The iRhom2/ADAM17 Axis Attenuates Bacterial Uptake by Phagocytes in a Cell Autonomous Manner

 ,
,
Abstract
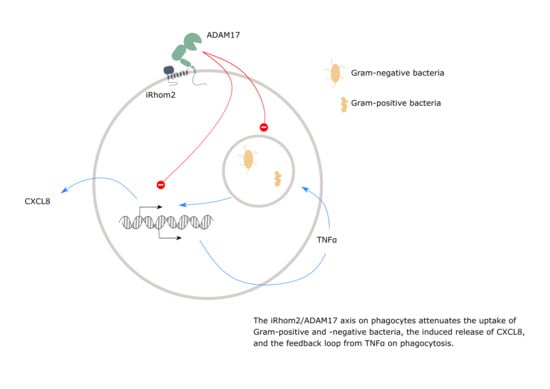
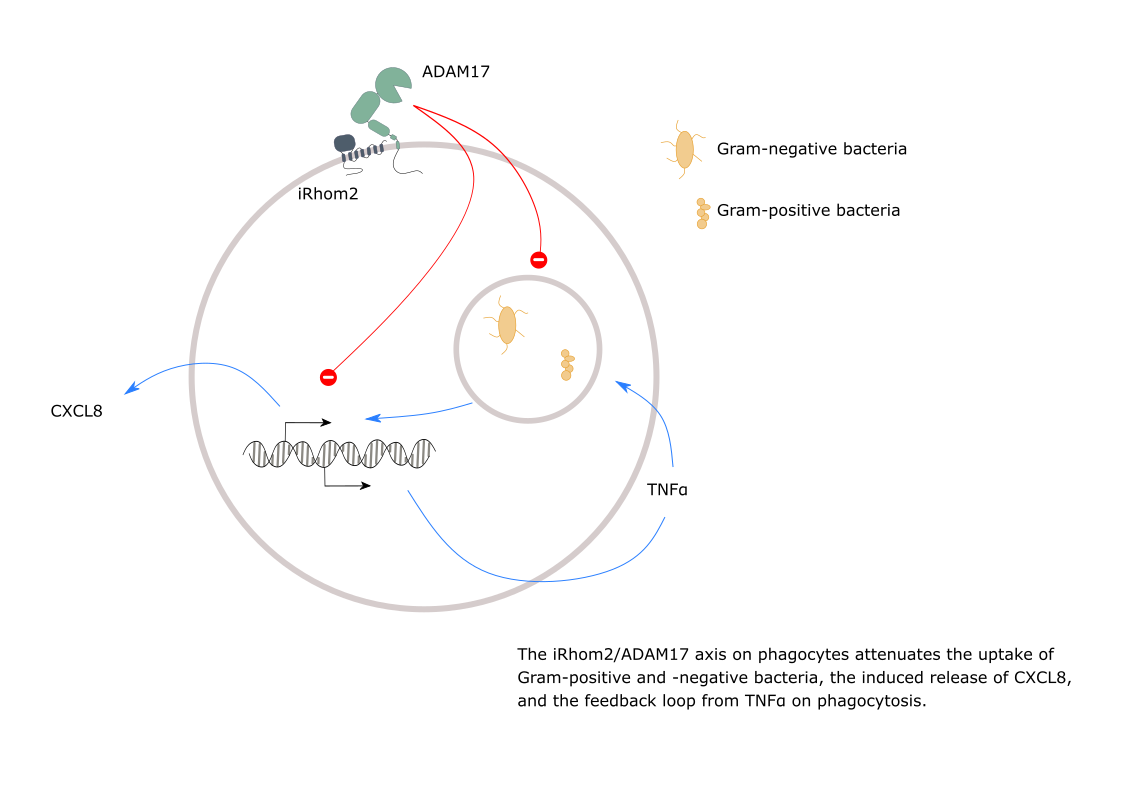
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
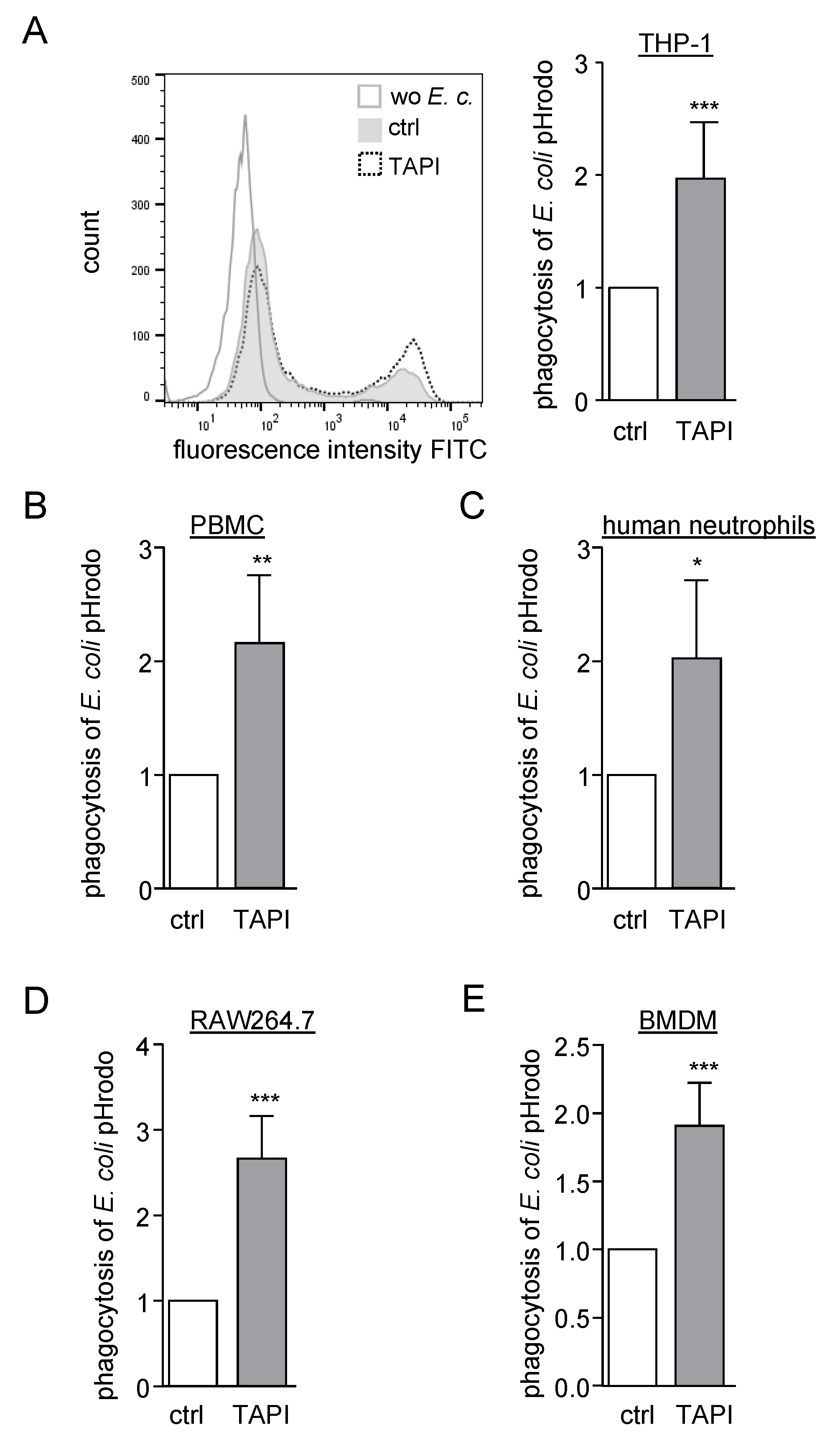
2.1. Inhibition of ADAM Proteases Increases Uptake of pHrodo Labelled Bacteria by Human and Murine Phagocytes
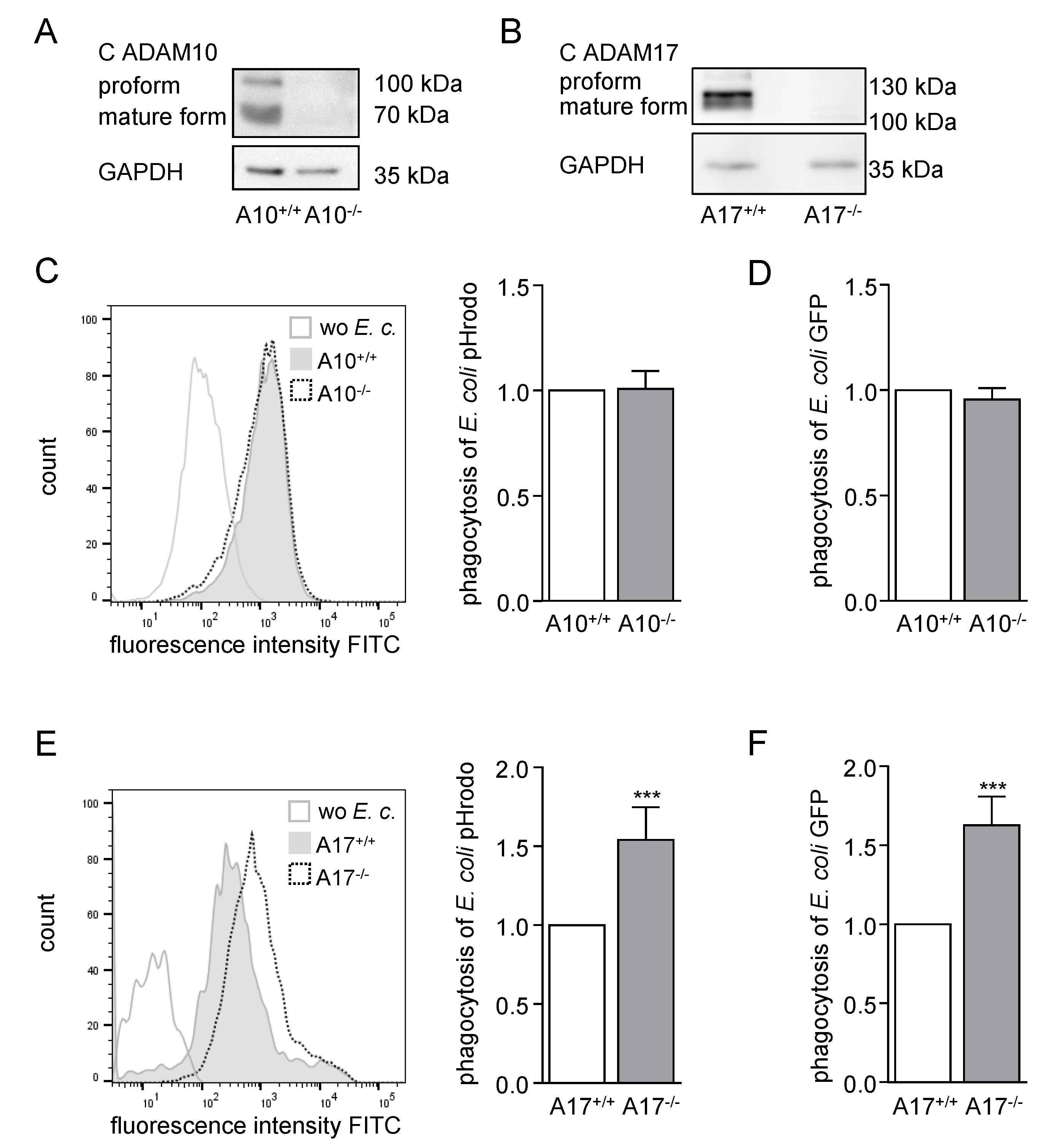
2.2. ADAM17 Deficiency in BMDMs Increases Phagocytosis of Bacteria Ex Vivo
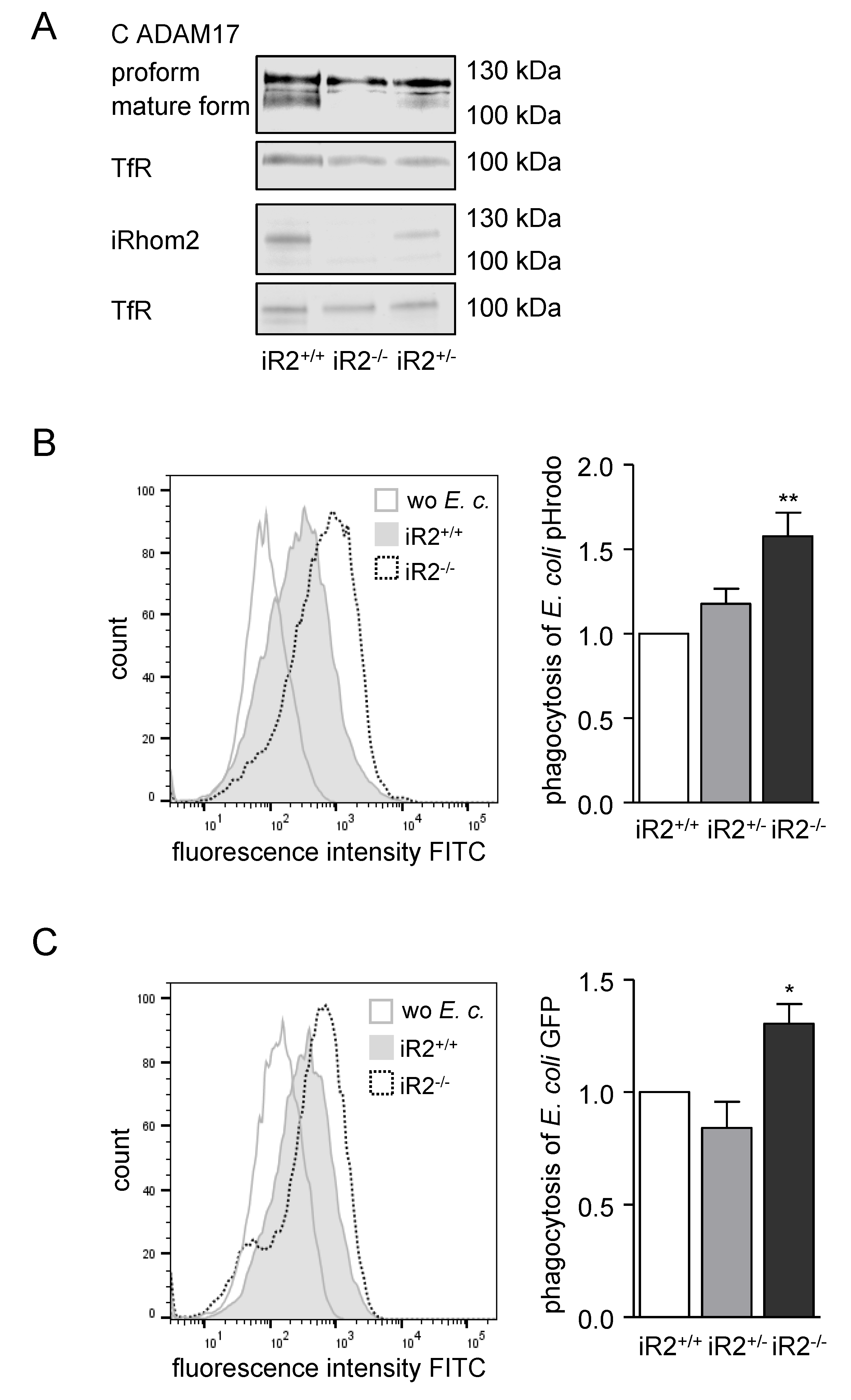
2.3. Deficiency or Inactivation of iRhom2 Leads to Enhanced Phagocytosis Ex Vivo
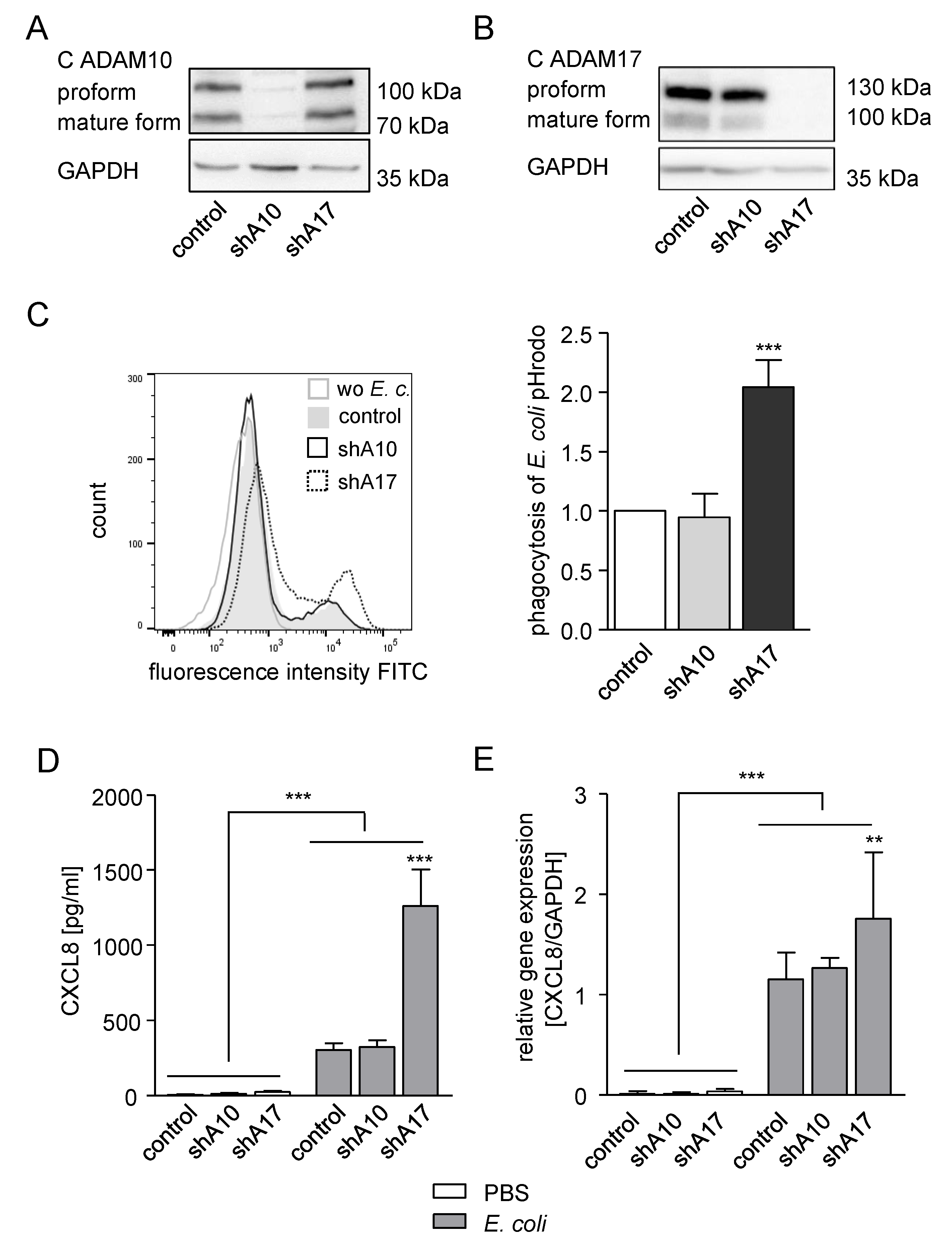
2.4. Knockdown of ADAM17 in THP-1 Cells Enhances Phagocytosis and CXCL8 Release
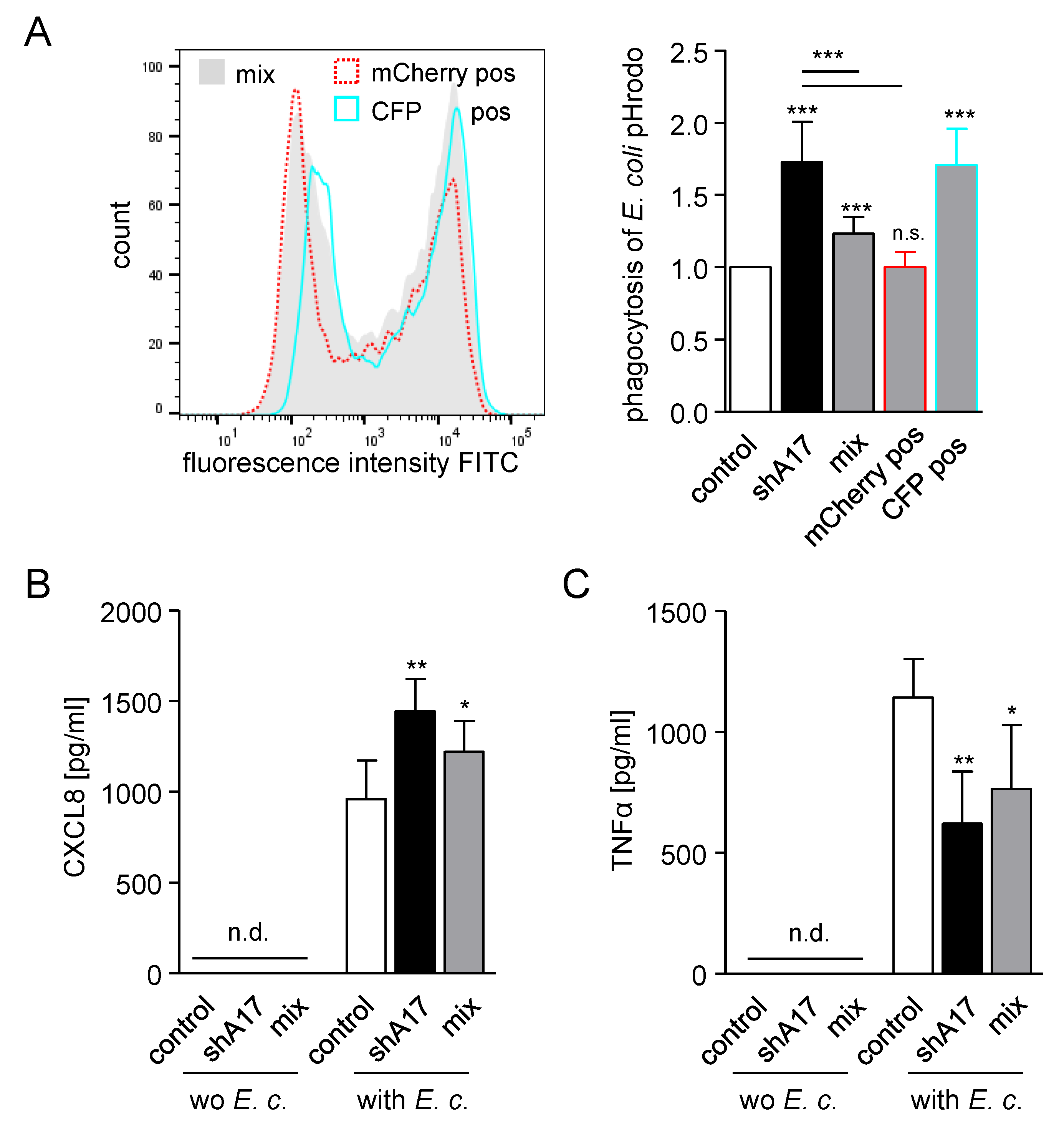
2.5. Knockdown of ADAM17 Increases Phagocytosis in a Cell Autonomous Manner
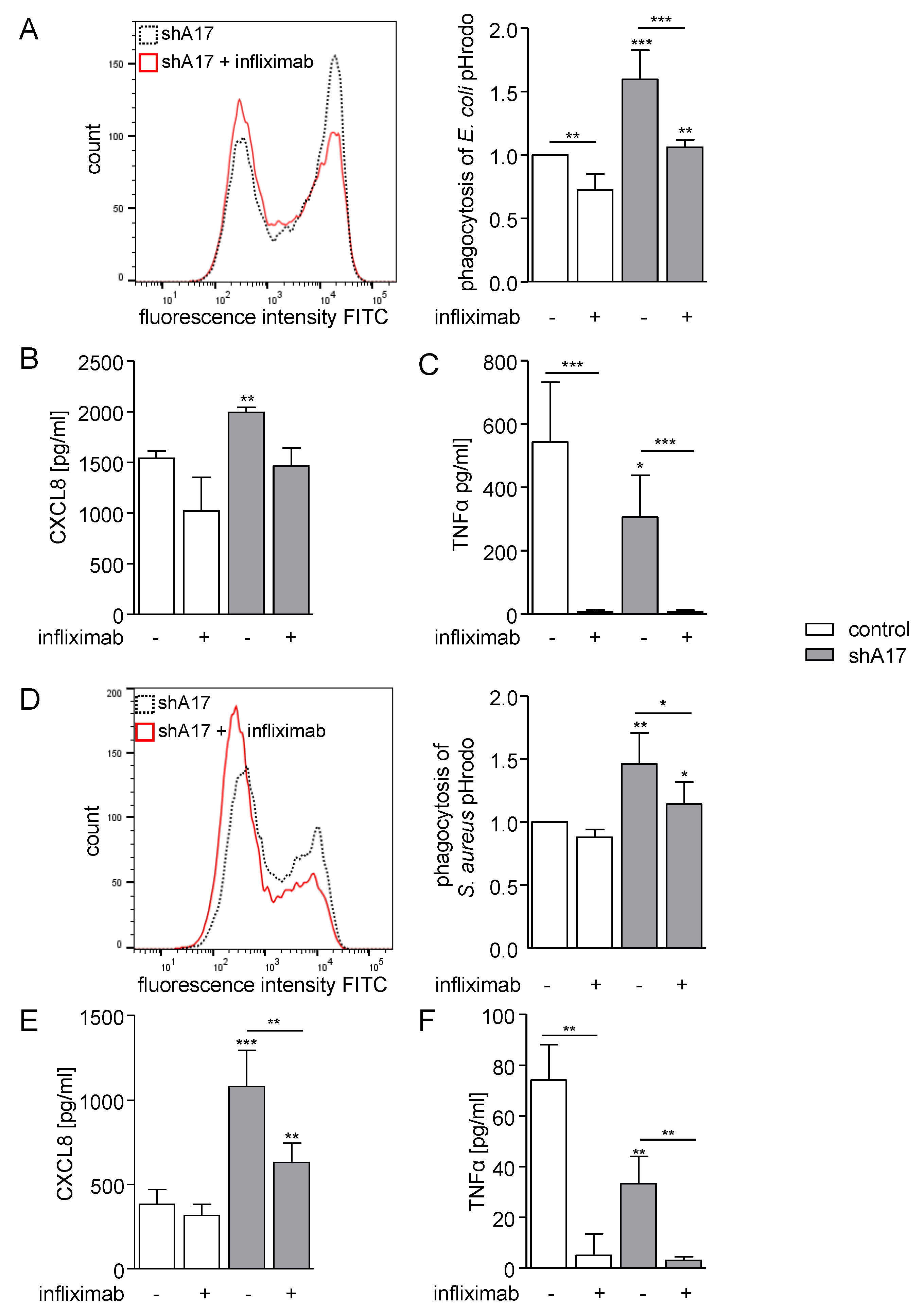
2.6. Inhibition of TNFα Diminishes Phagocytosis and CXCL8 Release
3. Discussion
4. Materials and Methods
4.1. Antibodies, Kits and Reagents
4.2. Cell Culture and Transduction
4.3. Cell Treatment and Phagocytosis Assay
4.4. Flow Cytometric Analysis
4.5. Cell Lysis and Western Blotting
4.6. ELISA
4.7. Quantitative PCR Analysis
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloproteinase |
| BMDM | bone marrow-derived macrophage |
| ctrl | control |
| CFP | cyan fluorescent protein |
| CXCL8 | C-X-C motif chemokine ligand 8 |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| E. coli (E. c.) | Escherichia coli |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| ER | endoplasmic reticulum |
| FBS | fetal bovine serum |
| FDR | false discovery rate |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GFP | green fluorescent protein |
