Á.; Kousteni, S. The bone marrow microenvironment in health and myeloid malignancy. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Itkin, T.; Gur-cohen, S.; Spencer, J.A.; Schajnovitz, A.; Saravana, K.; Ramasamy, M.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; et al. Distinct bone marrow blood vessels differentially regulate hematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Morikawa, T.; Takubo, K. Use of imaging techniques to illuminate dynamics of hematopoietic stem cells and their niches. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Galán-Díez, M.; Kousteni, S. The osteoblastic niche in hematopoiesis and hematological myeloid malignancies. Curr. Mol. Biol. Rep. 2017, 3, 53–62. [Google Scholar] [CrossRef]
- Klamer, S.; Voermans, C. The role of novel and known extracellular matrix and adhesion molecules in the homeostatic and regenerative bone marrow microenvironment. Cell Adhes. Migr. 2014, 8, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Walkley, C.R.; Shea, J.M.; Sims, N.A.; Purton, L.E.; Orkin, S.H. pRb Extrinsically Regulates Hematopoietic Stem Cells via myeloid cell-bone marrow microenvironment interactions. Cell 2007, 129, 1081–1095. [Google Scholar] [CrossRef] [Green Version]
- Walkley, C.R.; Olsen, G.H.; Dworkin, S.; Fabb, S.A.; Swann, J.; Mcarthur, G.A.; Westmoreland, S.V.; Chambon, P.; Scadden, T.; Purton, L.E. A Microenvironment-induced myeloproliferative syndrome caused by Rarγ deficiency. Cell 2007, 129, 1097–1110. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related cancer mutations associated with clonal hematopoietic expansion. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala-Torra, O.; Hanna, C.; Loken, M.R.; Flowers, M.E.D.; Maris, M.; Ladne, P.A.; Mason, J.R.; Senitzer, D.; Rodriguez, R.; Forman, S.J.; et al. Evidence of Donor-Derived Hematologic Malignancies after Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2006, 12, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Secreto, C.R.; Knox, T.R.; Ding, W.; Mukhopadhyay, D.; Kay, N.E. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: Implications for disease progression. Blood 2010, 115, 1755–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Chu, S.; Agarwal, P.; Campbell, V.L.; Hopcroft, L.; Jørgensen, H.G.; Lin, A.; Gaal, K.; Holyoake, T.L.; Bhatia, R. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood 2016, 128, 2671–2682. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 5, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Ruivo, N.; Foster, K.; et al. T cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522. [Google Scholar] [CrossRef] [Green Version]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef]
- Dias, S.; Hattori, K.; Zhu, Z.; Heissig, B.; Choy, M.; Lane, W.; Wu, Y.; Chadburn, A.; Hyjek, E.; Gill, M.; et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J. Clin. Investig. 2000, 106, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.P.; Rossi, R.M.; Neering, S.; Pei, S.; O’Dwyer, K.; Liesveld, J.L.; Brookes, P.S.; Becker, M.W.; et al. Bcl-2 inhibitor ABT-263 targets oxidative phosphorylation and selectively eradicates quiescent human Leukemia Stem Cells. Blood 2012, 120, 206. [Google Scholar] [CrossRef]
- Lutzny, G.; Kocher, T.; Schmidt-Supprian, M.; Rudelius, M.; Klein-Hitpass, L.; Finch, A.J.; Dürig, J.; Wagner, M.; Haferlach, C.; Kohlmann, A.; et al. Protein kinase C-β-dependent activation of NF-κB in stromal cells is indispensable for the survival of Chronic Lymphocytic Leukemia B cells in vivo. Cancer Cell 2013, 23, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Link, D.C. Concise Review: The Malignant Hematopoietic Stem Cell Niche. Stem Cells 2017, 35, 3–8. [Google Scholar] [CrossRef]
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008, 111, 1327–1333. [Google Scholar] [CrossRef]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef]
- Krause, D.S.; Lazarides, K.; Von Andrian, U.H.; Van Etten, R.A. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat. Med. 2006, 12, 1175–1180. [Google Scholar] [CrossRef]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.S.; Azab, F.; Leleu, X.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Jacamo, R.; Chen, Y.; Wang, Z.; Wencai, M.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; Ozaki, K.; et al. Bortezomib overcomes cell adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Bourgine, P.E.; Martin, I.; Schroeder, T. Engineering Human Bone Marrow Proxies. Stem Cell 2018, 22, 298–301. [Google Scholar] [CrossRef] [Green Version]
