Na+, K+-ATPase α Isoforms and Endogenous Cardiac Steroids in Prefrontal Cortex of Bipolar Patients and Controls

Abstract
:1. Introduction
2. Results
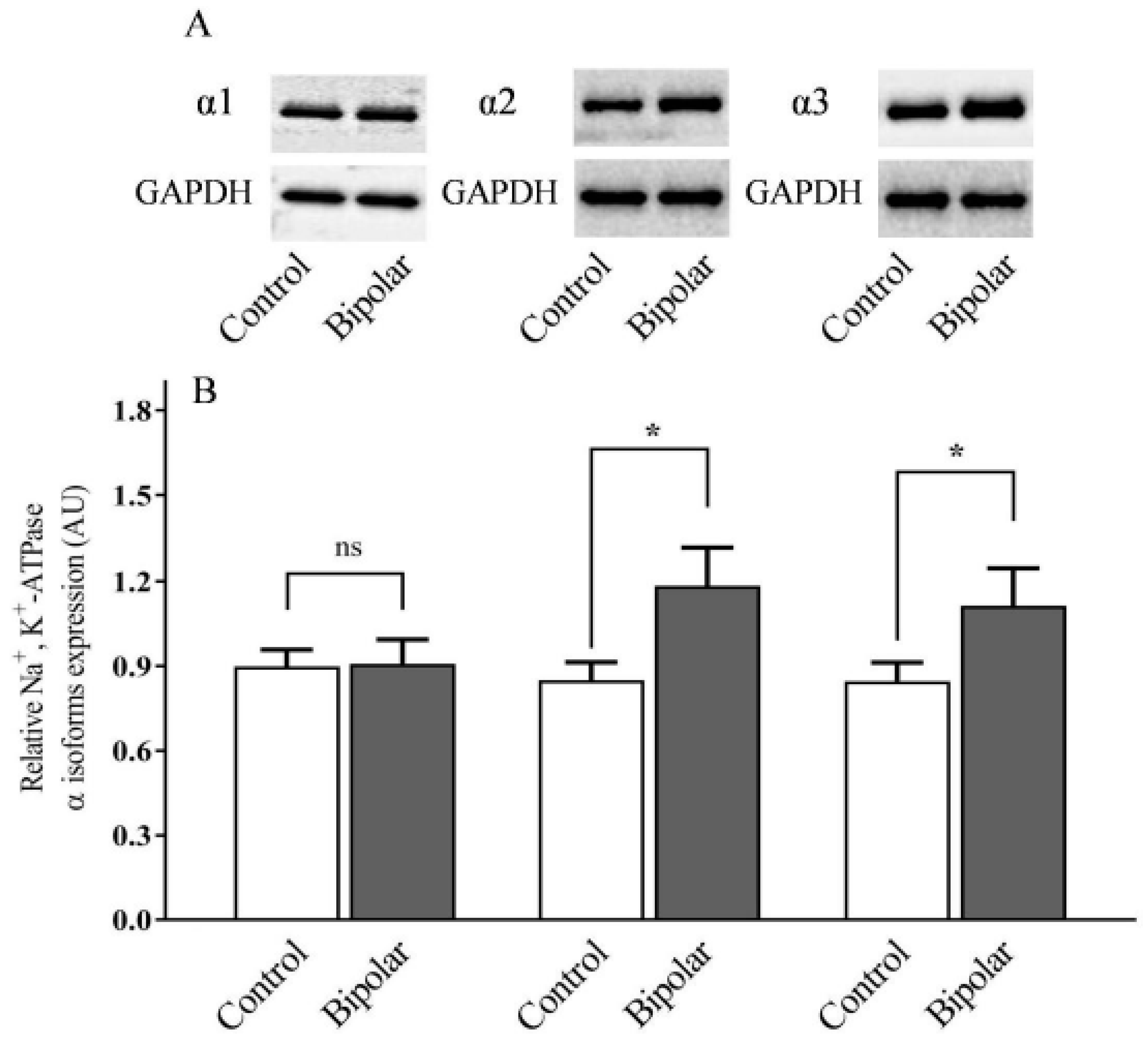
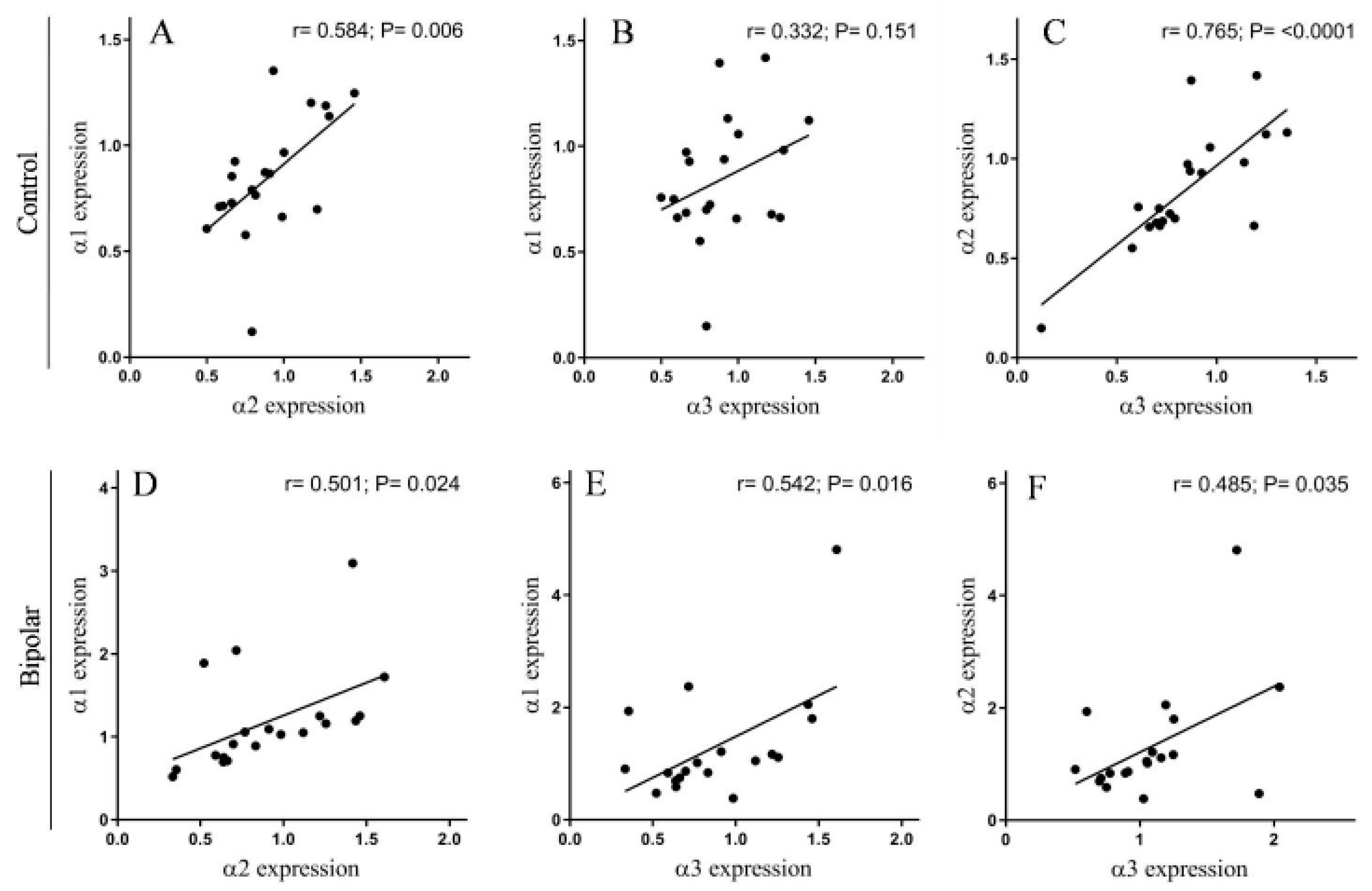
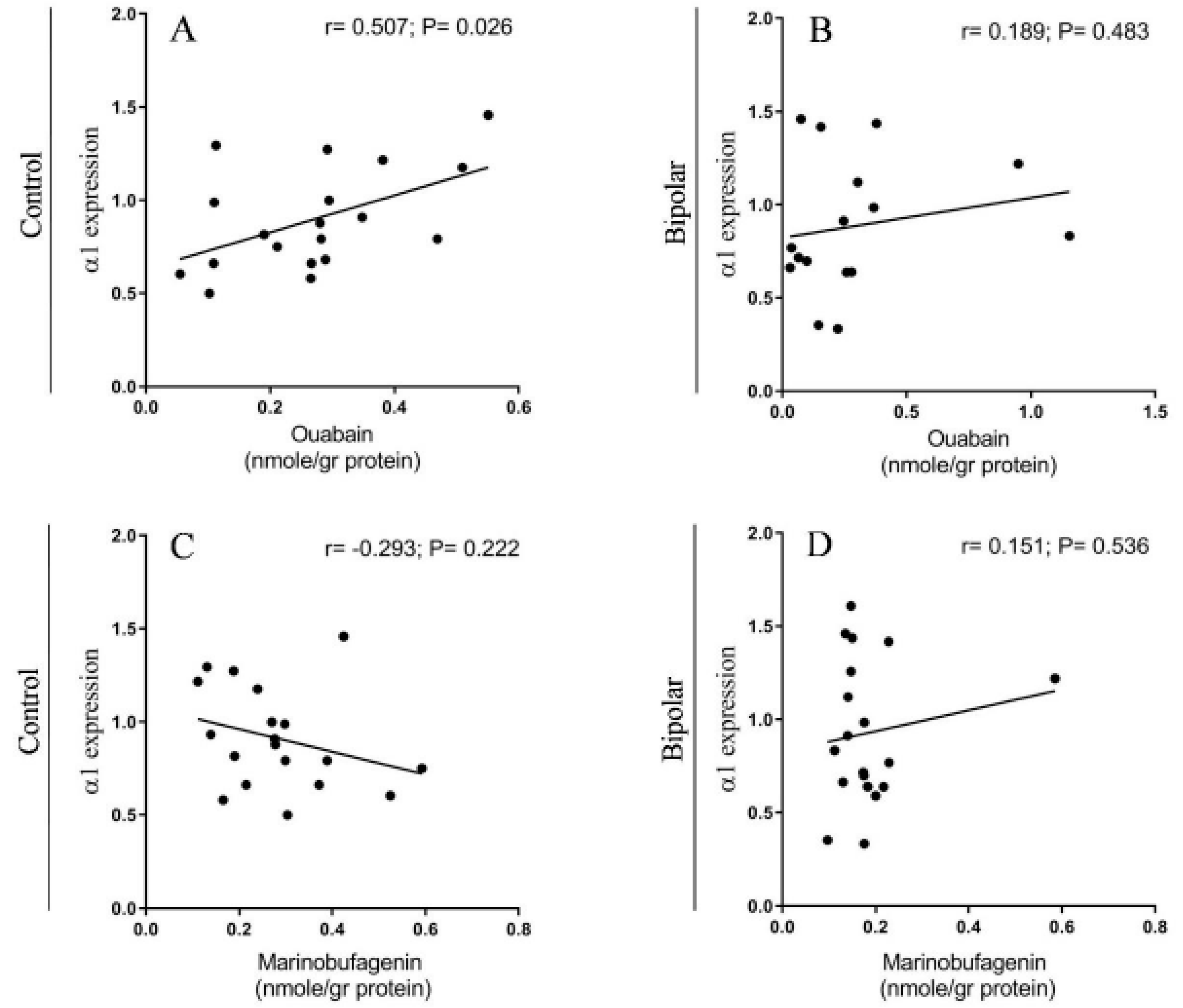
2.1. Na+, K+-ATPase α Isoforms in PFC of BD Patients and Controls
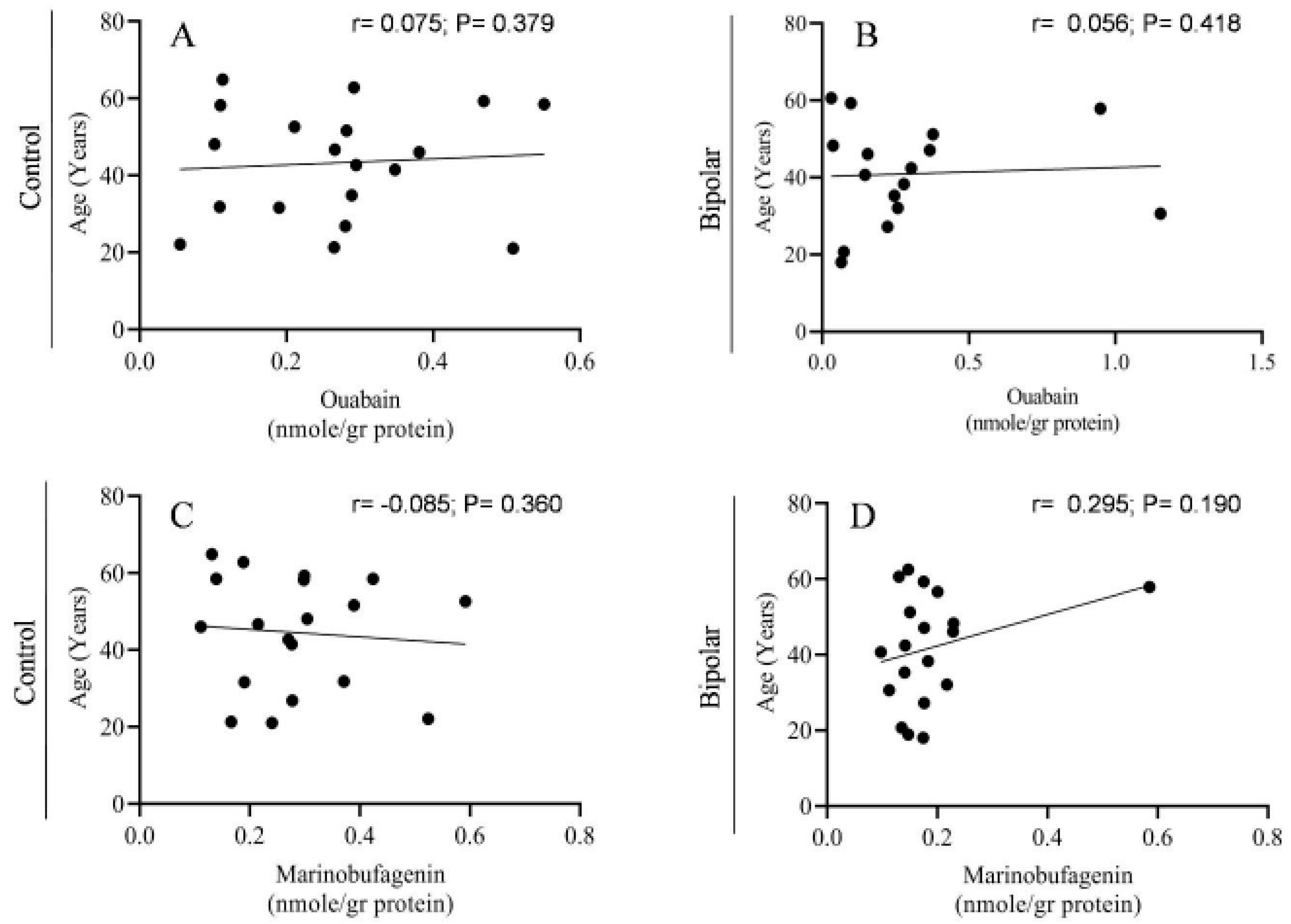
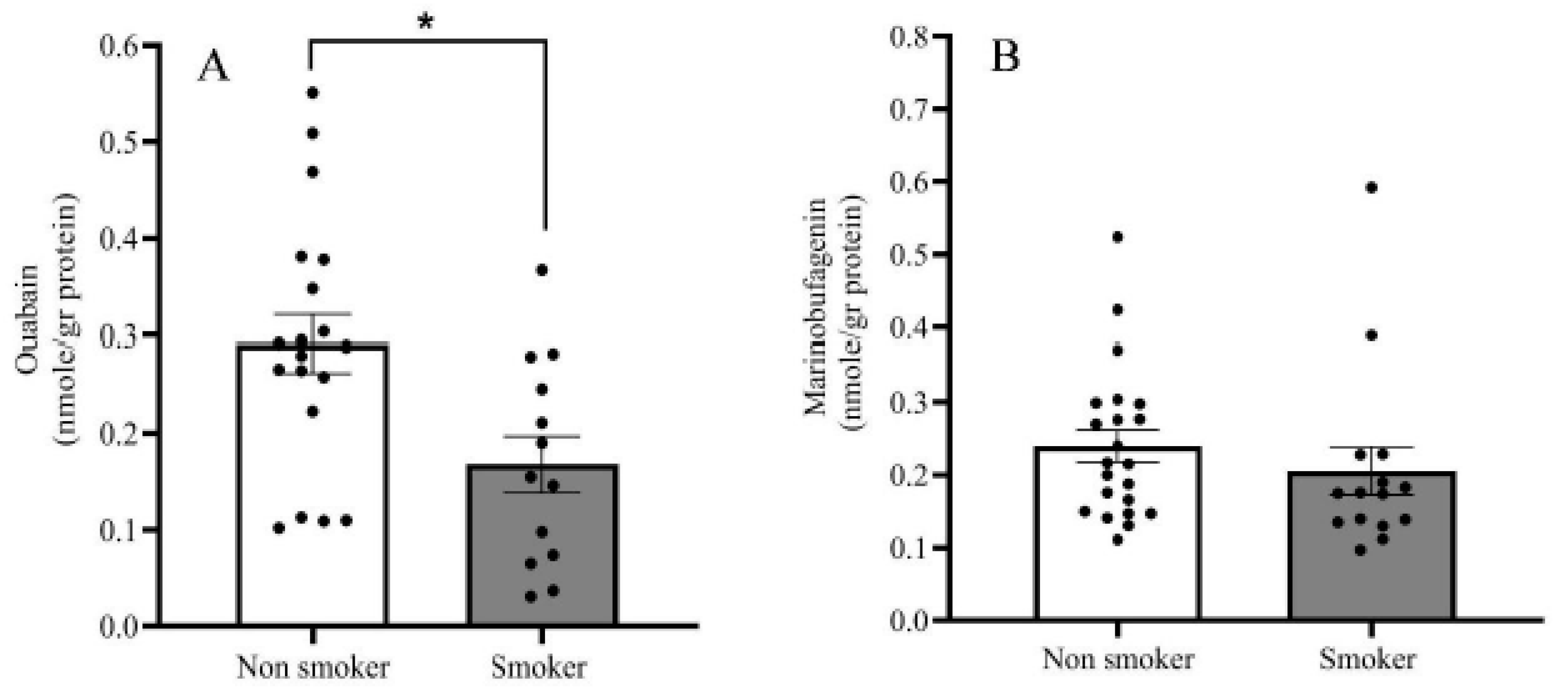
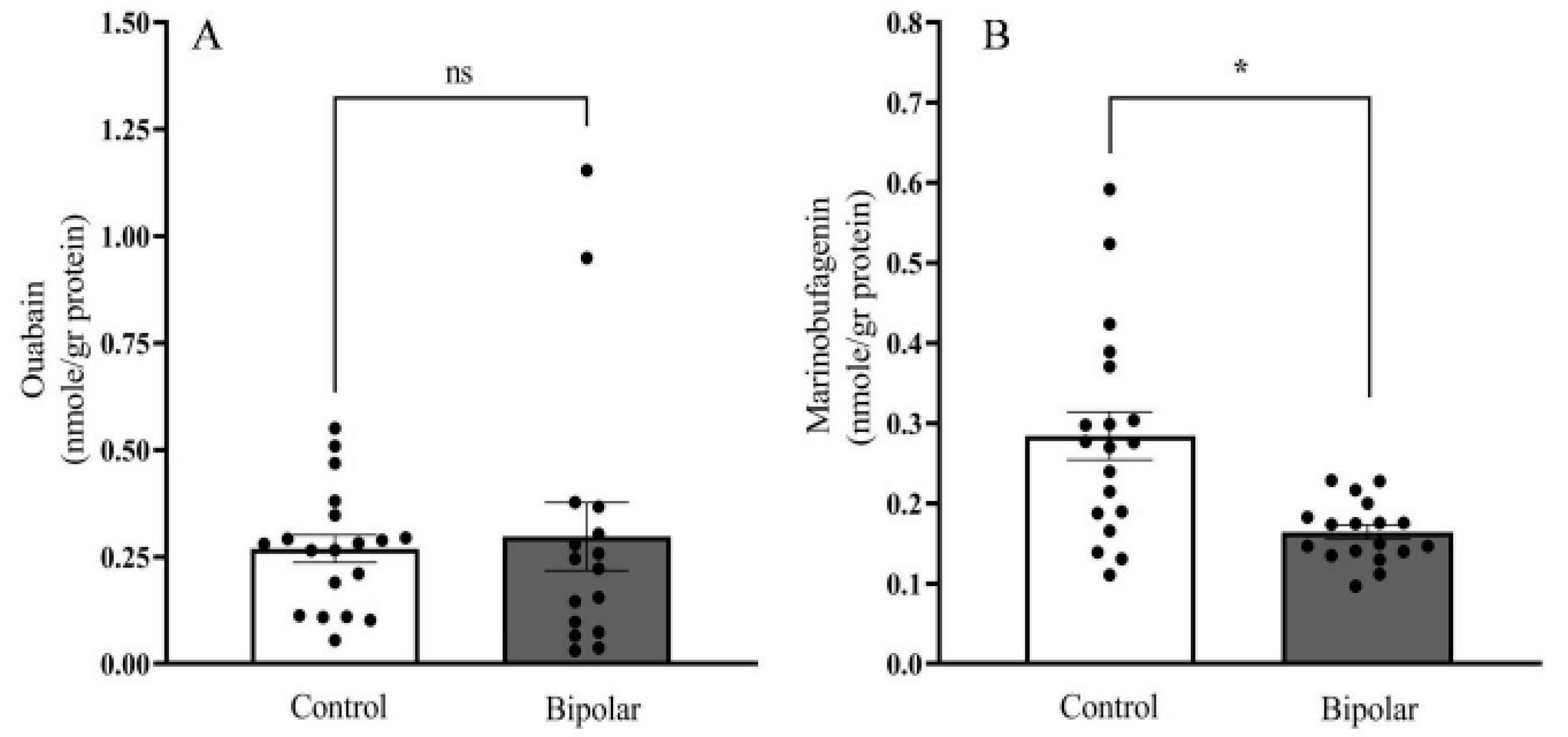
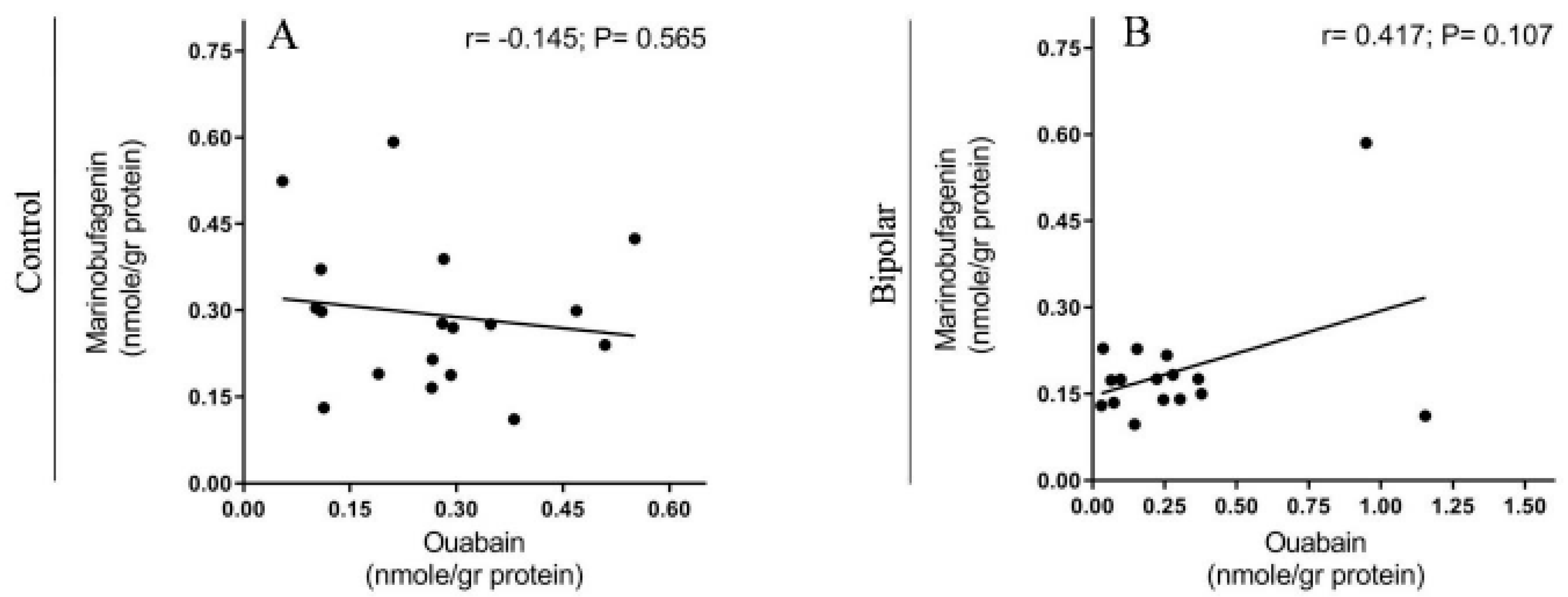
2.2. Endogenous OUA and MBG in the PFC of the BD Patients and the Controls
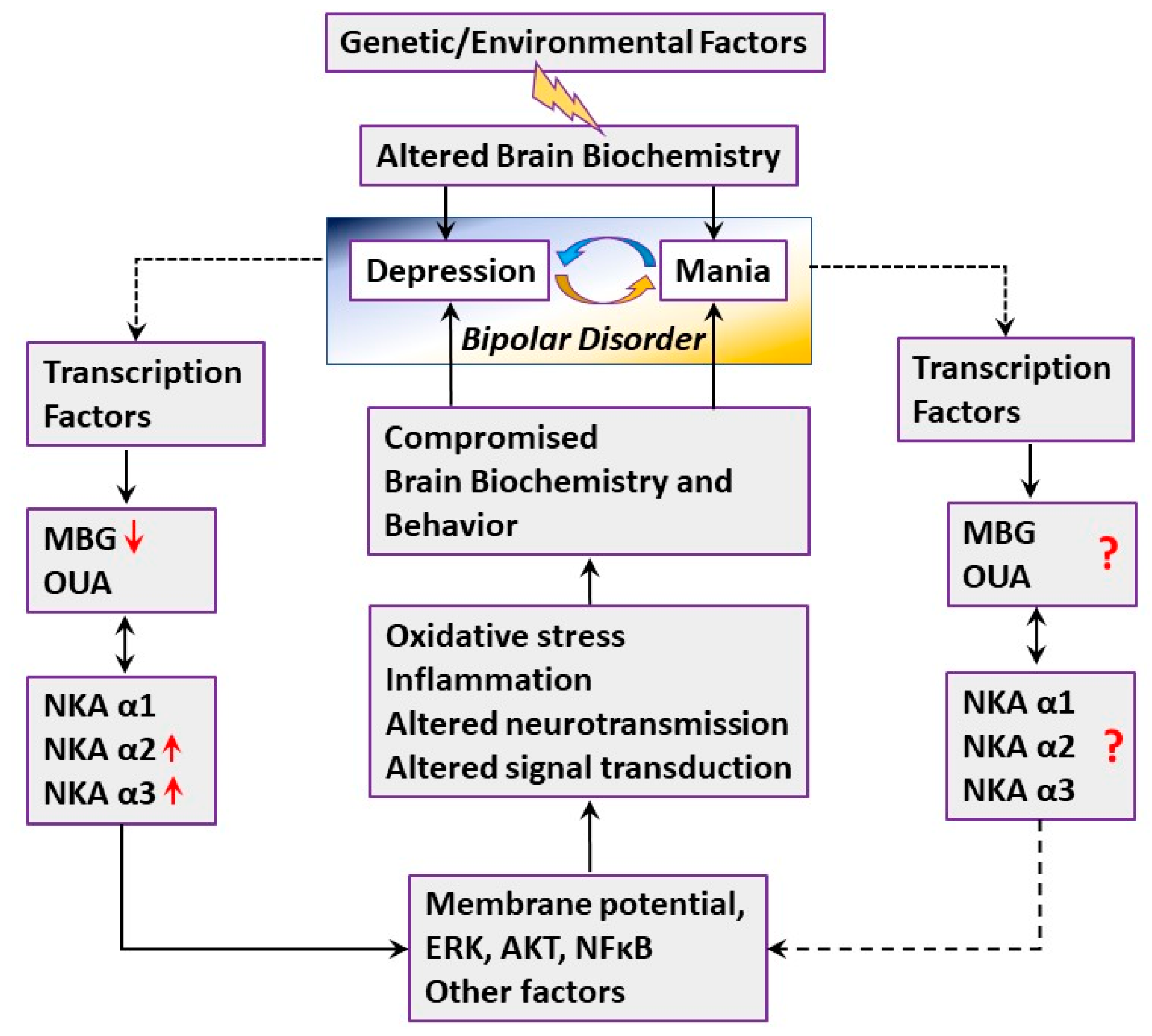
3. Discussion
4. Materials and Methods
4.1. Brain Samples
4.2. Quantification of Na+, K+-ATPase α Isoforms with Western Blot
4.3. Determination of Endogenous OUA and MBG in Brain Samples
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BD: | Bipolar Disorder |
| PFC: | Prefrontal Cortex |
| CS: | Cardiac Steroids |
| ECS: | Endogenous Cardiac Steroids |
| OUA: | Ouabain |
| MBG: | Marinobufagenin |
| Na+, K+-ATPase: | Sodium-Potassium-Activated Adenosine Triphosphatase |
| HBCC | Human Brain Collection Core |
References
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef]
- Nierenberg, A.A.; Sylvia, L.G.; Leon, A.C.; A Reilly-Harrington, N.; Shesler, L.W.; McElroy, S.L.; Friedman, E.S.; Thase, M.E.; Shelton, R.C.; Bowden, C.L.; et al. Clinical and Health Outcomes Initiative in Comparative Effectiveness for Bipolar Disorder (Bipolar CHOICE): A pragmatic trial of complex treatment for a complex disorder. Clin. Trials 2014, 11, 114–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whayne, T.F., Jr. Clinical Use of Digitalis: A State of the Art Review. Am. J. Cardiovasc. Drugs 2018, 18, 427–440. [Google Scholar] [CrossRef]
- Buckalew, V.M. Endogenous digitalis-like factors: An overview of the history. Front. Endocrinol. (Lausanne) 2015, 6, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leenen, F.H.H.; Blaustein, M.P.; Hamlyn, J.M. Update on angiotensin II: New endocrine connections between the brain, adrenal glands and the cardiovascular system. Endocr. Connect. 2017, 6, R131–R145. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Dvela, M.; Rosen, H.; Ben-Ami, H.C.; Lichtstein, D. Endogenous ouabain regulates cell viability. Am. J. Physiol. Cell Physiol. 2012, 302, C442–C452. [Google Scholar] [CrossRef] [Green Version]