| IL-6R | Interleukin-6 receptor |
| JAM-A | junctional adhesion molecule |
| KO | knockout |
| LPS | lipopolysaccharide |
| MPD | membrane-proximal domain |
| n.d. | not detected |
| S. aureus (S. a.) | Staphylococcus aureus |
| PBMC | peripheral blood mononuclear cell |
| PRR | pattern recognition receptor |
| shRNA | small hairpin RNA |
| TAPI | TAPI-1 |
| TIMP3 | tissue inhibitor of metalloproteinase 3 |
| TLR | toll-like receptor |
| TNFα | tumour necrosis factor-α |
| TNFR | TNF receptor |
| WT | wild type |
References
- Dreymueller, D.; Uhlig, S.; Ludwig, A. Adam-family metalloproteinases in lung inflammation: Potential therapeutic targets. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, 325–343. [Google Scholar] [CrossRef] [Green Version]
- Pruessmeyer, J.; Ludwig, A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 2009, 20, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Düsterhöft, S.; Babendreyer, A.; Giese, A.A.; Flasshove, C.; Ludwig, A. Status update on iRhom and ADAM17: It’s still complicated. Biochim. Biophys. Acta-Mol. Cell Res. 2019, 1866, 1567–1583. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science (80-) 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science (80-) 2012, 335, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science (80-) 2012, 335, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Maretzky, T.; Weskamp, G.; Monette, S.; Qing, X.; Issuree, P.D.A.; Crawford, H.C.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 6080–6085. [Google Scholar] [CrossRef] [Green Version]
- Christova, Y.; Adrain, C.; Bambrough, P.; Ibrahim, A.; Freeman, M. Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 2013, 14, 884–890. [Google Scholar] [CrossRef]
