Enhanced Heterologous Production of Glycosyltransferase UGT76G1 by Co-Expression of Endogenous prpD and malK in Escherichia coli and Its Transglycosylation Application in Production of Rebaudioside

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Enhanced Soluble Expression of UGT76G1 in E. coli BL21 (DE3) by Fusion Partners
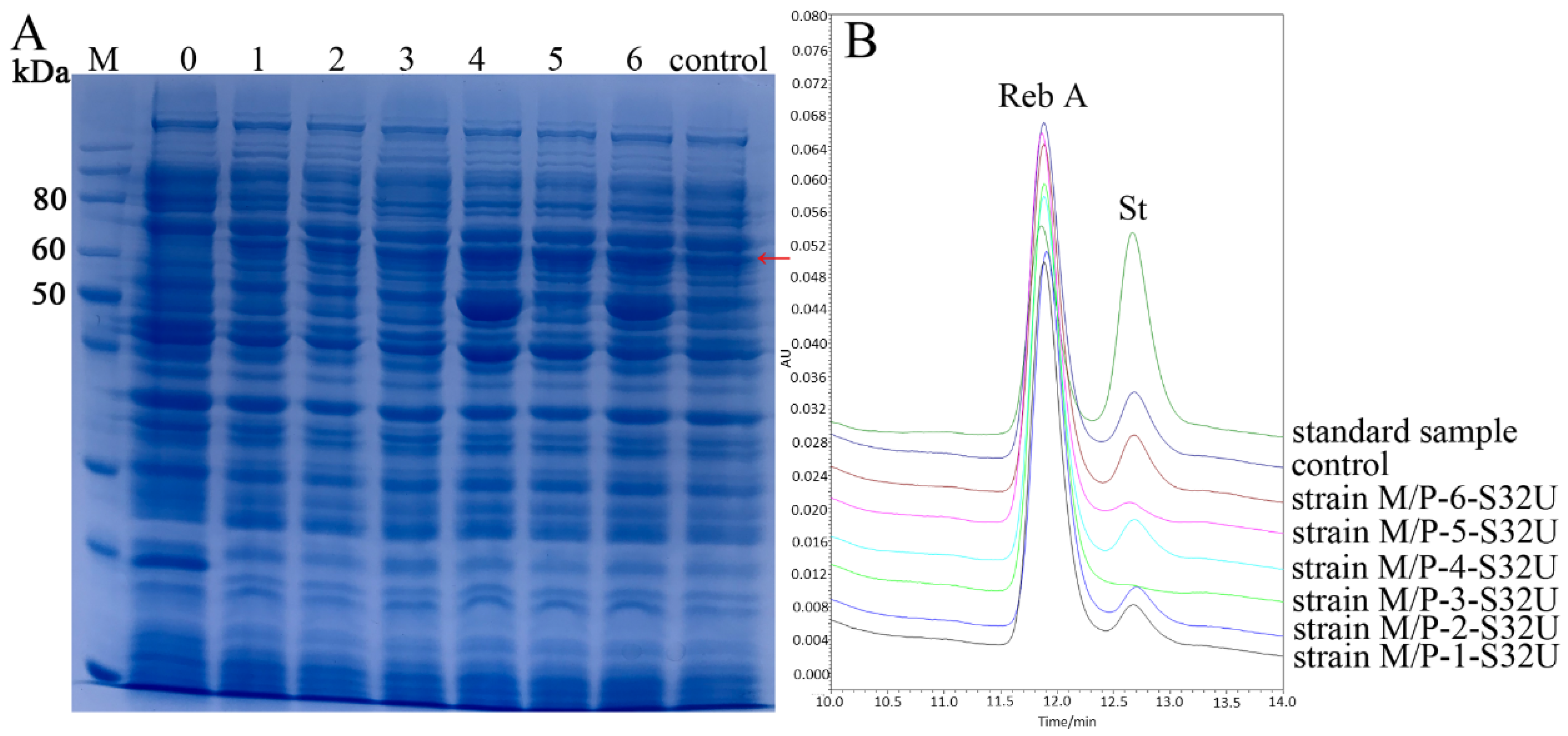
2.2. Overexpression of UGT76G1 by Co-Expression with prpD and malK
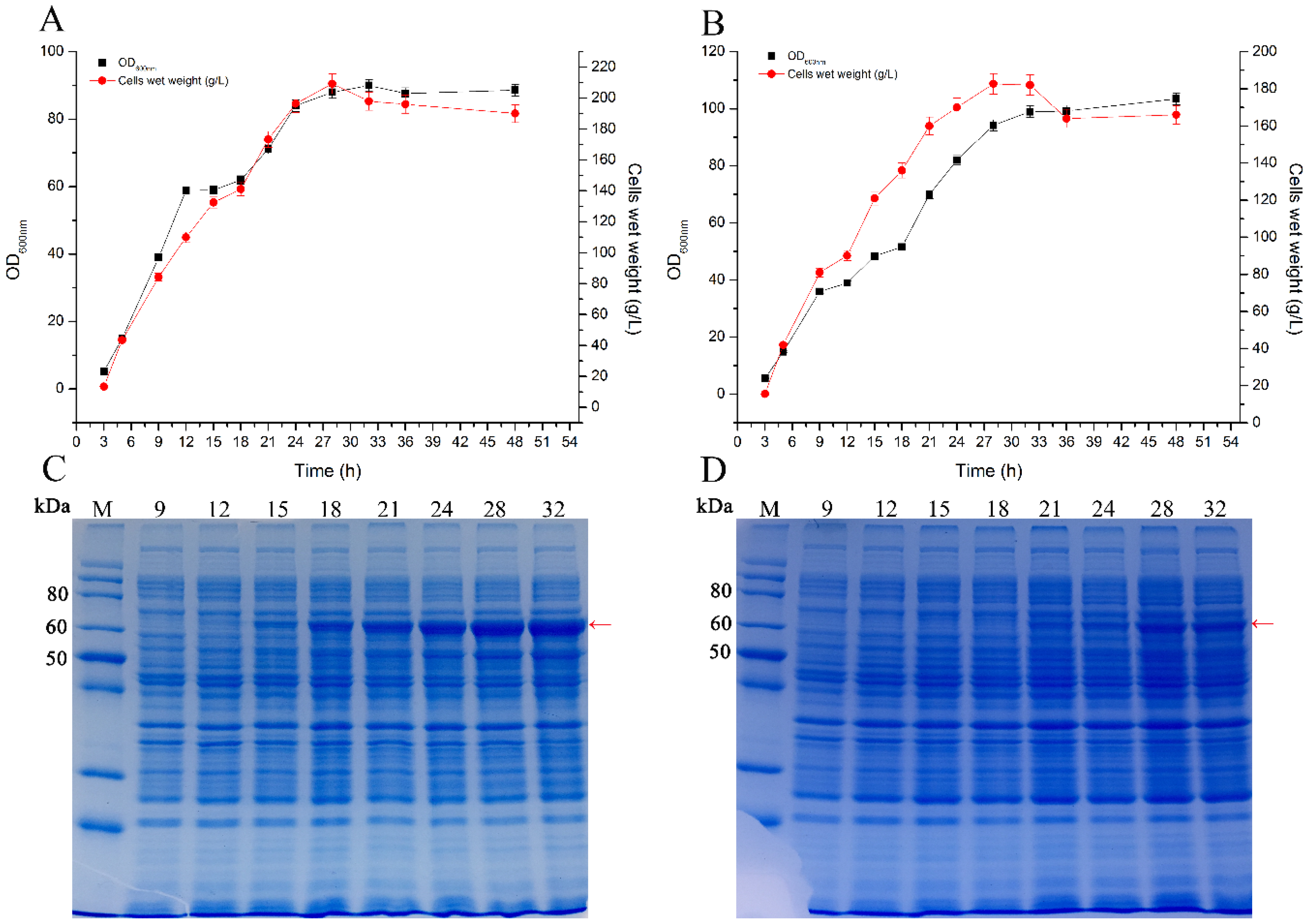
2.3. Scale-Up Production of Glycosyltransferase Smt3-UGT76G1 by Fed-Batch Fermentation in a 10 L Fermenter
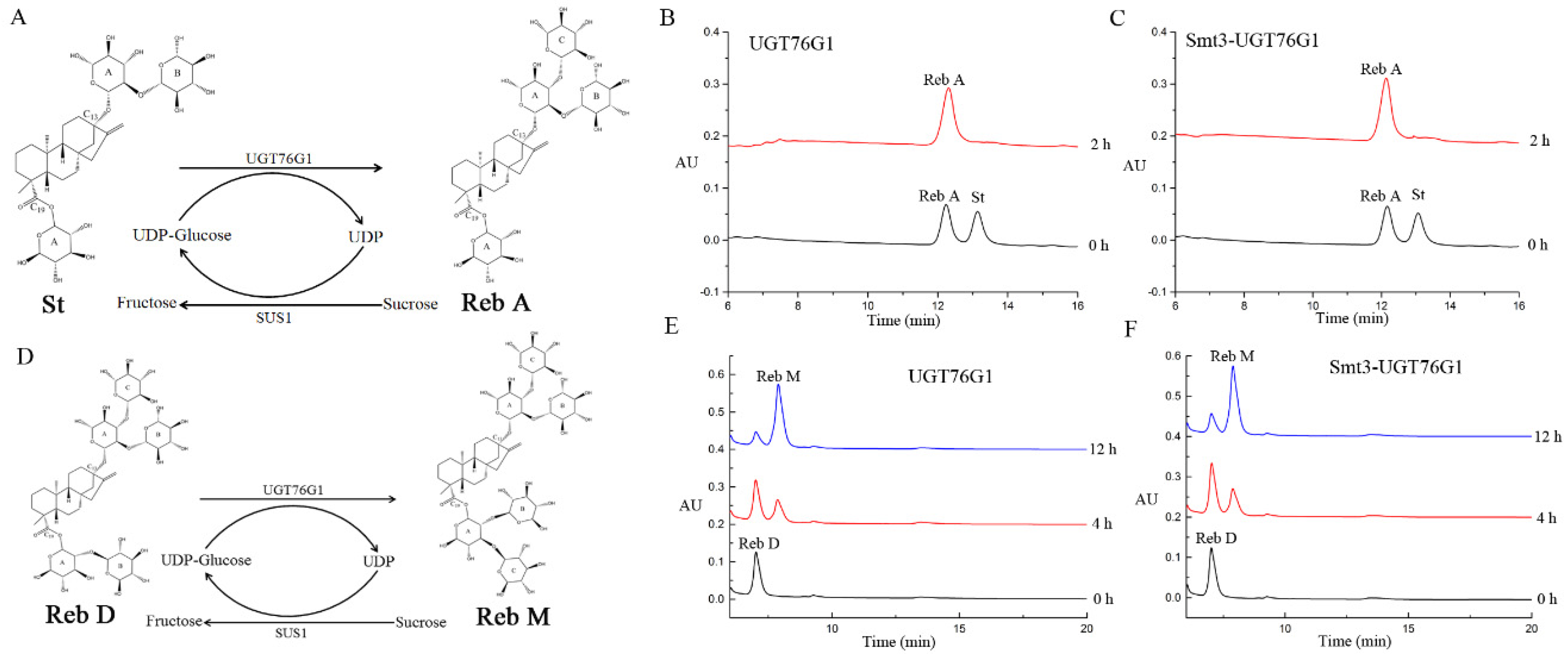
2.4. Biotransformation of SGs by the Recombinant UGT76G1 in Vitro Enzymatic Catalysis
3. Materials and Methods
3.1. Gene Cloning and Plasmids Construction for Fusion Expression
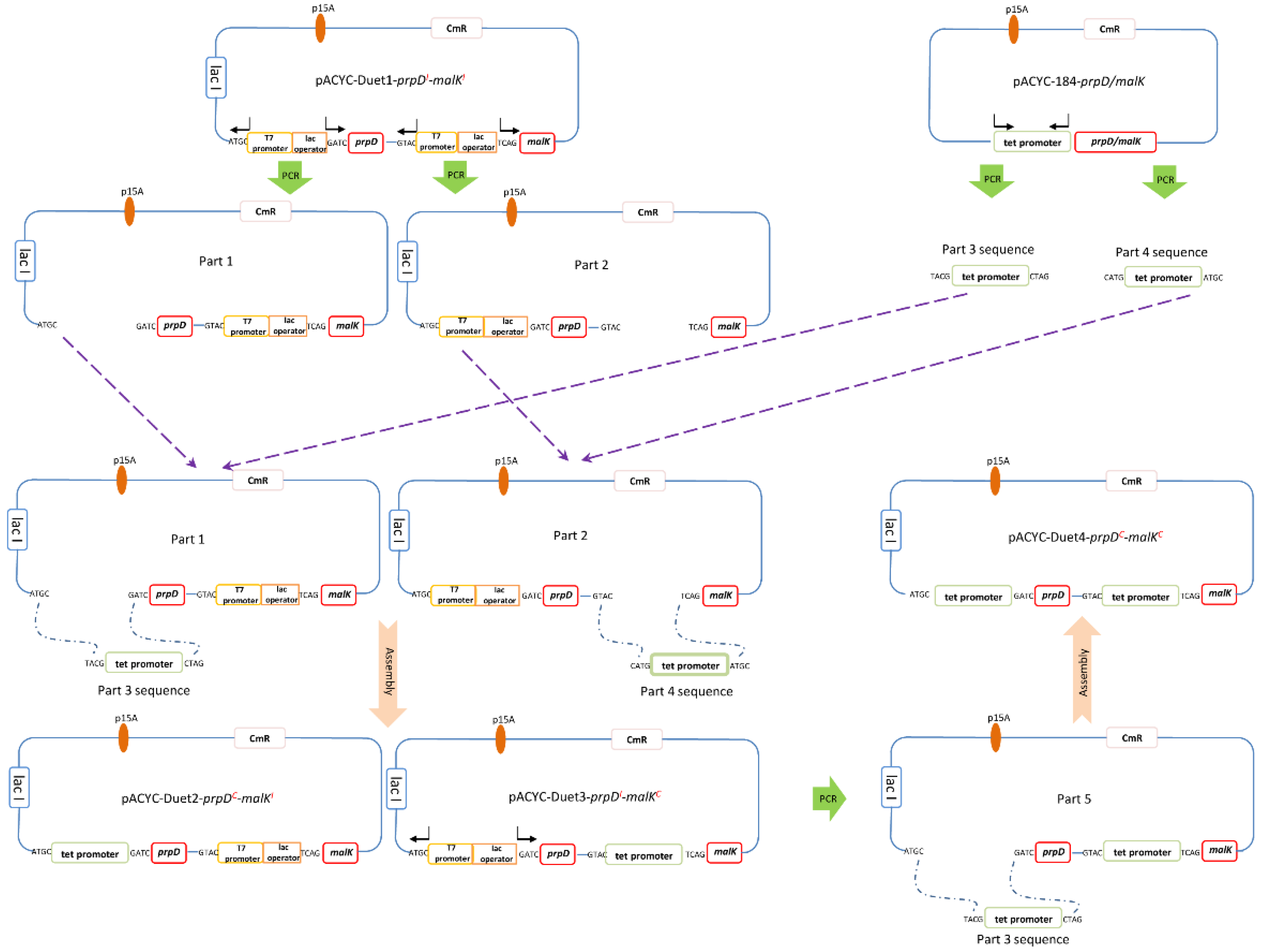
3.2. Construction of E. coli Overexpression System by Co-Expressing the Endogenous Genes prpD and malK
3.3. Cultivation of the Recombinant Strains
3.4. Preparation of the Recombinant Proteins and SDS-PAGE
3.5. Active Assay of Glycosyltransferase by HPLC
3.6. Enzymatic Biotransformation of SGs Using Recombinant UGT76G1 In Vitro
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| UGTs | Uridine diphosphate dependent glucosyltransferases |
| SGs | Steviol glycosides |
| MCSs | Multiple cloning sites |
| DO | Dissolved oxygen |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| IPTG | Isopropyl-β-d-thiogalactoside |
| PCR | Polymerase chain reaction |
| OD600nm | Optical density at 600 nm |