- Leenen, F.H.H.; Wang, H.W.; Hamlyn, J.M. Sodium pumps, ouabain and aldosterone in the brain: A neuromodulatory pathway underlying salt-sensitive hypertension and heart failure. Cell Calcium 2020, 86, 102151. [Google Scholar] [CrossRef]
- Fedorova, O.V.; Agalakova, N.I.; Talan, M.I.; Lakatta, E.G.; Bagrov, A.Y. Brain ouabain stimulates peripheral marinobufagenin via angiotensin II signalling in NaCl-loaded Dahl-S rats. J. Hypertens. 2005, 23, 1515–1523. [Google Scholar] [CrossRef]
- Lichtstein, D.; Rosen, H. Endogenous digitalis-like Na+, K+-ATPase inhibitors, and brain function. Neurochem. Res. 2001, 26, 971–978. [Google Scholar] [CrossRef]
- Hodes, A.; Lichtstein, D. Natriuretic hormones in brain function. Front. Endocrinol. (Lausanne) 2014, 5, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossier, B.C.; Baker, M.E.; Studer, R.A. Epithelial sodium transport and its control by aldosterone: The story of our internal environment revisited. Physiol. Rev. 2015, 95, 297–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- McGrail, K.M.; Phillips, J.M.; Sweadner, K.J. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: Both neurons and glia can express more than one Na,K-ATPase. J. Neurosci. 1991, 11, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Bottger, P.; Tracz, Z.; Heuck, A.; Nissen, P.; Romero-Ramos, M.; Lykke-Hartmann, K. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J. Comp. Neurol. 2011, 519, 376–404. [Google Scholar] [CrossRef]
- Blom, H.; Ronnlund, D.; Scott, L.; Spicarova, Z.; Widengren, J.; Bondar, A.; Aperia, A.; Brismar, H. Spatial distribution of Na+-K+-ATPase in dendritic spines dissected by nanoscale superresolution STED microscopy. BMC Neurosci. 2011, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, A.N.; Triller, A.; Melki, R. Cell biology and dynamics of Neuronal Na(+)/K(+)-ATPase in health and diseases. Neuropharmacology 2020, 169, 107461. [Google Scholar] [CrossRef] [PubMed]
- Madan, N.; Xu, Y.; Duan, Q.; Banerjee, M.; Larre, I.; Pierre, S.V.; Xie, Z. Src-independent ERK signaling through the rat alpha3 isoform of Na/K-ATPase. Am. J. Physiol. Cell Physiol. 2017, 312, C222–C232. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xie, Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 2017, 22, 990. [Google Scholar]
- Zhang, Y.; Wei, W.; Shilova, V.; Petrashevskaya, N.N.; Zernetkina, V.I.; Grigorova, Y.N.; Marshall, C.A.; Fenner, R.C.; Lehrmann, E.; Woodlll, W.H.; et al. Monoclonal Antibody to Marinobufagenin Downregulates TGFbeta Profibrotic Signaling in Left Ventricle and Kidney and Reduces Tissue Remodeling in Salt-Sensitive Hypertension. J. Am. Heart Assoc. 2019, 8, e012138. [Google Scholar] [CrossRef]