- Issuree, P.D.A.; Maretzky, T.; McIlwain, D.R.; Monette, S.; Qing, X.; Lang, P.A.; Swendeman, S.L.; Park-Min, K.H.; Binder, N.; Kalliolias, G.D.; et al. iRHOM2 is a critical pathogenic mediator of infammatory arthritis. J. Clin. Investig. 2013, 123, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Blaydon, D.C.; Etheridge, S.L.; Risk, J.M.; Hennies, H.C.; Gay, L.J.; Carroll, R.; Plagnol, V.; McRonald, F.E.; Stevens, H.P.; Spurr, N.K.; et al. RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. Am. J. Hum. Genet. 2012, 90, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukerman, P.; Eisenstein, E.M.; Chavkin, M.; Schmiedel, D.; Wong, E.; Werner, M.; Yaacov, B.; Averbuch, D.; Molho-Pessach, V.; Stepensky, P.; et al. Cytokine secretion and NK cell activity in human ADAM17 deficiency. Oncotarget 2015, 6, 44151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaydon, D.C.; Biancheri, P.; Di, W.L.; Plagnol, V.; Cabral, R.M.; Brooke, M.A.; Van Heel, D.A.; Ruschendorf, F.; Toynbee, M.; Walne, A.; et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 2011, 365, 1502–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Takaishi, H.; Okada, Y.; Toyama, Y.; Blobel, C.P. Cutting Edge: TNF-α-Converting Enzyme (TACE/ADAM17) Inactivation in Mouse Myeloid Cells Prevents Lethality from Endotoxin Shock. J. Immunol. 2007, 179, 2686–2689. [Google Scholar] [CrossRef] [Green Version]
- Dreymueller, D.; Martin, C.; Kogel, T.; Pruessmeyer, J.; Hess, F.M.; Horiuchi, K.; Uhlig, S.; Ludwig, A. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol. Med. 2012, 4, 412–423. [Google Scholar] [CrossRef]
