Blockade of Hemichannels Normalizes the Differentiation Fate of Myoblasts and Features of Skeletal Muscles from Dysferlin-Deficient Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
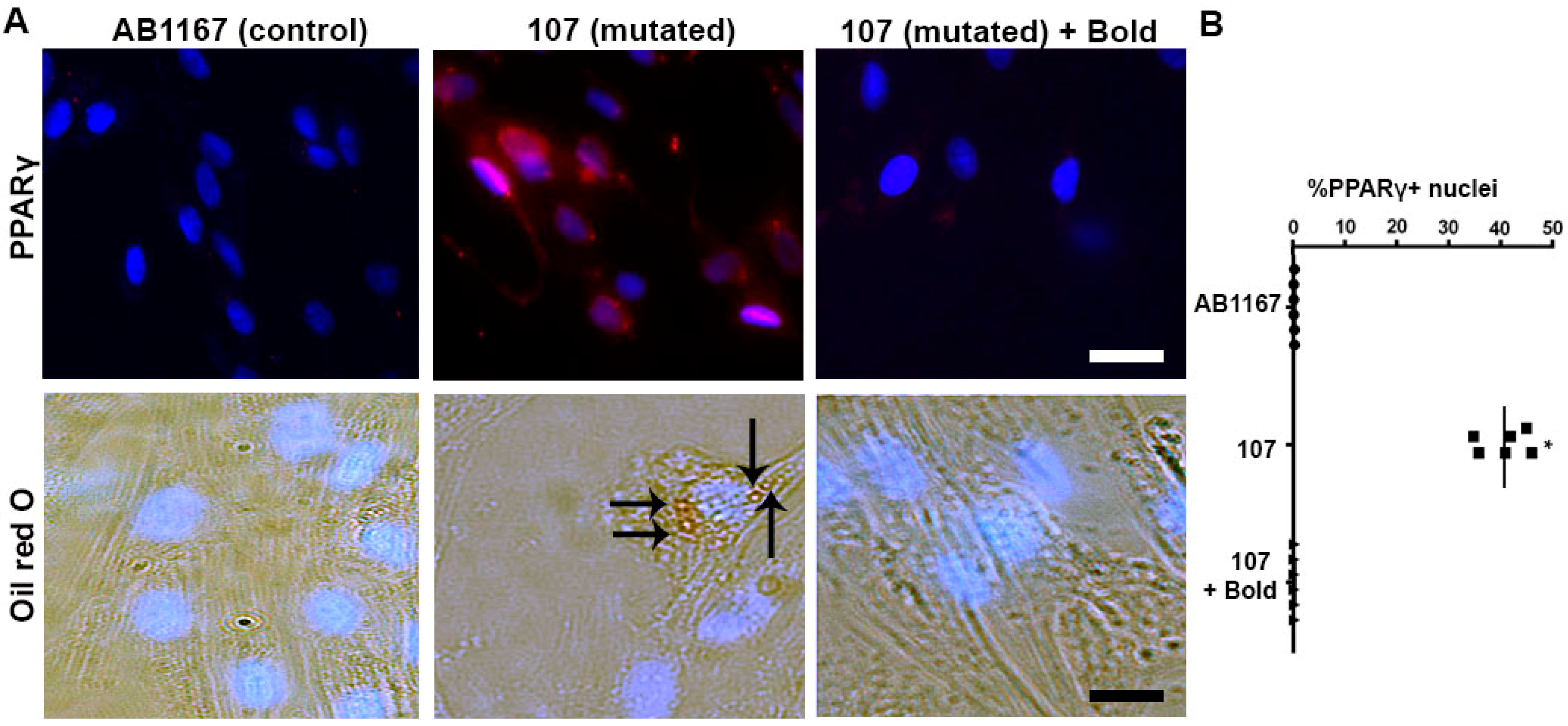
2.1. Boldine Rectifies the Aberrant Adipogenic Commitment of Dysferlin-Deficient Myoblasts
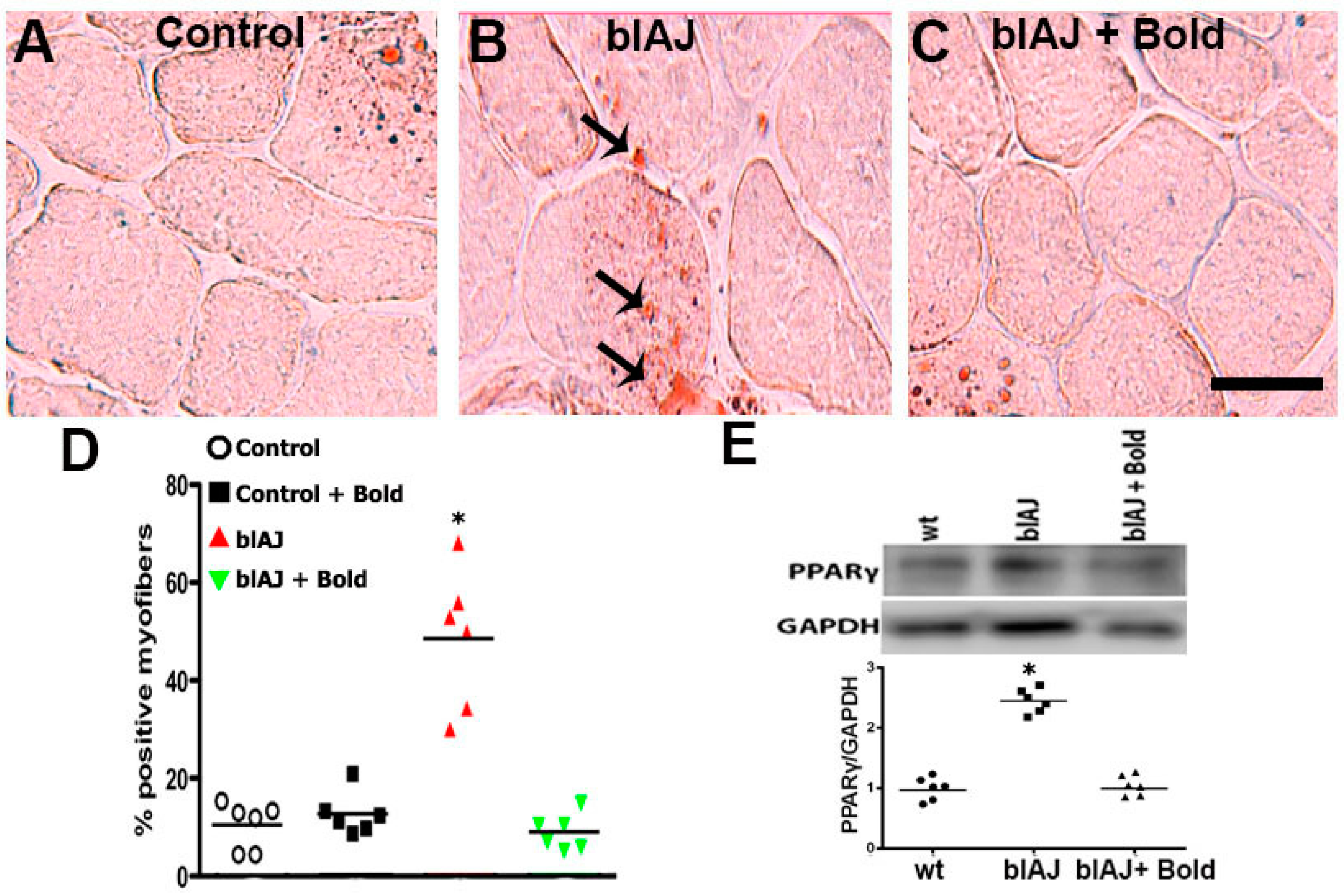
2.2. Connexin Hemichannels Are Involved in the Accumulation of Fat within the Skeletal Muscles of blAJ Mice