- Goldstein, I.; Lerer, E.; Laiba, E.; Mallet, J.; Mujaheed, M.; Laurent, C.; Rosen, H.; Ebstein, R.P.; Lichtstein, D. Association between sodium- and potassium-activated adenosine triphosphatase alpha isoforms and bipolar disorders. Biol. Psychiatry 2009, 65, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, G.S.; Clapcote, S.J.; Duffy, S.; Burgess, C.R.; Petersen, J.; Jarowek, K.J.; Yücel, Y.H.; Cortez, M.A.; Snead, O.C.; Vilsen, B.; et al. Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+,K+-ATPase {alpha}3 sodium pump. Proc. Natl. Acad. Sci. USA 2011, 108, 18144–18149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, T.H.; Lykke-Hartmann, K. Insights into the Pathology of the alpha3 Na(+)/K(+)-ATPase Ion Pump in Neurological Disorders; Lessons from Animal Models. Front. Physiol. 2016, 7, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Looney, S.W.; El-Mallakh, R.S. Meta-analysis of erythrocyte Na,K-ATPase activity in bipolar illness. Depress. Anxiety 1997, 5, 53–65. [Google Scholar] [CrossRef]
- El-Mallakh, R.S.; Stoddard, M.; Jortani, S.A.; El-Masri, M.A.; Sephton, S.; Valdes, R., Jr. Aberrant regulation of endogenous ouabain-like factor in bipolar subjects. Psychiatry Res. 2010, 178, 116–120. [Google Scholar] [CrossRef]
- Goldstein, I.; Levy, T.; Galili, D.; Ovadia, H.; Yirmiya, R.; Rosen, H.; Lichtstein, D. Involvement of Na(+), K(+)-ATPase and endogenous digitalis-like compounds in depressive disorders. Biol. Psychiatry 2006, 60, 491–499. [Google Scholar] [CrossRef]
- Hodes, A.; Rosen, H.; Deutsch, J.; Lifschytz, T.; Einat, H.; Lichtstein, D. Endogenous cardiac steroids in animal models of mania. Bipolar Disord. 2016, 18, 451–459. [Google Scholar] [CrossRef]
- Goldstein, I.; Lax, E.; Gispan-Herman, I.; Ovadia, H.; Rosen, H.; Yadid, G.; Lichtstein, D. Neutralization of endogenous digitalis-like compounds alters catecholamines metabolism in the brain and elicits anti-depressive behavior. Eur. Neuropsychopharmacol. 2012, 22, 72–79. [Google Scholar] [CrossRef]
- Hodes, A.; Lifschytz, T.; Rosen, H.; Cohen Ben-Ami, H.; Lichtstein, D. Reduction in endogenous cardiac steroids protects the brain from oxidative stress in a mouse model of mania induced by amphetamine. Brain Res. Bull. 2018, 137, 356–362. [Google Scholar] [CrossRef]