- Arndt, P.G.; Strahan, B.; Wang, Y.; Long, C.; Horiuchi, K.; Walcheck, B. Leukocyte adam17 regulates acute pulmonary inflammation. PLoS ONE 2011, 6, e19938. [Google Scholar] [CrossRef]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAMIO regulates endothelial permeability and T-cell transmigration by proteolysis of vascular endothelial cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef] [Green Version]
- Hundhausen, C.; Schulte, A.; Schulz, B.; Andrzejewski, M.G.; Schwarz, N.; von Hundelshausen, P.; Winter, U.; Paliga, K.; Reiss, K.; Saftig, P.; et al. Regulated Shedding of Transmembrane Chemokines by the Disintegrin and Metalloproteinase 10 Facilitates Detachment of Adherent Leukocytes. J. Immunol. 2007, 178, 8064–8072. [Google Scholar] [CrossRef] [Green Version]
- Dreymueller, D.; Martin, C.; Schumacher, J.; Groth, E.; Boehm, J.K.; Reiss, L.K.; Uhlig, S.; Ludwig, A. Smooth Muscle Cells Relay Acute Pulmonary Inflammation via Distinct ADAM17/ErbB Axes. J. Immunol. 2014, 192, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.H.; Herrera, A.H.; Li, Y.; Walcheck, B. Role of ADAM17 in the ectodomain shedding of TNF- and its receptors by neutrophils and macrophages. J. Leukoc. Biol. 2007, 82, 173–176. [Google Scholar] [CrossRef]
- Li, Y.; Brazzell, J.; Herrera, A.; Walcheck, B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood 2006, 108, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Maniecki, M.B.; Møller, K.; Møller, H.J.; Moestrup, S.K. Tumor necrosis factor α-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J. Leukoc. Biol. 2010, 88, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Kneidl, J.; Löffler, B.; Erat, M.C.; Kalinka, J.; Peters, G.; Roth, J.; Barczyk, K. Soluble CD163 promotes recognition, phagocytosis and killing of Staphylococcus aureus via binding of specific fibronectin peptides. Cell. Microbiol. 2012, 14, 914–936. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, W.S.; Vaisar, T.; Tang, J.; Wilson, C.L.; Raines, E.W. Macrophage ADAM17 deficiency augments CD36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circ. Res. 2013, 113, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, P.J.; Garton, K.J.; Wille, P.T.; Rychlewski, M.; Dempsey, P.J.; Raines, E.W. A Disintegrin and Metalloproteinase 10-Mediated Cleavage and Shedding Regulates the Cell Surface Expression of CXC Chemokine Ligand 16. J. Immunol. 2004, 172, 3678–3685. [Google Scholar] [CrossRef] [Green Version]
- Langjahr, P.; Díaz-Jiménez, D.; De La Fuente, M.; Rubio, E.; Golenbock, D.; Bronfman, F.C.; Quera, R.; Lez, M.J.G.; Hermoso, M.A.; Benjamim, C.F. Metalloproteinase-dependent TLR2 ectodomain shedding is involved in soluble toll-like receptor 2 (sTLR2) production. PLoS ONE 2014, 9, e104624. [Google Scholar] [CrossRef]
- Richmond, J.M.; Duffy, E.R.; Lee, J.; Kaboli, K.; Remick, D.G.; Kornfeld, H.; Cruikshank, W.W. Mannose-capped lipoarabinomannan from Mycobacterium tuberculosis induces soluble tumor necrosis factor receptor production through tumor necrosis factor alpha-converting enzyme activation. Infect. Immun. 2012, 80, 3858–3868. [Google Scholar] [CrossRef] [Green Version]
- Long, C.; Wang, Y.; Herrera, A.H.; Horiuchi, K.; Walcheck, B. In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli -mediated peritonitis. J. Leukoc. Biol. 2010, 87, 1097–1101. [Google Scholar] [CrossRef] [Green Version]
- Long, C.; Hosseinkhani, M.R.; Wang, Y.; Sriramarao, P.; Walcheck, B. ADAM17 activation in circulating neutrophils following bacterial challenge impairs their recruitment. J. Leukoc. Biol. 2012, 92, 667–672. [Google Scholar] [CrossRef]