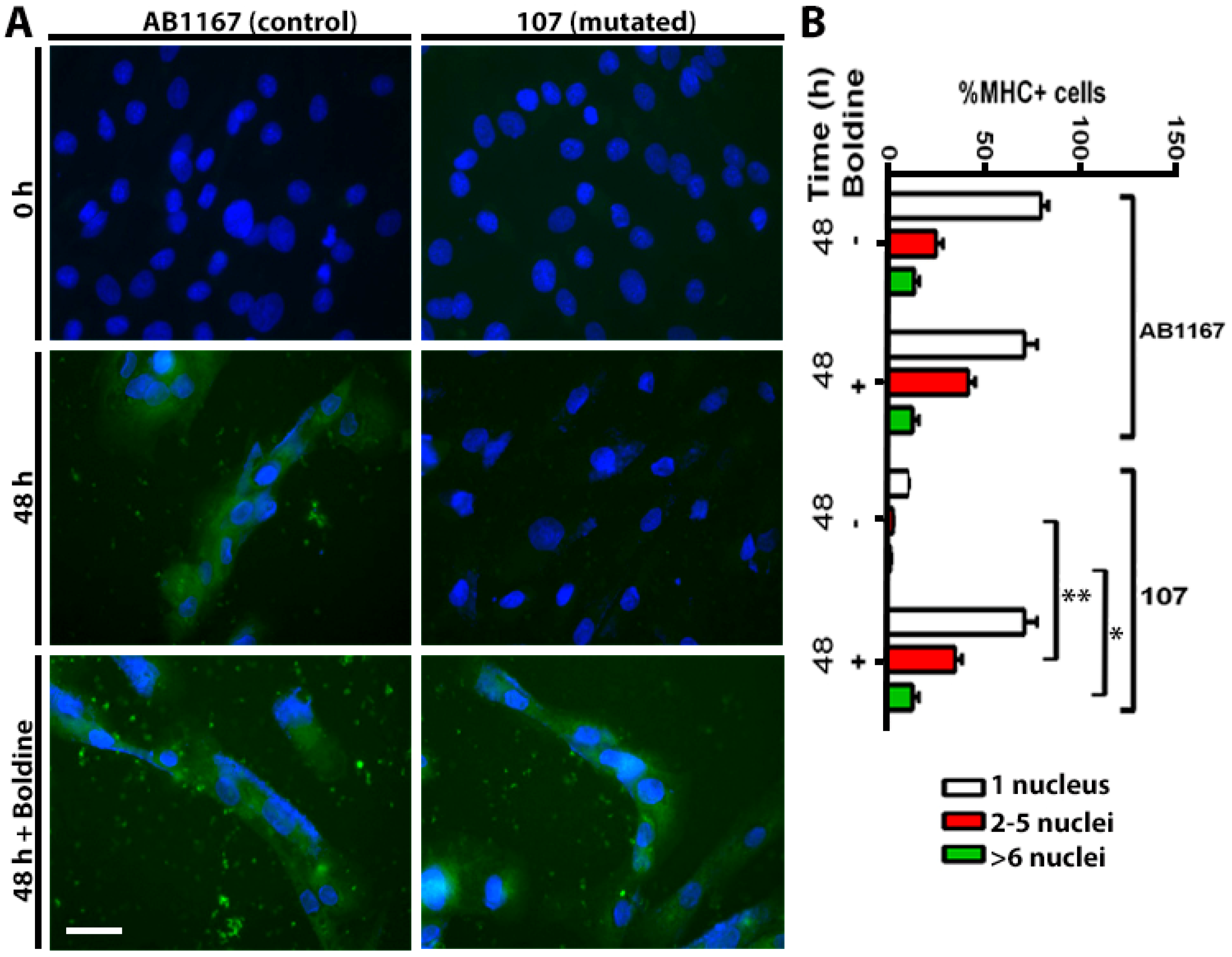
2.3. Boldine Favors Dysferlin-Deficient Myoblasts Differentiation to Myotubes
2.4. Blockade of Functional Connexin Hemichannels Restores the Sarcolemma Permeability Features of Skeletal Muscles from blAJ Mice
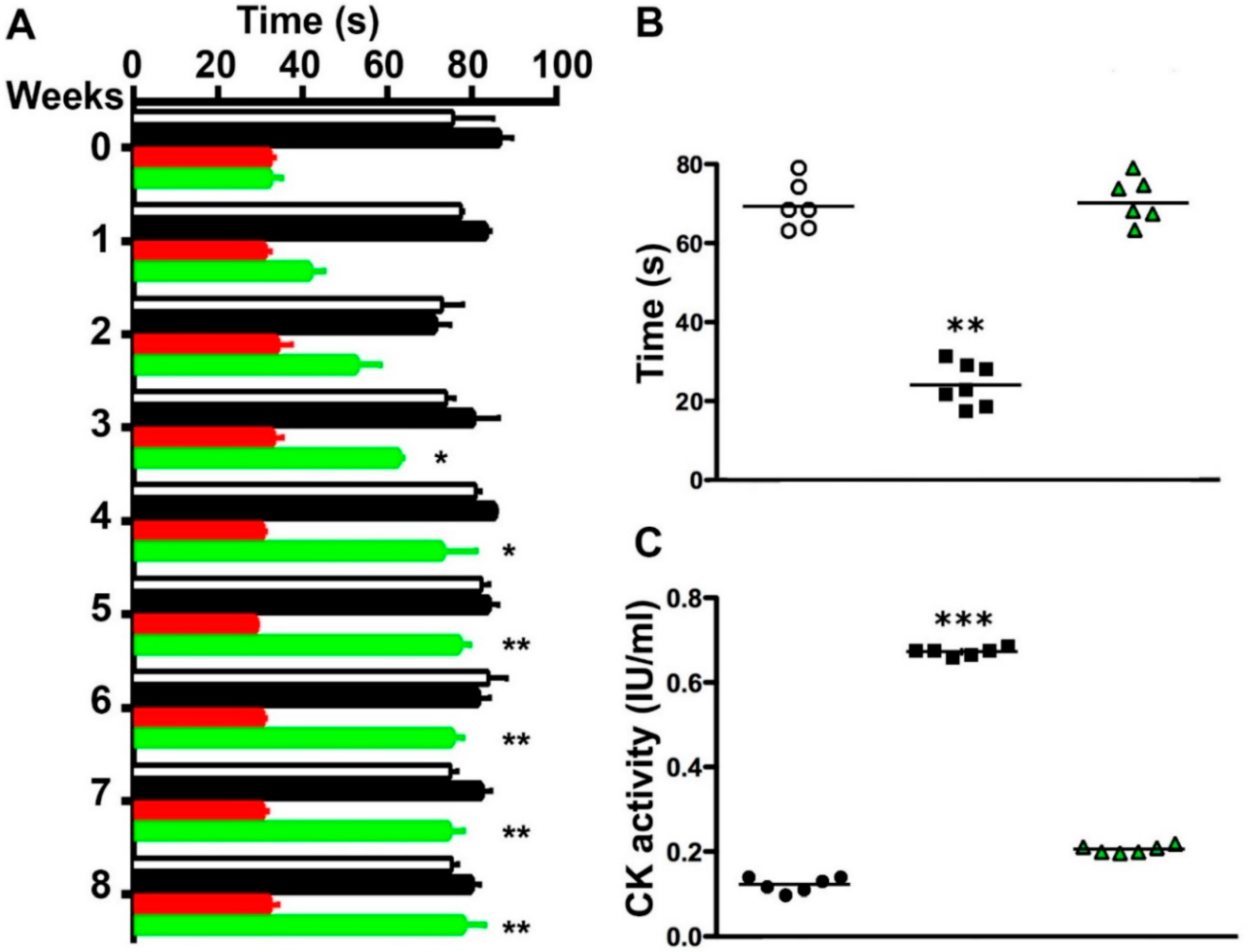
2.5. Boldine Induces Progressive Recovery of Motor Performance and Abrogates Muscle Damage in Symptomatic blAJ Mice
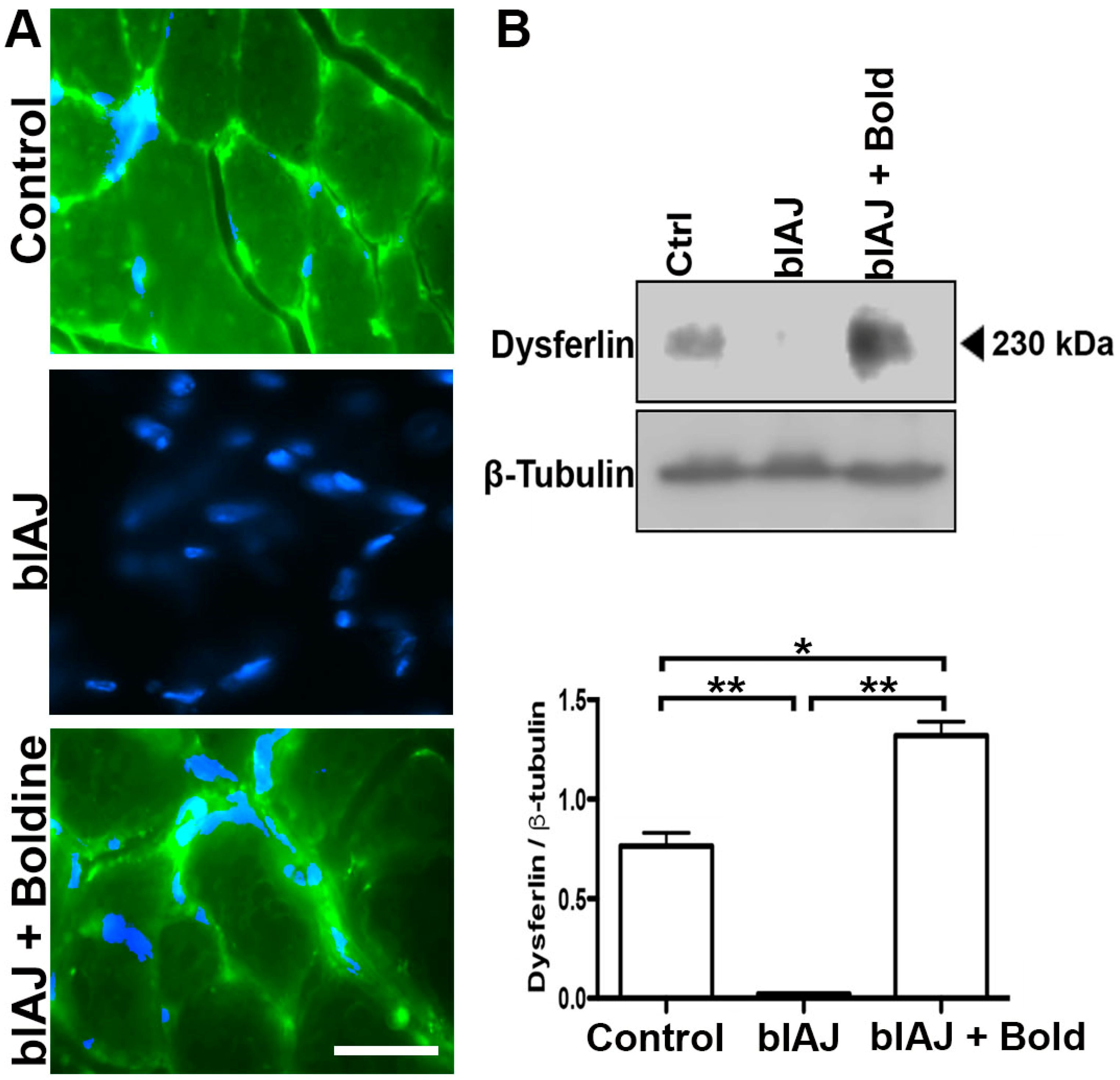