- Miller, E.K.; Cohen, J.D. An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 2001, 24, 167–202. [Google Scholar] [CrossRef] [Green Version]
- Fuster, J.M. The prefrontal cortex--an update: Time is of the essence. Neuron 2001, 30, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Geng, H.; Jiang, X.; Zhou, Q.; Chang, M.; Zhou, Y.; Xu, K.; Tang, Y.; Wang, F. Amygdala-prefrontal cortex resting-state functional connectivity varies with first depressive or manic episode in bipolar disorder. Neurosci. Lett. 2017, 641, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Andreazza, A.C.; Young, L.T. The neurobiology of bipolar disorder: Identifying targets for specific agents and synergies for combination treatment. Int. J. Neuropsychopharmacol. 2014, 17, 1039–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanford, L.C.; Nazarov, A.; Hall, G.B.; Sassi, R.B. Cortical thickness in bipolar disorder: A systematic review. Bipolar Disord. 2016, 18, 4–18. [Google Scholar] [CrossRef]
- Nenadic, I.; Maitra, R.; Dietzek, M.; Langbein, K.; Smesny, S.; Sauer, H.; Gaser, C. Prefrontal gyrification in psychotic bipolar I disorder vs. schizophrenia. J. Affect. Disord. 2015, 185, 104–107. [Google Scholar] [CrossRef]
- Smalheiser, N.R.; Lugli, G.; Zhang, H.; Rizavi, H.; Cook, E.H.; Dwivedi, Y. Expression of microRNAs and other small RNAs in prefrontal cortex in schizophrenia, bipolar disorder and depressed subjects. PLoS ONE 2014, 9, e86469. [Google Scholar] [CrossRef]
- Woo, T.U.; Kim, A.M.; Viscidi, E. Disease-specific alterations in glutamatergic neurotransmission on inhibitory interneurons in the prefrontal cortex in schizophrenia. Brain Res. 2008, 1218, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Hodes, A.; Rosen, H.; Ami, H.C.-B.; Lichtstein, D. Na(+), K(+)-ATPase alpha3 isoform in frontal cortex GABAergic neurons in psychiatric diseases. J. Psychiatr. Res. 2019, 115, 21–28. [Google Scholar]
- Rose, A.M.; Mellett, B.J.; Valdes, R., Jr.; Kleinman, J.E.; Herman, M.M.; Li, R.; El-Mallakh, R.S. Alpha 2 isoform of the Na,K-adenosine triphosphatase is reduced in temporal cortex of bipolar individuals. Biol. Psychiatry 1998, 44, 892–897. [Google Scholar] [CrossRef]