- Chramiec, A.; Vunjak-Novakovic, G. Tissue engineered models of healthy and malignant human bone marrow. Adv. Drug Deliv. Rev. 2019, 140, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Tavakol, D.N.; Tratwal, J.; Bonini, F.; Genta, M.; Campos, V.; Burch, P.; Hoehnel, S.; Béduer, A.; Alessandrini, M.; Naveiras, O.; et al. Injectable, scalable 3D tissue-engineered model of marrow hematopoiesis. Biomaterials 2020, 232, 119665. [Google Scholar] [CrossRef] [PubMed]
- Isern, J.; Martín-Antonio, B.; Ghazanfari, R.; Martín, A.M.; López, J.A.; del Toro, R.; Sánchez-Aguilera, A.; Arranz, L.; Martín-Pérez, D.; Suárez-Lledó, M.; et al. Self-Renewing Human Bone Marrow Mesenspheres Promote Hematopoietic Stem Cell Expansion. Cell Rep. 2012, 3, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Jing, D.; Fonseca, A.; Alakel, N.; Fierro, F.A.; Muller, K.; Bornhauser, M.; Ehninger, G.; Corbeil, D.; Ordemann, R. Hematopoietic stem cells in co-culture with mesenchymal stromal cells-modeling the niche compartments in vitro. Haematologica 2010, 95, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.M.; Gars, E.J.; James, D.J.; Nolan, D.J.; Scandura, J.M.; Rafii, S. Development of a vascular niche platform for expansion of repopulating human cord blood stem and progenitor cells. Blood 2012, 120, 1344–1347. [Google Scholar] [CrossRef] [Green Version]
- Leisten, I.; Kramann, R.; Ventura Ferreira, M.S.; Bovi, M.; Neuss, S.; Ziegler, P.; Wagner, W.; Knüchel, R.; Schneider, R.K. 3D co-culture of hematopoietic stem and progenitor cells and mesenchymal stem cells in collagen scaffolds as a model of the hematopoietic niche. Biomaterials 2012, 33, 1736–1747. [Google Scholar] [CrossRef]
- Raic, A.; Rödling, L.; Kalbacher, H.; Lee-Thedieck, C. Biomimetic macroporous PEG hydrogels as 3D scaffolds for the multiplication of human hematopoietic stem and progenitor cells. Biomaterials 2014, 35, 929–940. [Google Scholar] [CrossRef]
- Severn, C.E.; Macedo, H.; Eagle, M.J.; Rooney, P.; Mantalaris, A.; Toye, A.M. Polyurethane scaffolds seeded with CD34+ cells maintain early stem cells whilst also facilitating prolonged egress of haematopoietic progenitors. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mahadik, B.P.; Bharadwajb, N.A.K.; Ewoldtb, R.H.; Harley, B.A.C. Regulating dynamic signaling between hematopoietic stem cells and niche cells via a hydrogel matrix. Biomaterials 2017, 125, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, A.C.; Ishida, R.; Kikuchi, M.; Sudo, K.; Morita, M.; Crisostomo, R.V.; Yamamoto, R.; Loh, K.M.; Nakamura, Y.; Watanabe, M.; et al. Long-term ex vivo hematopoietic stem cell expansion affords nonconditioned transplantation. Nature 2019, 571, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Sieber, S.; Wirth, L.; Cavak, N.; Koenigsmark, M.; Marx, U.; Lauster, R.; Rosowski, M. Bone marrow-on-a-chip: Long-term culture of human haematopoietic stem cells in a three-dimensional microfluidic environment. J. Tissue Eng. Regen. Med. 2018, 12, 479–489. [Google Scholar] [CrossRef]
- Bourgine, P.E.; Klein, T.; Paczulla, A.M.; Shimizu, T.; Kunz, L.; Kokkaliaris, K.D.; Coutu, D.L.; Lengerke, C.; Skoda, R.; Schroeder, T.; et al. In vitro biomimetic engineering of a human hematopoietic niche with functional properties. Proc. Natl. Acad. Sci. USA 2018, 115, E5688–E5695. [Google Scholar] [CrossRef] [Green Version]
- De la Puente, P.; Muz, B.; Gilson, R.C.; Azab, F.; Luderer, M.; King, J.; Achilefu, S.; Vij, R.; Azab, A.K. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials 2015, 73, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Torisawa, Y.S.; Spina, C.S.; Mammoto, T.; Mammoto, A.; Weaver, J.C.; Tat, T.; Collins, J.J.; Ingber, D.E. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat. Methods 2014, 11, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Reinisch, A.; Hernandez, D.C.; Schallmoser, K.; Majeti, R. Generation and use of a humanized bone marrow ossicle niche for hematopoietic xenotransplantation into mice. Nat. Protoc. 2017, 12, 2169–2188. [Google Scholar] [CrossRef]