- Sommer, D.; Corstjens, I.; Sanchez, S.; Dooley, D.; Lemmens, S.; Van Broeckhoven, J.; Bogie, J.; Vanmierlo, T.; Vidal, P.M.; Rose-John, S.; et al. ADAM17-deficiency on microglia but not on macrophages promotes phagocytosis and functional recovery after spinal cord injury. Brain. Behav. Immun. 2019, 80, 129–145. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-∅ from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.; Taubitz, A.; Eltrich, N.; Mulay, S.R.; Allam, R.; Vielhauer, V. Analysis of TNF-mediated recruitment and activation of glomerular dendritic cells in mouse kidneys by compartment-specific flow cytometry. Kidney Int. 2013, 84, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.; Schuster, B.; Schütze, S.; Bussmeyer, I.; Ludwig, A.; Hundhausen, C.; Sadowski, T.; Saftig, P.; Hartmann, D.; Kallen, K.J.; et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 2003, 278, 38829–38839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalaris, A.; Gewiese, J.; Paliga, K.; Fleig, L.; Schneede, A.; Krieger, K.; Rose-John, S.; Scheller, J. ADAM17-mediated shedding of the IL6R induces cleavage of the membrane stub by γ-secretase. Biochim. Biophys. Acta-Mol. Cell Res. 2010, 1803, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Friedland, J.S.; Constantin, D.; Shaw, T.C.; Stylianou, E. Regulation of interleukin-8 gene expression after phagocytosis of zymosan by human monocytic cells. J. Leukoc. Biol. 2001, 70, 447–454. [Google Scholar] [CrossRef]
- Friedland, J.S.; Remick, D.G.; Shattock, R.; Griffin, G.E. Secretion of interleukin-8 following phagocytosis of Mycobacterium tuberculosis by human monocyte cell lines. Eur. J. Immunol. 1992, 22, 1373–1378. [Google Scholar] [CrossRef]