2.6. Dysferlin-Like Immunoreactivity Is Recovered in Skeletal Muscles of blAJ Mice Treated with Boldine
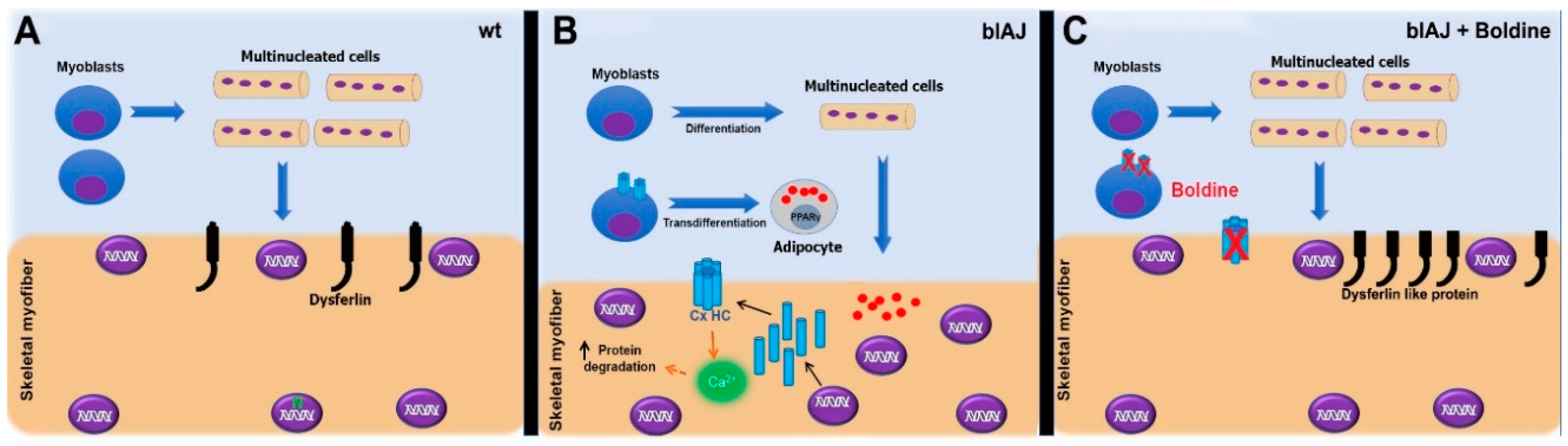
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Human Control and Dysferlin-Deficient Myoblast Cell Line
4.4. Isolation of Mouse Skeletal Myofibers
4.5. Boldine Treatment
4.6. Immunofluorescence Analysis
4.7. Oil Red O Staining
4.8. Western Blot Analysis
4.9. Evans Blue Uptake In Vivo
4.10. Time-Lapse Recording of Etd+ Uptake