- Theocharides, A.P.A.; Rongvaux, A.; Fritsch, K.; Flavell, R.A.; Manz, M.G. Humanized hemato-lymphoid system mice. Haematologica 2016, 101, 5–19. [Google Scholar] [CrossRef]
- Abarrategi, A.; Mian, S.A.; Passaro, D.; Rouault-Pierre, K.; Grey, W.; Bonnet, D. Modeling the human bone marrow niche in mice: From host bone marrow engraftment to bioengineering approaches. J. Exp. Med. 2018, 215, 729–743. [Google Scholar] [CrossRef] [Green Version]
- Rose-Zerilli, M.J.J.; Gibson, J.; Wang, J.; Tapper, W.; Davis, Z.; Parker, H.; Larrayoz, M.; McCarthy, H.; Walewska, R.; Forster, J.; et al. Longitudinal copy number, whole exome and targeted deep sequencing of “good risk” IGHV-mutated CLL patients with progressive disease. Leukemia 2016, 30, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinisch, A.; Thomas, D.; Corces, M.R.; Zhang, X.; Gratzinger, D.; Hong, W.J.; Schallmoser, K.; Strunk, D.; Majeti, R. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat. Med. 2016, 22, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Vaiselbuh, S.R.; Edelman, M.; Lipton, J.M.; Liu, J.M. Ectopic human mesenchymal stem cell-coated scaffolds in NOD/SCID mice: An in vivo model of the leukemia niche. Tissue Eng. Part C Methods 2010, 16, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Groen, R.W.J.; Noort, W.A.; Raymakers, R.A.; Prins, H.J.; Aalders, L.; Hofhuis, F.M.; Moerer, P.; Van Velzen, J.F.; Bloem, A.C.; Van Kessel, B.; et al. Reconstructing the human hematopoietic niche in immunodeficient mice: Opportunities for studying primary multiple myeloma. Blood 2012, 120, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jacamo, R.; Shi, Y.X.; Wang, R.Y.; Battula, V.L.; Konoplev, S.; Strunk, D.; Hofmann, N.A.; Reinisch, A.; Konopleva, M.; et al. Human extramedullary bone marrow in mice: A novel in vivo model of genetically controlled hematopoietic microenvironment. Blood 2012, 119, 4971–4980. [Google Scholar] [CrossRef] [Green Version]
- Holzapfel, B.M.; Hutmacher, D.W.; Nowlan, B.; Barbier, V.; Thibaudeau, L.; Theodoropoulos, C.; Hooper, J.D.; Loessner, D.; Clements, J.A.; Russell, P.J.; et al. Tissue engineered humanized bone supports human hematopoiesis in vivo. Biomaterials 2015, 61, 103–114. [Google Scholar] [CrossRef]
- Bourgine, P.E.; Fritsch, K.; Pigeot, S.; Takizawa, H.; Kunz, L.; Kokkaliaris, K.D.; Coutu, D.L.; Manz, M.G.; Martin, I.; Schroeder, T. Fate distribution and regulatory role of human mesenchymal stromal cells in engineered hematopoietic bone organs. Science 2019, 19, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Abarrategi, A.; Foster, K.; Hamilton, A.; Mian, S.A.; Passaro, D.; Gribben, J.; Mufti, G.; Bonnet, D. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J. Clin. Investig. 2017, 127, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Antonelli, A.; Noort, W.A.; Jaques, J.; De Boer, B.; De Jong-Koolar, R.; Brouwers-Vos, R.; Lubbers-Alders, L.; Van Velzen, J.F.; Bloem, A.C.; Yuan, H.; et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow-like scaffol-based niches. Blood 2016, 128, 2949–2959. [Google Scholar] [CrossRef] [Green Version]
- Nefedova, Y.; Landowski, T.H.; Dalton, W.S. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia 2003, 17, 1175–1182. [Google Scholar] [CrossRef] [Green Version]
- Ibraheem, A.; Attar-schneider, O.; Dabbah, M.; Jarchowsky, O.D.; Matalon, S.T.; Lishner, M.; Drucker, L.; Saba, K.; Aviv, T.E.L. BM-MSCs-derived ECM modifies multiple myeloma phenotype and drug response in a source-dependent manner. Transl. Res. 2019, 207, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lin, T.L.; Lipe, B.; Hopkins, R.A.; Shinogle, H.; Aljitawi, O.S. A novel extracellular matrix-based leukemia model supports leukemia cells with stem cell-like characteristics. Leuk. Res. 2018, 72, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Blanco, T.M.; Mantalaris, A.; Bismarck, A.; Panoskaltsis, N. The development of a three-dimensional scaffold for ex vivo biomimicry of human acute myeloid leukaemia. Biomaterials 2010, 31, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lin, T.L.; Zhang, D.; Li, L.; Hopkins, R.A.; Stehno-Bittel, L.; Aljitawi, O.S. Resistance to chemotherapy in leukemia cells grown on an extracellular matrix-based leukemia model derived from Wharton’s Jelly. Blood 2013, 201. [Google Scholar] [CrossRef]
