Repositioning Dequalinium as Potent Muscarinic Allosteric Ligand by Combining Virtual Screening Campaigns and Experimental Binding Assays

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Preliminary Virtual Screening Simulations
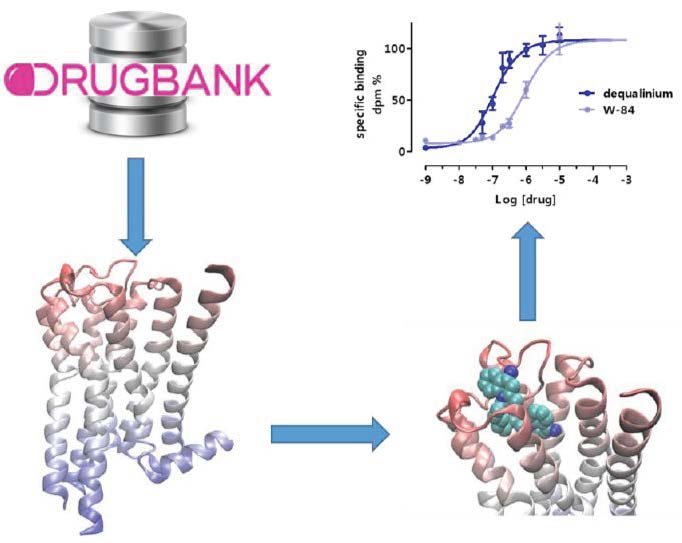
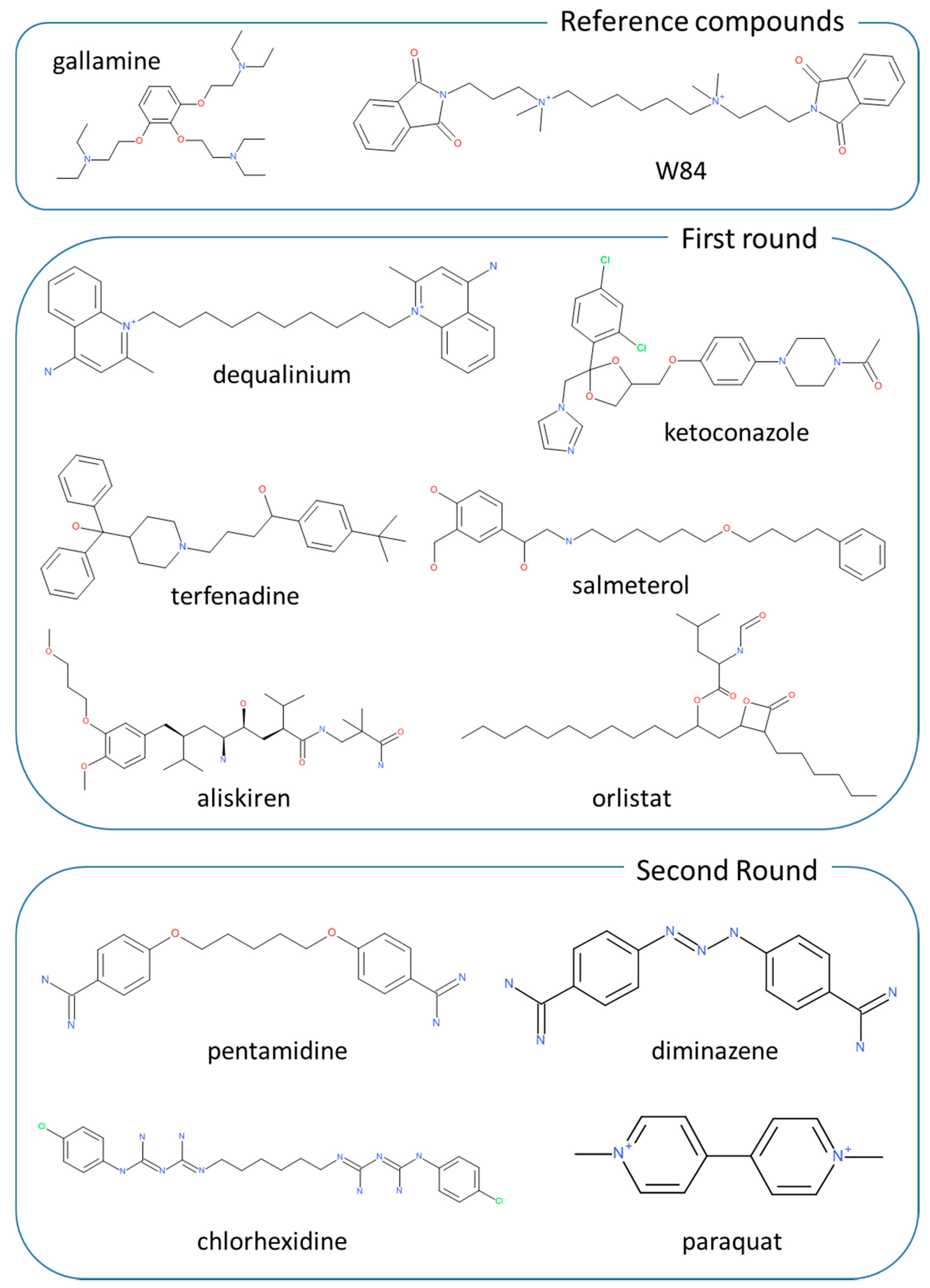
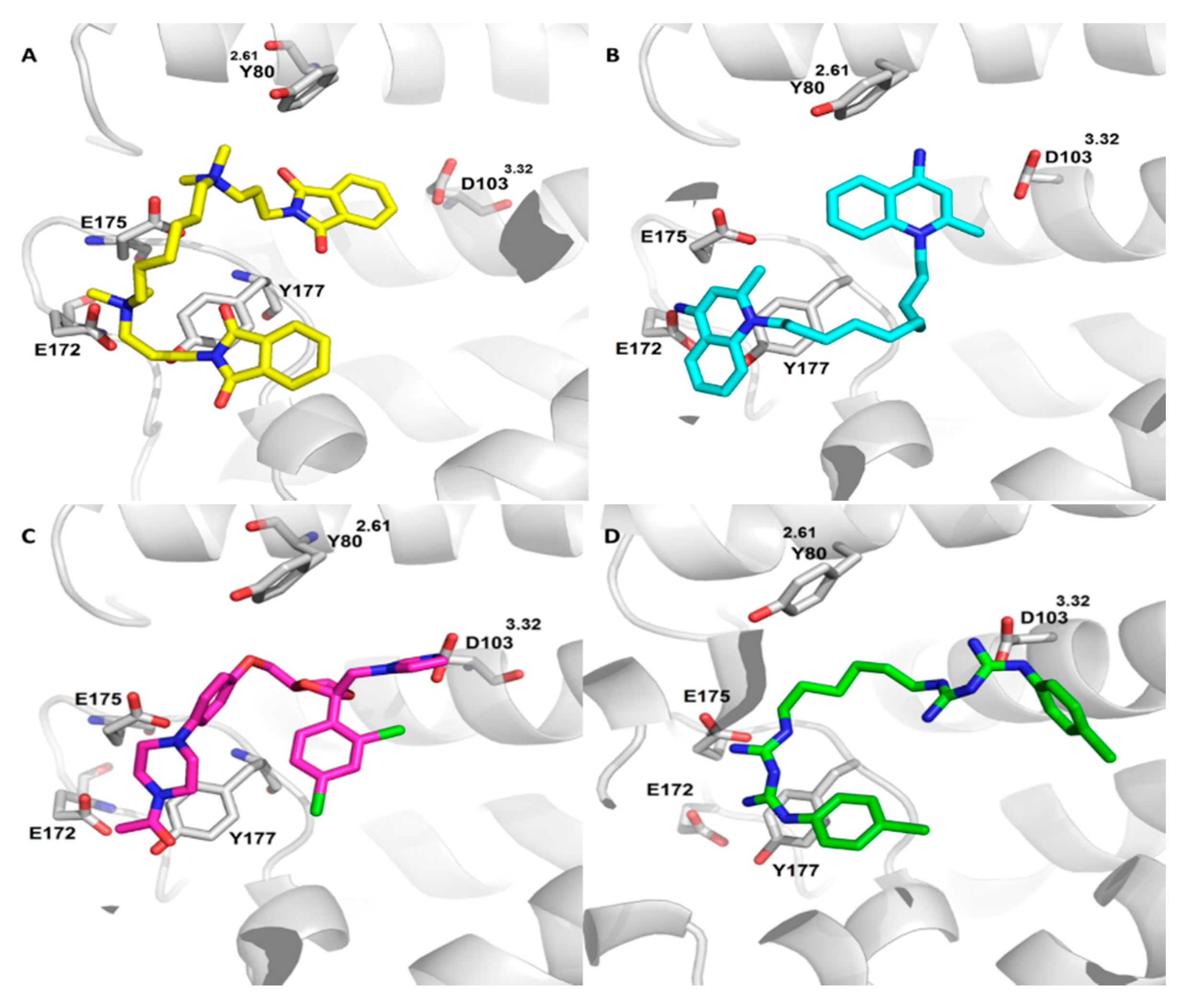
2.2. Virtual Screening for Drug Repositioning
2.3. Equilibrium and Kinetic Binding Studies
2.4. Second Targeted Screening Campaign
3. Materials and Methods
3.1. Preliminary Virtual Screening Simulations
3.2. Virtual Screening Simulations for Drug Repositioning
3.3. Biologic Studies
3.3.1. Equilibrium Binding Assays
3.3.2. Dissociation Kinetic Assay
Full time course
One point kinetic assays
3.3.3. Data Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EF | Enrichment factor |
| EFO | Enrichment factor optimization |
| GPCR | G protein-coupled receptor |
| hM | Human muscarinic acetylcholine receptor |
| hmAChR | Human muscarinic acetylcholine receptor |
| NMS | N-methyl scopolamine |
| QNB | Quinuclidinyl benzilate |
| VS | Virtual screening |
References
- Kumar, R.; Harilal, S.; Gupta, S.V.; Jose, J.; Parambi, D.G.T.; Uddin, S.; Shah, M.A.; Mathew, B.; Thomas, D.G. Exploring the new horizons of drug repurposing: A vital tool for turning hard work into smart work. Eur. J. Med. Chem. 2019, 182, 111602. [Google Scholar] [CrossRef] [PubMed]
- Gns, H.S.; Gr, S.; Murahari, M.; Krishnamurthy, M. An update on Drug Repurposing: Re-written saga of the drug’s fate. Biomed. Pharmacother. 2019, 110, 700–716. [Google Scholar] [CrossRef] [PubMed]
- Polamreddy, P.; Gattu, N. The drug repurposing landscape from 2012 to 2017: Evolution, challenges, and possible solutions. Drug Discov. Today 2019, 24, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.J.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Loiodice, S.; Da Costa, A.N.; Atienzar, F. Current trends in in silico, in vitro toxicology, and safety biomarkers in early drug development. Drug Chem. Toxicol. 2017, 42, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Yang, B. Rational application of drug promiscuity in medicinal chemistry. Futur. Med. Chem. 2018, 10, 1835–1851. [Google Scholar] [CrossRef]