- Kang, H.J.; Ha, J.M.; Kim, H.S.; Lee, H.; Kurokawa, K.; Lee, B.L. The role of phagocytosis in IL-8 production by human monocytes in response to lipoproteins on Staphylococcus aureus. Biochem. Biophys. Res. Commun. 2011, 406, 449–453. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Wang, J.; Dong, Z.; Mian, S.; Yu, F.S.X. Role of EGFR transactivation in preventing apoptosis in Pseudomonas aeruginosa-infected human corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2569–2576. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Kim, J.J.; Lee, M.J.; Lee, E.K.; Park, S.K. ADAM17-Mediated Ectodomain Shedding of Toll-Like Receptor 4 as a Negative Feedback Regulation in Lipopolysaccharide-Activated Aortic Endothelial Cells. Cell. Physiol. Biochem. 2018, 45, 1851–1862. [Google Scholar] [CrossRef]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef] [Green Version]
- Yoda, M.; Kimura, T.; Tohmonda, T.; Morioka, H.; Matsumoto, M.; Okada, Y.; Toyama, Y.; Horiuchi, K. Systemic Overexpression of TNFα-converting Enzyme Does Not Lead to Enhanced Shedding Activity In Vivo. PLoS ONE 2013, 8, e54412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düsterhöft, S.; Michalek, M.; Kordowski, F.; Oldefest, M.; Sommer, A.; Röseler, J.; Reiss, K.; Grötzinger, J.; Lorenzen, I. Extracellular Juxtamembrane Segment of ADAM17 Interacts with Membranes and Is Essential for Its Shedding Activity. Biochemistry 2015, 54, 5791–5801. [Google Scholar] [CrossRef] [PubMed]
- Pruessmeyer, J.; Hess, F.M.; Alert, H.; Groth, E.; Pasqualon, T.; Schwarz, N.; Nyamoya, S.; Kollert, J.; Van Der Vorst, E.; Donners, M.; et al. Leukocytes require ADAM10 but not ADAM17 for their migration and inflammatory recruitment into the alveolar space. Blood 2014, 123, 4077–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenen, R.R.; Pruessmeyer, J.; Soehnlein, O.; Fraemohs, L.; Zernecke, A.; Schwarz, N.; Reiss, K.; Sarabi, A.; Lindbom, L.; Hackeng, T.M.; et al. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood 2009, 113, 4799–4809. [Google Scholar] [CrossRef] [Green Version]