- Li, Z.W.; Dalton, W.S. Tumor microenvironment and drug resistance in hematologic malignancies. Blood Rev. 2006, 20, 333–342. [Google Scholar] [CrossRef]
- Khaldoyanidi, S.K.; Goncharova, V.; Mueller, B.; Schraufstatter, I.U. Hyaluronan in the healthy and malignant hematopoietic microenvironment. In Advances in Cancer Research; Elsevier Inc.: Amsterdam, The Netherlands, 2014; pp. 149–189. [Google Scholar] [CrossRef]
- Li, D.; Chiu, G.; Lipe, B.; Hopkins, R.A.; Lillis, J.; Ashton, J.M.; Paul, S.; Aljitawi, O.S. Decellularized Wharton jelly matrix: A biomimetic scaffold for ex vivo hematopoietic stem cell culture. Blood Adv. 2019, 3, 1011–1026. [Google Scholar] [CrossRef] [Green Version]
- Katz, B. Seminars in Cancer Biology Adhesion molecules—The lifelines of multiple myeloma cells. Semin. Cancer Biol. 2010, 20, 186–195. [Google Scholar] [CrossRef]
- Dabbah, M.; Attar-schneider, O.; Matalon, S.T.; Shefler, I.; Dolberg, O.J.; Lishner, M.; Drucker, L. Microvesicles derived from normal and multiple myeloma bone marrow mesenchymal stem cells differentially modulate myeloma cells ’ phenotype and translation initiation. Carcinogenesis 2017, 38, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Attar-schneider, O.; Zismanov, V.; Dabbah, M.; Tartakover-matalon, S. Multiple Myeloma and Bone Marrow Mesenchymal Stem Cells’ crosstalk: Effect on translation initiation. Mol. Carcinog. 2015, 1–12. [Google Scholar] [CrossRef]
- Marcus, H.; Attar-Schneider, O.; Dabbah, M.; Zismanov, V.; Matalon, S.T.; Lishner, M.; Drucker, L. Mesenchymal stem cells secretomes’ affect multiple myeloma translation initiation. Cell. Signal. 2016. [Google Scholar] [CrossRef]
- Jakubikova, J.; Cholujova, D.; Hideshima, T.; Gronesova, P.; Szalat, E.; Richardson, P.G.; Munshi, N.C.; Dorfman, D.M. A novel 3D mesenchymal stem cell model of the multiple myeloma bone marrow niche: Biologic and clinical applications. Oncotarget 2016, 7, 77326–77341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Wolanska, K.I.; Morgan, M.R. Fibronectin remodelling: Cell-mediated regulation of the microenvironment. Biochem. Soc. Trans. 2015, 43, 122–128. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, H.; Sahai, E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 2018, 20, 766–774. [Google Scholar] [CrossRef]
- Madl, C.M.; Lesavage, B.L.; Dewi, R.E.; Dinh, C.B.; Stowers, R.S.; Khariton, M.; Lampe, K.J.; Nguyen, D.; Chaudhuri, O.; Enejder, A.; et al. 3D hydrogels requires matrix remodelling. Nat. Mater. 2017, 16. [Google Scholar] [CrossRef] [Green Version]
- Clara-Trujillo, S.; Marín-Payá, J.C.; Cordón, L.; Sempere, A.; Gallego Ferrer, G.; Gómez Ribelles, J.L. Biomimetic microspheres for 3D mesenchymal stem cell culture and characterization. Colloids Surf. B Biointerfaces 2019, 177. [Google Scholar] [CrossRef]
- Salmerón-sánchez, M.; Rico, P.; Moratal, D.; Lee, T.T.; Schwarzbauer, J.E.; García, A.J. Role of material-driven fibronectin fibrillogenesis in cell differentiation. Biomaterials 2011, 32, 2099–2105. [Google Scholar] [CrossRef]
- Karamichos, D.; Skinner, J.; Brown, R.; Mudera, V. Matrix stiffness and serum concentration effects matrix remodelling and ECM regulatory genes of human bone marrow stem cells. J. Tissue Eng. Regen. Med. 2008, 97–105. [Google Scholar] [CrossRef]
- Haase, K.; Kamm, R.D. Advances in on-chip vascularization. Regen. Med. 2017, 12, 285–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, R.Y.; Salacinski, H.J.; Sales, K.; Butler, P.; Seifalian, A.M. The roles of tissue engineering and vascularisation in the development of micro-vascular networks: A review. Biomaterials 2005, 26, 1857–1875. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Sudo, R.; Mack, P.J.; Wan, C.; Kamm, R.D. Cell migration into scaffolds under co-culture conditions in a microfluidic platform. Lab Chip 2009, 9, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Moya, M.L.; Hsu, Y.; Lee, A.P.; Hughes, C.C.W.; George, S.C. In vitro perfused human capillary networks. Tissue Eng. Part C Methods 2013, 19, 730–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, L.; Angonin, R.; Regnard, J.; Fellmann, D.; Hospital, A.M.; Pathologique, A.; Unite, P.C. Human bone marrow angiogenesis: In vitro modulation by substance P and neurokinin A. Br. J. Hematol. 