- Wang, T.; Liu, X.H.; Guan, J.; Ge, S.; Wu, M.B.; Lin, J.P.; Yang, L.R. Advancement of multi-target drug discoveries and promising applications in the field of Alzheimer’s disease. Eur J Med Chem. 2019, 169, 200–223. [Google Scholar] [CrossRef]
- Artasensi, A.; Pedretti, A.; Vistoli, G.; Fumagalli, L. Type 2 Diabetes Mellitus: A Review of Multi-Target Drugs. Molecules 2020, 25, 1987. [Google Scholar] [CrossRef]
- Karaman, B.; Sippl, W. Computational Drug Repurposing: Current Trends. Curr. Med. Chem. 2019, 26, 5389–5409. [Google Scholar] [CrossRef]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2018, 18, 59–82. [Google Scholar] [CrossRef]
- Burger, W.A.; Sexton, P.M.; Christopoulos, A.; Thal, D.M. Toward an understanding of the structural basis of allostery in muscarinic acetylcholine receptors. J. Gen. Physiol. 2018, 150, 1360–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, A.C.; Hu, J.; Kobilka, B.K.; Wess, J. Muscarinic acetylcholine receptor X-ray structures: Potential implications for drug development. Curr. Opin. Pharmacol. 2014, 16, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Vuckovic, Z.; Gentry, P.R.; Berizzi, A.E.; Hirata, K.; Varghese, S.; Thompson, G.; Van Der Westhuizen, E.T.; Burger, W.A.C.; Rahmani, R.; Valant, C.; et al. Crystal structure of the M5muscarinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2019, 116, 26001–26007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, K.; Kruse, A.C.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; I Weis, W.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, A.C.; Ring, A.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Opportunities and Challenges in the Discovery of Allosteric Modulators of GPCRs. Methods Mol. Biol. 2018, 1705, 297–319. [Google Scholar] [CrossRef]
- Bock, A.; Schrage, R.; Mohr, K. Allosteric modulators targeting CNS muscarinic receptors. Neuropharmacology 2018, 136, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Vistoli, G. Rescoring and Linearly Combining: A Highly Effective Consensus Strategy for Virtual Screening Campaigns. Int. J. Mol. Sci. 2019, 20, 2060. [Google Scholar] [CrossRef] [Green Version]