- Gigase, F.A.J.; Snijders, G.; Boks, M.P.; de Witte, L.D. Neurons and glial cells in bipolar disorder: A systematic review of postmortem brain studies of cell number and size. Neurosci. Biobehav. Rev. 2019, 103, 150–162. [Google Scholar] [CrossRef]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.H.; Zhen, X.C. Glial pathology in bipolar disorder: Potential therapeutic implications. CNS Neurosci. Ther. 2015, 21, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M. Glial cells as key elements in the pathophysiology and treatment of bipolar disorder. Acta Neuropsychiatr. 2017, 29, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.B.; Clausen, T. The Na+/K(+)-pump protects muscle excitability and contractility during exercise. Exerc. Sport Sci. Rev. 2000, 28, 159–164. [Google Scholar] [PubMed]
- Radzyukevich, T.L.; Neumann, J.C.; Rindler, T.N.; Oshiro, N.; Goldhamer, D.J.; Lingrel, J.B.; Heiny, J.A. Tissue-specific role of the Na,K-ATPase alpha2 isozyme in skeletal muscle. J. Biol. Chem. 2013, 288, 1226–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, N.; Siegel, G. Na,K-ATPase: Increases in alpha1-messenger RNA and decreases in alpha3-messenger RNA levels in aging rat cerebral cortex. Neuroscience 1997, 78, 7–11. [Google Scholar]
- Fedorova, O.V.; Talan, M.I.; Agalakova, N.I.; Lakatta, E.G.; Bagrov, A.Y. Coordinated shifts in Na/K-ATPase isoforms and their endogenous ligands during cardiac hypertrophy and failure in NaCl-sensitive hypertension. J. Hypertens. 2004, 22, 389–397. [Google Scholar] [CrossRef]
- Pimentel, V.C.; Zanini, D.; Cardoso, A.M.; Schmatz, R.; Bagatini, M.D.; Gutierres, J.M.; Carvalho, F.B.; Gomes, J.L.; Rubin, M.A.; Morsch, V.M.; et al. Hypoxia-ischemia alters nucleotide and nucleoside catabolism and Na+,K+-ATPase activity in the cerebral cortex of newborn rats. Neurochem. Res. 2013, 38, 886–894. [Google Scholar] [CrossRef]
- Bystriansky, J.S.; Ballantyne, J.S. Gill Na+-K+-ATPase activity correlates with basolateral membrane lipid composition in seawater- but not freshwater-acclimated Arctic char (Salvelinus alpinus). Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1043–R1051. [Google Scholar] [CrossRef]
- Nagafuji, T.; Koide, T.; Takato, M. Neurochemical correlates of selective neuronal loss following cerebral ischemia: Role of decreased Na+,K(+)-ATPase activity. Brain Res. 1992, 571, 265–271. [Google Scholar] [CrossRef]
- Yamada, H.; Ihara, N.; Takahashi, H.; Yoshimura, M.; Sano, Y. Distribution of the endogenous digitalis-like substance (EDLS)-containing neurons labeled by digoxin antibody in hypothalamus and three circumventricular organs of dog and macaque. Brain Res. 1992, 584, 237–243. [Google Scholar] [CrossRef]