2002, 119, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.S.; Kaushansky, K.; Zhan, H. JAK2V617F-mutant vascular niche contributes to JAK2V617F clonal expansion in myeloproliferative neoplasms. Blood Cells Mol. Dis. 2016, 62, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Stouffer, R.L.; Wolf, D.P. Creation of long-lasting blood vessels. Nature 2004, 428, 138–139. [Google Scholar] [CrossRef]
- Laroche, M.; Brousset, P.; Ludot, I.; Mazières, B.; Thiechart, M.; Attal, M. Increased vascularization in myeloma. Eur. J. Haematol. 2001, 66, 89–93. [Google Scholar] [CrossRef]
- Lim, S.T.; Levine, A.M. Angiogenesis and hematological malignancies. Hematology 2013, 8454. [Google Scholar] [CrossRef]
- De la Puente, P.; Azab, A.K. 3D tissue-engineered bone marrow: What does this mean for the treatment of multiple myeloma? Future Oncol. 2016, 12, 1545–1547. [Google Scholar] [CrossRef]
- Ouyang, L.; Armstrong, J.P.K.; Salmeron-sanchez, M.; Stevens, M.M. Assembling living building blocks to engineer complex tissues. Adv. Funct. Mater. 2020, 1909009, 1–22. [Google Scholar] [CrossRef]
- Costa, M.H.G.; de Soure, A.M.; Cabral, J.M.S.; Ferreira, F.C.; da Silva, C.L. Hematopoietic niche–exploring biomimetic cues to improve the functionality of Hematopoietic Stem/ Progenitor Cells. Biotechnol. J. 2017, 13. [Google Scholar] [CrossRef]
- Mahadik, B.P.; Pedron Haba, S.; Skertich, L.J.; Harley, B.A.C. The use of covalently immobilized stem cell factor to selectively affect hematopoietic stem cell activity within a gelatin hydrogel. Biomaterials 2015, 67, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschling, S.; Saffrich, R.; Seckinger, A.; Krause, U.; Horsch, K.; Miesala, K.; Ho, A. Human Mesenchymal stromal cells regulate initial self-renewing divisions of hematopoietic progenitor cells by a β 1-Integrin. Stem Cells 2007, 25, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Sart, S.; Agathos, S.N.; Li, Y. Engineering Stem cell fate with biochemical and biomechanical properties of microcarriers. Biotechnol. Prog. 2013, 29, 1354–1366. [Google Scholar] [CrossRef]
- Birmingham, E.; Niebur, G.L.; McHugh, P.E.; Shaw, G.; Barry, F.P.; McNamara, L.M. Osteogenic diferentiation of mesenchymal stem cells is regulated by osteocytte and osteoblast cells in simplified bone niche. Eur. Cells Mater. 2012, 23. [Google Scholar] [CrossRef]
- Persson, M.; Lehenkari, P.P.; Berglin, L.; Turun, S.; Finnilä, M.A.J.; Riste, J.; Skrifvars, M.; Tuukkanen, J. Osteogenic differentiation of human mesenchymal stem cells in a 3D woven scaffold. Sci. Rep. 2018, 1–12. [Google Scholar] [CrossRef]
- Anderson, H.J.; Sahoo, J.K.; Ulijn, R.V.; Dalby, M.J.; Anderson, H.J. Mesenchymal Stem Cell fate: Applying biomaterials for control of stem cell behavior. Front. Bioeng. Biotechnol. 2016, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Curran, J.M.; Stokes, R.; Irvine, E.; Graham, D.; Amro, N.A.; Sanedrin, R.G.; Jamil, H.; Hunt, J.A. Introducing dip pen nanolithography as a tool for controlling stem cell behaviour: Unlocking the potential of the next generation of smart materials in regenerative medicine. Lab Chip 2010, 10, 1662–1670. [Google Scholar] [CrossRef]
- Dalby, M.J.; Gadegaard, N.; Tare, R.; Andar, A.; O Riehle, M.; Herzyk, P.; Wilkinson, C.D.W.; Oreffo, R.O.C. The control of human mesenchymal cell differentiation using nanoscale symmetry and disorder. Nat. Mater. 2007, 6, 997–1003. [Google Scholar] [CrossRef]
- Fares, I.; Chagraoui, J.; Gareau, Y.; Gingras, S.; Ruel, R.; Csaszar, E.; Knapp, D.J.H.F.; Miller, P.; Ngom, M.; Imren, S.; et al. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 2014, 345, 1509–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawitan, J.A. Prospect of stem cell conditioned medium in regenerative medicine. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamir, M.; Bar-on, Y.; Phillips, R.; Milo, R. SnapShot: Timescales in Cell Biology. Cell 2016, 164, 1302. [Google Scholar] [CrossRef] [PubMed]