References
- Yang, T.; Zhang, J.; Ke, D.; Yang, W.; Tang, M.; Jiang, J.; Cheng, G.; Li, J.; Cheng, W.; Wei, Y.; et al. Hydrophobic recognition allows the glycosyltransferase UGT76G1 to catalyze its substrate in two orientations. Nat. Commun. 2019, 10, 3214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wang, L.; He, J.; Bi, Y.; Li, M.; Wang, T.; Wang, L.; Jiang, Y.; Dai, M.; Lu, J.; et al. Prevalence and control of diabetes in Chinese adults. JAMA 2013, 310, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of Childhood and Adult Obesity in the United States, 2011–2012. JAMA J. Am. Med. Assoc. 2014, 311, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelsalam, N.R.; Botros, W.A.; Khaled, A.E.; Ghonema, M.A.; Hussein, S.G.; Ali, H.M.; Elshikh, M.S. Comparison of uridine diphosphate-glycosyltransferase UGT76G1 genes from some varieties of Stevia rebaudiana Bertoni. Sci. Rep. 2019, 9, 8559. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.M.; Fetahu, I.S.; Wu, F.Z.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.Y.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Smith, M.; Tokuda, M. The role of artificial and natural sweeteners in reducing the consumption of table sugar: A narrative review. Clin. Nutr. ESPEN 2017, 18, 1–8. [Google Scholar] [CrossRef]
- Philippaert, K.; Pironet, A.; Mesuere, M.; Sones, W.; Vermeiren, L.; Kerselaers, S.; Pinto, S.; Segal, A.; Antoine, N.; Gysemans, C.; et al. Steviol glycosides enhance pancreatic beta-cell function and taste sensation by potentiation of TRPM5 channel activity. Nat. Commun. 2017, 8, 14733. [Google Scholar] [CrossRef]