- Pruessmeyer, J.; Martin, C.; Hess, F.M.; Schwarz, N.; Schmidt, S.; Kogel, T.; Hoettecke, N.; Schmidt, B.; Sechi, A.; Uhlig, S.; et al. A Disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J. Biol. Chem. 2010, 285, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Babendreyer, A.; Molls, L.; Simons, I.M.; Dreymueller, D.; Biller, K.; Jahr, H.; Denecke, B.; Boon, R.A.; Bette, S.; Schnakenberg, U.; et al. The metalloproteinase ADAM15 is upregulated by shear stress and promotes survival of endothelial cells. J. Mol. Cell. Cardiol. 2019, 134, 51–61. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seifert, A.; Wozniak, J.; Düsterhöft, S.; Kasparek, P.; Sedlacek, R.; Dreschers, S.; Orlikowsky, T.W.; Yildiz, D.; Ludwig, A. The iRhom2/ADAM17 Axis Attenuates Bacterial Uptake by Phagocytes in a Cell Autonomous Manner. Int. J. Mol. Sci. 2020, 21, 5978. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175978
Seifert A, Wozniak J, Düsterhöft S, Kasparek P, Sedlacek R, Dreschers S, Orlikowsky TW, Yildiz D, Ludwig A. The iRhom2/ADAM17 Axis Attenuates Bacterial Uptake by Phagocytes in a Cell Autonomous Manner. International Journal of Molecular Sciences. 2020; 21(17):5978. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175978
Chicago/Turabian StyleSeifert, Anke, Justyna Wozniak, Stefan Düsterhöft, Petr Kasparek, Radislav Sedlacek, Stephan Dreschers, Thorsten W. Orlikowsky, Daniela Yildiz, and Andreas Ludwig. 2020. "The iRhom2/ADAM17 Axis Attenuates Bacterial Uptake by Phagocytes in a Cell Autonomous Manner" International Journal of Molecular Sciences 21, no. 17: 5978. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175978