- Talarico, C.; Gervasoni, S.; Manelfi, C.; Pedretti, A.; Vistoli, G.; Beccari, A.R. Combining Molecular Dynamics and Docking Simulations to Develop Targeted Protocols for Performing Optimized Virtual Screening Campaigns on the hTRPM8 Channel. Int. J. Mol. Sci. 2020, 21, 2265. [Google Scholar] [CrossRef] [Green Version]
- Mazzolari, A.; Vistoli, G.; Testa, B.; Pedretti, A. Prediction of the Formation of Reactive Metabolites by A Novel Classifier Approach Based on Enrichment Factor Optimization (EFO) as Implemented in the VEGA Program. Molecules 2018, 23, 2955. [Google Scholar] [CrossRef] [Green Version]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ’Omics’ research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vistoli, G.; Mazzolari, A.; Testa, B.; Pedretti, A. Binding Space Concept: A New Approach To Enhance the Reliability of Docking Scores and Its Application to Predicting Butyrylcholinesterase Hydrolytic Activity. J. Chem. Inf. Model. 2017, 57, 1691–1702. [Google Scholar] [CrossRef]
- Kubo, N.; Shirakawa, O.; Kuno, T.; Tanaka, C. Antimuscarinic effects of antihistamines: Quantitative evaluation by receptor-binding assay. Jpn. J. Pharmacol. 1987, 43, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Fluman, N.; Adler, J.; Rotenberg, S.A.; Brown, M.H.; Bibi, E. Export of a single drug molecule in two transport cycles by a multidrug efflux pump. Nat. Commun. 2014, 5, 4615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matucci, R.; Bellucci, C.; Martino, M.V.; Nesi, M.; Manetti, D.; Welzel, J.; Bartz, U.; Holze, J.; Tränkle, C.; Mohr, K.; et al. Carbachol dimers with primary carbamate groups as homobivalent modulators of muscarinic receptors. Eur. J. Pharmacol. 2020, 173183. [Google Scholar] [CrossRef]
- Zlotos, D.P.; Buller, S.; Stiefl, N.; Baumann, K.; Mohr, K. Probing the Pharmacophore for Allosteric Ligands of Muscarinic M2Receptors: SAR and QSAR Studies in a Series of Bisquaternary Salts of Caracurine V and Related Ring Systems. J. Med. Chem. 2004, 47, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- De Amici, M.; Traenkle, C.; Dallanoce, C.; Holzgrabe, U.; Mohr, K. ChemInform Abstract: Allosteric Ligands for G Protein-Coupled Receptors: A Novel Strategy with Attractive Therapeutic Opportunities. Med. Res. Rev. 2010, 41, 463–549. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC? A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Beccari, A.R.; Gemei, M.; Monte, M.L.; Menegatti, N.; Fanton, M.; Pedretti, A.; Bovolenta, S.; Nucci, C.; Molteni, A.; Rossignoli, A.; et al. Novel selective, potent naphthyl TRPM8 antagonists identified through a combined ligand- and structure-based virtual screening approach. Sci. Rep. 2017, 7, 10999. [Google Scholar] [CrossRef] [Green Version]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Pedretti, A.; Granito, C.; Mazzolari, A.; Vistoli, G. Structural Effects of Some Relevant Missense Mutations on the MECP2-DNA Binding: A MD Study Analyzed by Rescore+, a Versatile Rescoring Tool of the VEGA ZZ Program. Mol. Inform. 2016, 35, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Dei, S.; Angeli, P.; Bellucci, C.; Buccioni, M.; Gualtieri, F.; Marucci, G.; Manetti, D.; Matucci, R.; Romanelli, M.N.; Scapecchi, S.; et al. Muscarinic subtype affinity and functional activity profile of 1-methyl-2-(2-methyl-1,3-dioxolan-4-yl)pyrrolidine and 1-methyl-2-(2-methyl-1,3-oxathiolan-5-yl)pyrrolidine derivatives. Biochem. Pharmacol. 2005, 69, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Matucci, R.; Nesi, M.; Martino, M.V.; Bellucci, C.; Manetti, D.; Ciuti, E.; Mazzolari, A.; Dei, S.; Guandalini, L.; Teodori, E.; et al. Carbachol dimers as homobivalent modulators of muscarinic receptors. Biochem. Pharmacol. 2016, 108, 90–101. [Google Scholar] [CrossRef]
- Lazareno, S.; Birdsall, N.J. Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: Interactions of strychnine and acetylcholine at muscarinic receptors. Mol. Pharmacol. 1995, 48, 362–378. [Google Scholar] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Wold, E.A.; Chen, J.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem. 2018, 62, 88–127. [Google Scholar] [CrossRef]