- Plaza-Diaz, J.; Pastor-Villaescusa, B.; Rueda-Robles, A.; Abadia-Molina, F.; Ruiz-Ojeda, F.J. Plausible Biological Interactions of Low- and Non-Calorie Sweeteners with the Intestinal Microbiota: An Update of Recent Studies. Nutrients 2020, 12, 1153. [Google Scholar] [CrossRef]
- Adari, B.R.; Alavala, S.; George, S.A.; Meshram, H.M.; Tiwari, A.K.; Sarma, A.V. Synthesis of rebaudioside-A by enzymatic transglycosylation of stevioside present in the leaves of Stevia rebaudiana Bertoni. Food Chem. 2016, 200, 154–158. [Google Scholar] [CrossRef]
- Chen, L.; Sun, P.; Zhou, F.; Li, Y.; Chen, K.; Jia, H.; Yan, M.; Gong, D.; Ouyang, P. Synthesis of rebaudioside D, using glycosyltransferase UGTSL2 and in situ UDP-glucose regeneration. Food Chem. 2018, 259, 286–291. [Google Scholar] [CrossRef]
- Nidetzky, B.; Gutmann, A.; Zhong, C. Leloir Glycosyltransferases as Biocatalysts for Chemical Production. ACS Catal. 2018, 8, 6283–6300. [Google Scholar] [CrossRef]
- Yin, Q.G.; Han, X.Y.; Han, Z.X.; Chen, Q.F.; Shi, Y.H.; Gao, H.; Zhang, T.Y.; Dong, G.Q.; Xiong, C.; Song, C.; et al. Genome-wide analyses reveals a glucosyltransferase involved in rutin and emodin glucoside biosynthesis in tartary buckwheat. Food Chem. 2020, 318, 126478. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Liu, F.; Shao, W.; Chu, J.; Wu, B.; He, B. Efficient Synthesis of Crocins from Crocetin by a Microbial Glycosyltransferase from Bacillus subtilis 168. J. Agric. Food Chem. 2018, 66, 11701–11708. [Google Scholar] [CrossRef] [PubMed]