- Morgan, K.; Lewis, M.D.; Spurlock, G.; Collins, P.A.; Foord, S.M.; Southgate, K.; Scanlon, M.F.; Mir, M.A. Characterization and partial purification of the sodium-potassium-ATPase inhibitor released from cultured rat hypothalamic cells. J. Biol. Chem. 1985, 260, 13595–13600. [Google Scholar] [PubMed]
- Wang, J.G.; Staessen, J.A.; Messaggio, E.; Nawrot, T.; Fagard, R.; Hamlyn, J.M.; Bianchi, G.; Manunta, P. Salt, endogenous ouabain and blood pressure interactions in the general population. J. Hypertens. 2003, 21, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, G.; Citterio, L.; Kouznetsova, T.; Lanzani, C.; Florio, M.; Modica, R.; Messaggio, E.; Hamlyn, J.M.; Zagato, L.; Bianchi, G.; et al. Steroid biosynthesis and renal excretion in human essential hypertension: Association with blood pressure and endogenous ouabain. Am. J. Hypertens. 2009, 22, 357–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, H.; Toyama, K.; Pratt, P.F.; Gutterman, D.D. Cigarette smoking impairs Na+-K+-ATPase activity in the human coronary microcirculation. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H109–H117. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, J.M.; Wootton, R.E.; Treur, J.L.; Sallis, H.; Jones, H.J.; Zammit, S.; Brink, W.V.D.; Goodwin, G.M.; De Haan, L.; Munafò, M.R. Smoking and the risk for bipolar disorder: Evidence from a bidirectional Mendelian randomisation study. Br. J. Psychiatry 2019, 1–7. [Google Scholar] [CrossRef]
- Lichtstein, D.; Steinitz, M.; Gati, I.; Samuelov, S.; Deutsch, J.; Orly, J. Biosynthesis of digitalis-like compounds in rat adrenal cells: Hydroxycholesterol as possible precursor. Life Sci. 1998, 62, 2109–2126. [Google Scholar] [CrossRef]
- Fedorova, O.V.; Zernetkina, V.I.; Shilova, V.Y.; Grigorova, Y.N.; Juhasz, O.; Wei, W.; Marshall, C.A.; Lakatta, E.G.; Bagrov, A.Y. Synthesis of an Endogenous Steroidal Na Pump Inhibitor Marinobufagenin, Implicated in Human Cardiovascular Diseases, Is Initiated by CYP27A1 via Bile Acid Pathway. Circ. Cardiovasc. Genet. 2015, 8, 736–745. [Google Scholar] [CrossRef]
- Qi, X.R.; Luchetti, S.; Verwer, R.W.H.; Sluiter, A.A.; Mason, M.R.J.; Zhou, J.-N.; Swaab, D.F. Alterations in the steroid biosynthetic pathways in the human prefrontal cortex in mood disorders: A post-mortem study. Brain Pathol. 2018, 28, 536–547. [Google Scholar] [CrossRef]
- Smolyaninova, L.V.; Shiyan, A.A.; Kapilevich, L.V.; Lopachev, A.V.; Fedorova, T.N.; Klementieva, T.S.; Moskovtsev, A.A.; Kubatiev, A.A.; Orlov, S.N. Transcriptomic changes triggered by ouabain in rat cerebellum granule cells: Role of alpha3- and alpha1-Na+,K+-ATPase-mediated signaling. PLoS ONE 2019, 14, e0222767. [Google Scholar] [CrossRef] [Green Version]