- Foster, D.; Conn, P.J. Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Mohr, K.; Tränkle, C.; Kostenis, E.; Barocelli, E.; De Amici, M.; Holzgrabe, U. Rational design of dualsteric GPCR ligands: Quests and promise. Br. J. Pharmacol. 2010, 159, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, K.; Lüllmann, H.; Mohr, K.; Pfeffer, J. Allosteric Stabilization of3H-N-Methylscopolamine Binding in Guinea-Pig Myocardium by an Antidote against Organophosphate Intoxication. Pharmacol. Toxicol. 1988, 63, 163–168. [Google Scholar] [CrossRef]
- Fish, I.; Stößel, A.; Eitel, K.; Valant, C.; Albold, S.; Huebner, H.; Möller, D.; Clark, M.J.; Sunahara, R.K.; Christopoulos, A.; et al. Structure-Based Design and Discovery of New M2 Receptor Agonists. J. Med. Chem. 2017, 60, 9239–9250. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Utilized Protein(s) (PDB Id) | Scores of Best Pose Only | Score Averages of 10 Poses | Score Averages and Ranges of 10 Poses | |||
|---|---|---|---|---|---|---|
| EF 1% Mean a | Best EF 1% | EF 1% Mean a | Best EF 1% | EF 1% Mean a | Best EF 1% | |
| 3UON | 18.1 | 24.1 | 42.8 | 48.2 | 47.5 | 51.6 |
| 4MQT | 59.4 | 65.4 | 54.4 | 65.4 | 64.2 | 68.8 |
| 3UON + 4MQT b | 60.7 | 65.4 | 59.0 | 65.4 | 66.1 | 68.8 |
| Utilized Protein (PDB Id) | Score Types | Equation | EF 1% |
|---|---|---|---|
| 3UON | Best scores | 1.00 ContactsNORM_HEVATMS_Best + 0.031 ChemPLP_Best − 0.014 PLP_Best a | 24.09 |
| 3UON | Mean scores | 1.00 ChemPLP_Mean − 0.75 PLP_Mean − 3.42 PLP95NORM_HEVATMS_Mean | 48.18 |
| 3UON | Means + ranges | 1.00 MLPINS_Range + 0.08847263 ChemPLP_Mean − 1.51 PLP95NORM_HEVATMS_Mean | 51.62 |
| 4MQT | Best scores | 1.00 ChemPLPNORM_HEVATMS_Best + 0.0073 PLP_Best − 4.00 PLP95NORM_HEVATMS_Best | 65.39 |
| 4MQT | Mean scores | 1.00 ChemPLPNORM_HEVATMS_Mean − 4.13 PLP95NORM_HEVATMS_Mean + 1.60 XScore_HM_Mean | 65.39 |
| 4MQT | Means + ranges | 1.00 ContactsNORM_WEIGHT_Range + 0.37 ChemPLPNORM_WEIGHT_Mean − 0.030 PLP95NORM_HEVATMS_Mean | 68.83 |
| Compound | Charge | logP | Known Bioactivity |
|---|---|---|---|
| adefovir dipivoxil | 0 | 1.5 | reverse transcriptase inhibitor |
| aliskiren | 1 | 3.3 | renin inhibitor |
| almitrine | 1 | 4.1 | Na/K-transporting ATPase subunit alpha-1 agonist |
| ambenonium | 2 | 2.3 | cholinesterase inhibitor |
| bepridil | 1 | 5.2 | calcium channel blocker |