- Speeckaert, N.; Adamou, N.M.; Hassane, H.A.; Baldacci-Cresp, F.; Mol, A.; Goeminne, G.; Boerjan, W.; Duez, P.; Hawkins, S.; Neutelings, G.; et al. Characterization of the UDP-glycosyltransferase UGT72 Family in Poplar and Identification of Genes Involved in the Glycosylation of Monolignols. Int. J. Mol. Sci. 2020, 21, 5018. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, J.G.; Mu, S.C.; Shang, N.; Liu, C.; Zhu, Y.M.; Cai, Y.; Liu, P.; Lin, J.P.; Liu, W.D.; et al. Efficient O-Glycosylation of Triterpenes Enabled by Protein Engineering of Plant Glycosyltransferase UGT74AC1. ACS Catal. 2020, 10, 3629–3639. [Google Scholar] [CrossRef]
- Dewitte, G.; Walmagh, M.; Diricks, M.; Lepak, A.; Gutmann, A.; Nidetzky, B.; Desmet, T. Screening of recombinant glycosyltransferases reveals the broad acceptor specificity of stevia UGT-76G1. J. Biotechnol. 2016, 233, 49–55. [Google Scholar] [CrossRef]
- Lu, J.; Yao, L.; Li, J.X.; Liu, S.J.; Hu, Y.Y.; Wang, S.H.; Liang, W.X.; Huang, L.Q.; Dai, Y.J.; Wang, J.; et al. Characterization of UDP-Glycosyltransferase Involved in Biosynthesis of Ginsenosides Rg1 and Rb1 and Identification of Critical Conserved Amino Acid Residues for Its Function. J. Agric. Food Chem. 2018, 66, 9446–9455. [Google Scholar] [CrossRef]
- Olsson, K.; Carlsen, S.; Semmler, A.; Simon, E.; Mikkelsen, M.D.; Moller, B.L. Microbial production of next-generation stevia sweeteners. Microb. Cell Factories 2016, 15, 207. [Google Scholar] [CrossRef] [Green Version]