- Albeck, J.G.; Burke, J.M.; Spencer, S.L.; Lauffenburger, D.A.; Sorger, P.K. Modeling a Snap-Action, Variable-Delay Switch Controlling Extrinsic Cell Death. PLoS Biol. 2008, 6, 2831–2852. [Google Scholar] [CrossRef]
- Morgan, M.M.; Johnson, B.P.; Livingston, M.K.; Schuler, L.A.; Alarid, E.T.; Sung, K.E.; Beebe, D.J. Personalized in vitro cancer models to predict therapeutic response: Challenges and a framework for improvement. Pharmacol. Ther. 2016, 165, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Letai, A. Functional precision cancer medicine—Moving beyond pure genomics. Nat. Med. 2017, 23, 1028–1035. [Google Scholar] [CrossRef]
- Snijder, B.; Vladimer, G.I.; Krall, N.; Miura, K.; Schmolke, A.; Kornauth, C.; Lopez, O.; Fuente, D.; Ringler, A.; Sabler, M.; et al. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: Interim results from a single-arm, open-label, pilot study. Lancet Haematol. 2017, 4, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Pemovska, T.; Kontro, M.; Yadav, B.; Edgren, H.; Eldfors, S.; Szwajda, A.; Almusa, H.; Bespalov, M.M.; Ellonen, P.; Elonen, E.; et al. Individualized Systems Medicine Strategy to Tailor Treatments for Patients with Chemorefractory Acute Myeloid Leukemia. Cancer Discov. 2013, 1416–1429. [Google Scholar] [CrossRef] [Green Version]
- de Campos, C.B.; Meurice, N.; Petit, J.L.; Polito, A.N.; Zhu, Y.X.; Wang, P.; Bruins, L.A.; Wang, X.; Armenta, I.D.L.; Darvish, S.A.; et al. “Direct to Drug” screening as a precision medicine tool in multiple myeloma. Blood Cancer J. 2020. [Google Scholar] [CrossRef]
- Silva, A.; Silva, M.C.; Sudalagunta, P.; Distler, A.; Jacobson, T.; Collins, A.; Nguyen, T.; Song, J.; Chen, D.; Chen, L.; et al. An Ex Vivo Platform for the Prediction of Clinical Response in Multiple Myeloma. Cancer Res. 2017, 77, 3336–3352. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Farach-Carson, M.C.; Jia, X. Three-dimensional in vitro tumor models for cancer research and drug evaluation. Biotechnol. Adv. 2014, 32, 1256–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic. Nat. Rev. Cancer 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- McMurray, R.J.; Gadegaard, N.; Tsimbouri, P.M.; Burgess, K.V.; Mcnamara, L.E.; Tare, R.; Murawski, K.; Kingham, E.; Oreffo, R.O.C.; Dalby, M.J. Nanoscale surfaces for the long-term maintenance of mesenchymal stem cell phenotype and multipotency. Nat. Mater. 2011, 10. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Type of Model | Material and ECM Mimicry | Tumor Type (Cell Line/Cell Source) | Application or Description | Conclusions |
|---|---|---|---|---|---|
| [15] 2016 | Scaffold-free. Spheroids. | Methylcellulose (for spheroid formation). | Colorectal cancer. (Cell line CaCo2). | Comparative study of tumor related pathway signaling in planar vs. spheroid culture. | Spheroids present diminished AKT–mTOR–S6K signaling. mTOR activity and crosstalk between AKT–mTOR–S6K signaling and the MAPK pathway is altered in 3D cultures. Spheroids present in vivo-like mTOR-S6 signaling gradients. |
| [19] 2017 | Scaffold-free. Patient-derived organoids (tumoroids). | Basement membrane extract. Matrigel. | Primary liver cancer. (Cell source: patient derived cells from 3 tumor subtypes; hepatocellular carcinoma, cholangiocarcinoma and combined). | Validation of patient-derived organoids as preclinical personalized cancer model. Identification of potential prognostic biomarkers and patient-specific drug sensitivities. | Primary liver cancer-derived organoids preserve features of native tumor and subtype in vitro. Tumorigenic and metastatic potential are preserved in vivo (xenograft implantation). Amenable systems for biomarker identification and drug testing. Identification of the ERK inhibitor SCH772984 as a potential therapeutic agent. |
| [21] 2017 | Scaffold-based. | Poly-ether- urethane foam. | Breast cancer. Bone metastasis model. (Cell source: human ADSC and MCFS). | In vitro model to recapitulate the metastatic spreading of breast cancer in bone tissue. | Importance of osteoblasts in mediating adhesion and growth of breast cancer cells. MCFS proliferate and form aggregates in the co-culture biomimetic model. MCFS affects Ca and P deposition in the bone mimetic tissue. |