| bimatoprost | 0 | 3.4 | structural analogs of prostaglandin |
| carvedilol | 1 | 3.1 | beta adrenoceptor blocker |
| cetirizine | 0 | 2.8 | histamine H1 antagonist |
| deferoxamine | 1 | 0.9 | chelating agent |
| demecarium | 2 | 0.6 | cholinesterase inhibitor |
| dequalinium | 2 | 0.2 | antiseptic and disinfectant agent |
| dinoprostone | −1 | 2.8 | naturally occurring prostaglandin derivative |
| fexofenadine | 1 | 5.0 | histamine H1 antagonist |
| hexafluronium | 2 | 1.8 | neuromuscular blocking agent |
| iloprost | −1 | 4.2 | synthetic analog of prostacyclin |
| ketoconazole | 0 | 4.4 | imidazole antifungal agent |
| lapatinib | 1 | 5.2 | tyrosine kinases inhibitor |
| latanoprost | 0 | 4.2 | prodrug analog of prostaglandin |
| mupirocin | −1 | 2.2 | antibacterial agent |
| orlistat | 0 | 7.5 | pancreatic lipase inhibitor |
| oxybutynin | 1 | 4.3 | antimuscarinic agent |
| phytonadione | 0 | 9.3 | vitamin K1 |
| pimozide | 1 | 6.3 | antipsychotic agent |
| salmeterol | 1 | 3.8 | beta2-adrenergic receptor agonist |
| silodosin | 1 | 3.0 | α1-adrenoceptor antagonist |
| terconazole | 0 | 4.5 | imidazole antifungal agent |
| terfenadine | 1 | 5.9 | histamine H1 antagonist |
| travoprost | 0 | 4.6 | synthetic prostaglandin analog |
| vilazodone | 1 | 4.2 | serotoninergic agent |
| ximelagatran | 1 | 1.4 | Anticoagulant agent |
| Compound | pKi hM1 | pKi hM2 | pKi hM3 | pKi hM4 | pKi hM5 | Log Kocc hM2 |
|---|---|---|---|---|---|---|
| Reference Ligands | ||||||
| W84 | 6.05 ± 0.43 | 5.21 ± 0.15 | 5.26 ± 0.25 | 5.30 ± 0.13 | 5.04 ± 0.16 | 6.54 ± 0.13 |
| gallamine | 6.38 ± 0.16 | 6.91 ± 0.10 | 5.69 ± 0.33 | 6.10 ± 0.12 | 5.66 ± 0.06 | 5.19 ± 0.06 |
| First round | ||||||
| dequalinium | 7.38 ± 0.32 | 6.18 ± 0.16 | 6.77 ± 0.27 | 7.16 ± 0.14 | 6.80 ± 0.10 | 7.72 ± 0.26 |
| terfenadine | 6.11 ± 0.14 | 4.8 ± 0.04 | 5.12 ± 0.21 | 4.96 ± 0.15 | 5.19 ± 0.20 | 4.74 ± 0.06 |
| ketoconazole | 5.43 ± 0.04 | 5.12 ± 0.17 | 5.01 ± 0.12 | 5.34 ± 0.10 | 5.1 ± 0.23 | 4.21 ± 0.08 |
| salmeterol | 5.49 ± 0.01 | 4.98 ± 0.13 | 4.92 ± 0.2 | 5.01 ± 0.12 | 4.9 ± 0.14 | 4.06 ± 0.07 |
| aliskiren | <4 | <4 | <4 | <4 | <4 | 3.60 ± 0.10 |