- Ghaheri, M.; Kahrizi, D.; Bahrami, G.; Mohammadi-Motlagh, H.R. Study of gene expression and steviol glycosides accumulation in Stevia rebaudiana Bertoni under various mannitol concentrations. Mol. Biol. Rep. 2019, 46, 7–16. [Google Scholar] [CrossRef]
- Lee, S.G.; Salomon, E.; Yu, O.; Jez, J.M. Molecular basis for branched steviol glucoside biosynthesis. Proc. Natl. Acad. Sci. USA 2019, 116, 13131–13136. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Sun, P.; Li, Y.; Yan, M.; Xu, L.; Chen, K.; Ouyang, P. A fusion protein strategy for soluble expression of Stevia glycosyltransferase UGT76G1 in Escherichia coli. 3 Biotech 2017, 7, 356. [Google Scholar] [CrossRef] [PubMed]
- Nik-Pa, N.I.M.; Sobri, M.F.M.; Abd-Aziz, S.; Ibrahim, M.F.; Kamal Bahrin, E.; Mohammed Alitheen, N.B.; Ramli, N. Combined Optimization of Codon Usage and Glycine Supplementation Enhances the Extracellular Production of a beta-Cyclodextrin Glycosyltransferase from Bacillus sp. NR5 UPM in Escherichia coli. Int. J. Mol. Sci. 2020, 21, 3919. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.B. Soluble expression of archaeal proteins in Escherichia coli by using fusion-partners. Protein Expr. Purif. 2008, 62, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.K.M.; Ki, M.R.; Son, R.G.; Pack, S.P. The NT11, a novel fusion tag for enhancing protein expression in Escherichia coli. Appl. Microbiol. Biot. 2019, 103, 2205–2216. [Google Scholar] [CrossRef]