| [22] 2019 | Hydrogel-based. 3D bioprinting. | Gelatin methacryloyl UV cross-linked. | Bladder cancer. (Cell lines: 5637 and T24). | Development of a 3D environment for tumor formation and chemotherapy response characterization. | 3D cultures showed higher cell proliferation and cell-cell interactions (E and N-cadherin expression). 3D cultures showed diminished response to rapamycin and Bacillus Calmette-Guérin. |
| [24] 2019 | Scaffold-based. Spheroids. Microfluidics. | Polystyrene scaffold. Microfluidics. Poly-l-lysine and laminin-1 coating. | Breast and lung carcinoma. (Cell source: 3D tumor spheroids displaying CSC-like features from breast (MCF-7) and lung (A549) cancer cell lines). | Reproduction of the adhesion process of CSC to a target tissue by using a 3D dynamic cell culture system. | Development of a 3D dynamic model to study metastasis processes, such as formation of premetastatic niche and attachment of circulating tumor cells. |
| [23] 2019 | Tumor-on-a-chip. Microfluidics. | PDMS. Matrigel. | Colorectal cancer. (Cell source: human colon cancer cell line HCT-116 and HCoMECs). | In vitro 3D microfluidic cell culture for studying onco-nanomedicine efficacy. | Validation of model with tumor core supported by adjacent microvasculature compatible with real-time image analysis, gradient-like response on cancer cells, supports a stable and viable co-culture of HCoMECs and HCT- 116 and gene expression analysis. |
| Reference | Factors of Mimicry | Cellular Component | Biomaterial | Achievements |
|---|---|---|---|---|
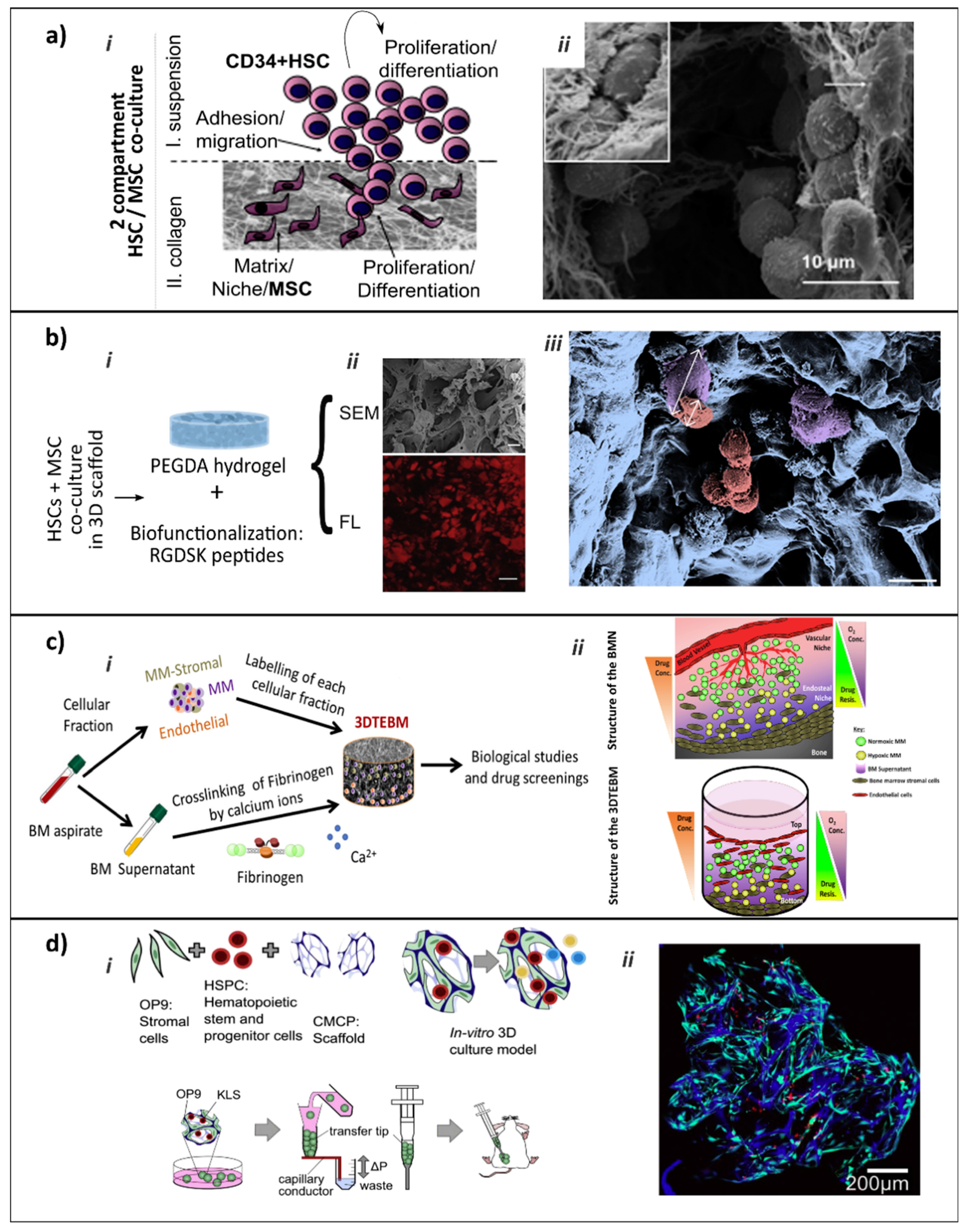
| [69] (Figure 4a) | Cell-cell and cell-ECM interactions. BMN Compartmentalization. | Human MSCs from UC or BM. HSPCs from UC blood. | COL I /III based hydrogels. | 3D co-culture system resembles the EBMN and dissects two sub-populations of HSPCs: (I) highly proliferative with tendency to lineage commitment and (II) with clonal expansion and immature phenotype with self-renewal and repopulation capacity. |
| [70] (Figure 4b) | Cell-cell and cell-ECM interactions. | Human HSPCs from UC blood. BMN-forming cells: MSCs (from BM and UC) and OBs cell line CAL-72. | PEGDA hydrogel mimicking trabecular bone. Adhesive peptides. | Co-culture showed more pronounced positive effect of MSCs on preservation of HSPCs stemness in 3D than 2D. Bio-functionalization offers adhesive sites, supplemented medium provides soluble factors, MSCs reflect the supporting stromal cell compartment. |
| [71] | Cell-ECM interactions. BMN Compartmentalization. | Human CD34+ cells from adult peripheral blood. | PU scaffold with honeycomb structure. | Compartmentalized scaffolds allow harvest HSCs across longer periods. Continuous egress of cells with an erythroid progenitor phenotype over a 28 days period. Maintenance of CD34+ population, while facilitating egress of increasingly differentiated cells. |
| [72] | Cell-cell and cell-ECM interactions. Biotransport. | Murine BM derived Lin−Sca1+cKit+ (LSK) sub-fraction and Lin+ BMN-forming cells. | Cell-laden COL I hydrogels with varying densities. | Co-variation of hydrogel diffusivity and BMN-forming cell density controls HSCs proliferation vs. differentiation by varying autocrine vs. paracrine signaling. Biotransport limitations in 3D models as critical design element. |
| [73] | Cell-ECM interactions. Soluble factors improved presentation. | Murine ckit+ enriched HSCs cells from BM. | PVA. FN as 2D coating for HSCs retention. Soluble TPO and SCF. | Ex vivo platform for long-term HSCs expansion. Affords 1-month expansion of functional HSCs. Cultures derived robustly engrafted in recipients without requirement for toxic pre-conditioning, suggesting new approaches for HSC transplantation. |
| Reference | Approach | Achievements |
|---|---|---|
| [74] | Hydroxyapatite-coated ceramic cancellous bone mimicking scaffold, pre-culture with primary BM-MSCs inducing ECM deposition and factor secretion. Co-culture with HSPCs from UC blood developed in the MOC platform [69]. | Long-term culture of HSPCs. MSCs generated microenvironment conducts HSC maintenance. HSPCs remain their native state after 4-weeks culture in dynamic conditions in the perfused MOC and retain multi-lineage differentiation potential. MOC platform allows co-culture with different organoids in adjacent chambers |