| orlistat | <4 | <4 | <4 | <4 | <4 | <3 |
| Second Round | ||||||
| chlorhexidine | 6.30 ± 0.10 | 5.42 ± 0.27 | 5.67 ± 0.04 | 5.79 ± 0.06 | 5.65 ± 0.05 | 5.24 ± 0.02 |
| pentamidine | 5.60 ± 0.11 | 6.04 ± 0.08 | 5.67 ± 0.12 | 5.88 ± 0.03 | 5.76 ± 0.05 | 4.73 ± 0.06 |
| diminazene | 5.5 ± 0.18 | 5.25 ± 0.12 | 5.32 ± 0.08 | 5.05 ± 0.05 | 5.32 ± 0.10 | 3.81 ± 0.28 |
| paraquat | 4.43 ± 0.09 | 4.63 ± 0.13 | 4.70 ± 0.07 | <4 | 4.83 ± 0.13 | 3.12 + 0.31 |
| Compound | hM2 | hM1 | hM5 | hM2/hM1 | hM2/hM5 |
|---|---|---|---|---|---|
| W84 | 6.46 ± 0.09 | 5.79 ± 0.09 | 4.74 ± 0.27 | 4.6 | 52.5 |
| Dequalinium | 7.72 ± 0.26 | 6.69 ± 0.10 | 5.48 ± 0.16 | 10.7 | 174 |
| Chlorhexidine | 5.24 ± 0.02 | 4.09 ± 0.11 | 3.73 ± 0.17 | 14 | 32.4 |
| Pentamidine | 4.73 ± 0.06 | 4.18 ± 0.15 | 3.74 ± 0.19 | 3.5 | 9.8 |
| Koff (min−1) (±SEM) | t1/2 (min) (95% C.I.) | |
|---|---|---|
| Control (NMS) | 0.16 ± 0.02 | 4.41 (3.53–5.88) |
| Dequalinium 1 nM | 0.15 ± 0.02 | 4.51 (3.62–5.99) |
| Dequalinium 0.1 µM | 0.033 ± 0.003 | 21.02 (17.95–25.37) |
| Dequalinium 0.3 µM | 0.009 ± 0.001 | 76.54 (64.83–93.42) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzolari, A.; Gervasoni, S.; Pedretti, A.; Fumagalli, L.; Matucci, R.; Vistoli, G. Repositioning Dequalinium as Potent Muscarinic Allosteric Ligand by Combining Virtual Screening Campaigns and Experimental Binding Assays. Int. J. Mol. Sci. 2020, 21, 5961. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175961
Mazzolari A, Gervasoni S, Pedretti A, Fumagalli L, Matucci R, Vistoli G. Repositioning Dequalinium as Potent Muscarinic Allosteric Ligand by Combining Virtual Screening Campaigns and Experimental Binding Assays. International Journal of Molecular Sciences. 2020; 21(17):5961. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175961
Chicago/Turabian StyleMazzolari, Angelica, Silvia Gervasoni, Alessandro Pedretti, Laura Fumagalli, Rosanna Matucci, and Giulio Vistoli. 2020. "Repositioning Dequalinium as Potent Muscarinic Allosteric Ligand by Combining Virtual Screening Campaigns and Experimental Binding Assays" International Journal of Molecular Sciences 21, no. 17: 5961. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175961