- Yu, Z.; Zheng, H.; Zhao, X.; Li, S.; Xu, J.; Song, H. High level extracellular production of a recombinant alkaline catalase in E. coli BL21 under ethanol stress and its application in hydrogen peroxide removal after cotton fabrics bleaching. Bioresour. Technol. 2016, 214, 303–310. [Google Scholar] [CrossRef]
- Zheng, H.; Yu, Z.; Shu, W.; Fu, X.; Zhao, X.; Yang, S.; Tan, M.; Xu, J.; Liu, Y.; Song, H. Ethanol effects on the overexpression of heterologous catalase in Escherichia coli BL21 (DE3). Appl. Microbiol. Biotechnol. 2019, 103, 1441–1453. [Google Scholar] [CrossRef]
- Zhen, J.; Tan, M.; Fu, X.P.; Shu, W.J.; Zhao, X.Y.; Yang, S.B.; Xu, J.Y.; Ma, Y.H.; Zheng, H.C.; Song, H. High-level extracellular production of an alkaline pectate lyase in E. coli BL21 (DE3) and its application in bioscouring of cotton fabric. 3 Biotech 2020, 10, 49. [Google Scholar] [CrossRef]
- Pei, J.; Chen, A.; Zhao, L.; Cao, F.; Ding, G.; Xiao, W. One-Pot Synthesis of Hyperoside by a Three-Enzyme Cascade Using a UDP-Galactose Regeneration System. J. Agric. Food Chem. 2017, 65, 6042–6048. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Harboured Plasmids |
|---|---|
| E. coli BL21 (DE3) | |
| Strain M/P-1 | pACYC184-malK |
| Strain M/P-2 | pACYC184-prpD |
| Strain M/P-3 | pACYCDuet1-prpDI-malKI |
| Strain M/P-4 | pACYCDuet2-prpDC-malKI |
| Strain M/P-5 | pACYCDuet3-prpDI-malKC |
| Strain M/P-6 | pACYCDuet4-prpDC-malKC |
| Strain M/P-1-S32U | pACYC184-malK; pET-Smt3-32-UGT76G1 |
| Strain M/P-2-S32U | pACYC184-prpD; pET-Smt3-32-UGT76G1 |
| Strain M/P-3-S32U | pACYCDuet1-prpDI-malKI; pET-Smt3-32-UGT76G1 |