| [75] | Bone-like ceramic scaffold, functionalized by human stromal cells and by the ECM they deposited during perfusion culture in bioreactors. | Perfusion-based bioreactor system, partially recapitulating structural, compositional and organizational features of EBMN. Support of HSPCs maintenance and expansion in vitro with preserved multilineage reconstitution potential. Functional compartmentalization. Possibility to exploit the system for study BMN with customized molecular signatures. |
| [76] (Figure 4c) | Micro-scale 3DTEBM cultures derived from the BM supernatant of MM patients. Different BM cellular components (MM cells, BMN cells, and ECs). Cross-linked fibrinogen scaffold. | 3DTEBM cultures allowed proliferation of MM cells, recapitulated their interaction with the microenvironment, recreated 3D aspects of BMN (such as oxygen gradients), and induced drug resistance in MM cells. |
| [77] | PDMS hollow compartment with a COL I gel containing bone-inducing DBP, BMP2 and BMP4, implanted subcutaneously in mouse. Posteriorly explanted and maintained in vitro in a microfluidic device for 4 or 7 days. Functionality and responsiveness of the tested by exposing it to γ-radiation to determine whether this method could be used as an in vitro model for radiation toxicity. | Formation a cylindrical disk of cortical and trabecular bone containing marrow with a hematopoietic cell composition nearly identical to that of natural BM. Presence of key cellular and molecular components of BMN. During posterior 1-week in vitro culture retained morphology and molecular patterns, enabled maintenance of a significantly higher proportion of long-term HSCs while effectively maintaining distribution of mature blood cells. Mimicked physiological response to clinically relevant doses of γ-radiation. |
| [78] | Generation of humanized heterotopically localized bone organoid, “ossicles”, recapitulating normal BMN morphology and function. Ossicles formed in-situ by BM-MSCs ectopically implantation in mice. HSCs can subsequently be transplanted into the ossicle. Transplantation of normal and malignant HSCs. | Robust and reproducible in vivo methodology to study human normal and malignant hematopoiesis in a physiologic setting. Effectively engraftment of primary patient-derived AML and myelofibrosis cells in mice. Although bone, cartilage, and MSCs within the ossicle are of human origin, the vasculature is mouse derived. Limited applicability of the model to human specific questions. |
| [65] (Figure 4d) | Cryogel-based COL coated carboxymethylcellulose micro-scaffold seeded with murine BMN-forming cell line OP9 to generate a living, injectable stroma supportive for hematopoiesis, and with murine HPSCs. Seeded scaffolds act as microcarriers, enabling culture in vivo, when implanted ectopically in mice for vascularization. | Scaffolds promote hematopoietic cell proliferation over time, amenable to live, high-resolution imaging. Co-culture on chemically defined scaffold microcarriers. Simple and scalable. No exogenous cytokine supplementation. Stromal and hematopoietic cells able to survive in vivo for 12 weeks, showing incorporation into the native tissue via de novo vascularization. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clara-Trujillo, S.; Gallego Ferrer, G.; Gómez Ribelles, J.L. In Vitro Modeling of Non-Solid Tumors: How Far Can Tissue Engineering Go? Int. J. Mol. Sci. 2020, 21, 5747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165747
Clara-Trujillo S, Gallego Ferrer G, Gómez Ribelles JL. In Vitro Modeling of Non-Solid Tumors: How Far Can Tissue Engineering Go? International Journal of Molecular Sciences. 2020; 21(16):5747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165747
Chicago/Turabian StyleClara-Trujillo, Sandra, Gloria Gallego Ferrer, and José Luis Gómez Ribelles. 2020. "In Vitro Modeling of Non-Solid Tumors: How Far Can Tissue Engineering Go?" International Journal of Molecular Sciences 21, no. 16: 5747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165747