- Amarelle, L.; Katzen, J.; Shigemura, M.; Welch, L.C.; Cajigas, H.; Peteranderl, C.; Celli, D.; Herold, S.; Lecuona, E.; Sznajder, J.I. Cardiac glycosides decrease influenza virus replication by inhibiting cell protein translational machinery. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1094–L1106. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61 (Suppl. 6), 4–6. [Google Scholar] [PubMed]
- Blacker, C.J.; Lewis, C.P.; Frye, M.A.; Veldic, M. Metabotropic glutamate receptors as emerging research targets in bipolar disorder. Psychiatry Res. 2017, 257, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Jeon, W.J.; Dean, B.; Scarr, E.; Gibbons, A. The Role of Muscarinic Receptors in the Pathophysiology of Mood Disorders: A Potential Novel Treatment? Curr. Neuropharmacol. 2015, 13, 739–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lener, M.S.; Niciu, M.J.; Ballard, E.D.; Park, M.; Park, L.T.; Nugent, A.C.; Zarate, C.A. Glutamate and Gamma-Aminobutyric Acid Systems in the Pathophysiology of Major Depression and Antidepressant Response to Ketamine. Biol. Psychiatry 2017, 81, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Walder, K.; McGee, S.L.; Dean, O.M.; Tye, S.J.; Maes, M.; Berk, M. A model of the mitochondrial basis of bipolar disorder. Neurosci. Biobehav. Rev. 2017, 74, 1–20. [Google Scholar] [CrossRef]
- Hamdani, N.; Doukhan, R.; Kurtlucan, O.; Tamouza, R.; Leboyer, M. Immunity, inflammation, and bipolar disorder: Diagnostic and therapeutic implications. Curr. Psychiatry Rep. 2013, 15, 387. [Google Scholar] [CrossRef]
- Vizi, E.S.; Oberfrank, F. Na+/K(+)-ATPase, its endogenous ligands and neurotransmitter release. Neurochem. Int. 1992, 20, 11–17. [Google Scholar] [CrossRef]
- Li, Q.; Pogwizd, S.M.; Prabhu, S.D.; Zhou, L. Inhibiting Na+/K+ ATPase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS ONE 2014, 9, e93928. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante-Silva, L.H.A.; Lima, E.A.; Carvalho, D.C.M.; De Sales-Neto, J.M.; Alves, A.K.D.A.; Galvão, J.G.F.M.; Silva, J.S.D.F.D.; Rodrigues-Mascarenhas, S. Much More than a Cardiotonic Steroid: Modulation of Inflammation by Ouabain. Front. Physiol. 2017, 8, 895. [Google Scholar] [CrossRef] [Green Version]