| Strain M/P-4-S32U | pACYCDuet2-prpDC-malKI; pET-Smt3-32-UGT76G1 |
| Strain M/P-5-S32U | pACYCDuet3-prpDI-malKC; pET-Smt3-32-UGT76G1 |
| Strain M/P-6-S32U | pACYCDuet4-prpDC-malKC; pET-Smt3-32-UGT76G1 |
| Strain S32U | pET-Smt3-32-UGT76G1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, W.; Zheng, H.; Fu, X.; Zhen, J.; Tan, M.; Xu, J.; Zhao, X.; Yang, S.; Song, H.; Ma, Y. Enhanced Heterologous Production of Glycosyltransferase UGT76G1 by Co-Expression of Endogenous prpD and malK in Escherichia coli and Its Transglycosylation Application in Production of Rebaudioside. Int. J. Mol. Sci. 2020, 21, 5752. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165752
Shu W, Zheng H, Fu X, Zhen J, Tan M, Xu J, Zhao X, Yang S, Song H, Ma Y. Enhanced Heterologous Production of Glycosyltransferase UGT76G1 by Co-Expression of Endogenous prpD and malK in Escherichia coli and Its Transglycosylation Application in Production of Rebaudioside. International Journal of Molecular Sciences. 2020; 21(16):5752. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165752
Chicago/Turabian StyleShu, Wenju, Hongchen Zheng, Xiaoping Fu, Jie Zhen, Ming Tan, Jianyong Xu, Xingya Zhao, Shibin Yang, Hui Song, and Yanhe Ma. 2020. "Enhanced Heterologous Production of Glycosyltransferase UGT76G1 by Co-Expression of Endogenous prpD and malK in Escherichia coli and Its Transglycosylation Application in Production of Rebaudioside" International Journal of Molecular Sciences 21, no. 16: 5752. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165752