- Lichtstein, D.; Ilani, A.; Rosen, H.; Horesh, N.; Singh, S.V.; Buzaglo, N.; Hodes, A. Na(+), K(+)-ATPase Signaling and Bipolar Disorder. Int. J. Mol. Sci. 2018, 19, 2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The National Institute of Mental Health. Human Brain Collection Core (HBCC). Available online: https://www.nimh.nih.gov/research/research-conducted-at-nimh/research-areas/research-support-services/hbcc/index.shtml (accessed on 16 August 2020).
- Lopatin, D.A.; Ailamazian, E.K.; Dmitrieva, R.I.; Shpen, V.M.; Fedorova, O.V.; Doris, P.A.; Bagrov, A.Y. Circulating bufodienolide and cardenolide sodium pump inhibitors in preeclampsia. J. Hypertens. 1999, 17, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Simbirtsev, A.S.; Kolodkin, N.I.; Kotov, A.Y.; Agalakova, N.I.; Kashkin, V.; Tapilskaya, N.I.; Bzhelyansky, A.; Reznik, V.A.; Frolova, E.V.; et al. Monoclonal antibody to an endogenous bufadienolide, marinobufagenin, reverses preeclampsia-induced Na/K-ATPase inhibition and lowers blood pressure in NaCl-sensitive hypertension. J. Hypertens. 2008, 26, 2414–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 20) | Bipolar Patients (n = 20) | |
|---|---|---|
| Age at death (years) | 43.12 ± 3.21 | 42.25 ± 3.23 |
| Cause of death (Suicide/other) | 0/20 | 15/5 |
| Brain weight (g) | 1382.75 ± 23.08 | 1511.75 ± 37.58 |
| Postmortem interval (h) | 30.70 ± 3.03 | 28.97 ± 3.94 |
| Brain tissue pH | 6.44 ± 0.05 | 6.34 ± 0.05 |
| Sex (Men/Women) | 14/6 | 14/6 |
| Alcohol history (none/positive) | 4/16 | 5/15 |
| Use of Psychoactive drugs (none/positive) | 14/6 | 0/20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.V.; Fedorova, O.V.; Wei, W.; Rosen, H.; Horesh, N.; Ilani, A.; Lichtstein, D. Na+, K+-ATPase α Isoforms and Endogenous Cardiac Steroids in Prefrontal Cortex of Bipolar Patients and Controls. Int. J. Mol. Sci. 2020, 21, 5912. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165912
Singh SV, Fedorova OV, Wei W, Rosen H, Horesh N, Ilani A, Lichtstein D. Na+, K+-ATPase α Isoforms and Endogenous Cardiac Steroids in Prefrontal Cortex of Bipolar Patients and Controls. International Journal of Molecular Sciences. 2020; 21(16):5912. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165912
Chicago/Turabian StyleSingh, Shiv Vardan, Olga V. Fedorova, Wen Wei, Haim Rosen, Noa Horesh, Asher Ilani, and David Lichtstein. 2020. "Na+, K+-ATPase α Isoforms and Endogenous Cardiac Steroids in Prefrontal Cortex of Bipolar Patients and Controls" International Journal of Molecular Sciences 21, no. 16: 5912. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165912