4.11. Intracellular Ca2+ Signal
4.12. Cross-Sectional Area (CSA) Measurements
4.13. Creatine Kinase (CK) Activity
4.14. Physical Exercise Tests (Rotarod Test and Four Limbs Hanging Test)
5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PBS | Phosphate-buffered saline |
| Cx | Connexin |
| Cx43 HC | Connexin43 hemichannel |
| Cx45 HC | Connexin45 hemichannel. |
| Cx39 HCs | Connexin39 hemichannels. |
| EB4- | Evans blue |
| Etd+ | Ethidium bromide |
| CSA | Cross-sectional area |
| CK | Creatine Kinase |
| Cbx | Carbenoxolone |
| GC | Gastrocnemius |
| MHC | Myosin Heavy Chain |
| SEM | Standard error of the mean |
References
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Lostal, W.; Bartoli, M.; Roudaut, C.; Bourg, N.; Krahn, M.; Pryadkina, M.; Borel, P.; Suel, L.; Roche, J.A.; Stockholm, D.; et al. Lack of correlation between outcomes of membrane repair assay and correction of dystrophic changes in experimental therapeutic strategy in dysferlinopathy. PLoS ONE 2012, 7, e38036. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Andrés, D.; Díaz, J.; Munell, F.; Sánchez-Montáñez, Á.; Pulido-Valdeolivas, I.; Suazo, L.; Garrido, C.; Quijano-Roy, S.; Bevilacqua, J.A. Disease duration and disability in dysferlinopathy can be described by muscle imaging using heatmaps and random forests. Muscle Nerve 2019, 59, 436–444. [Google Scholar] [CrossRef]
- Haynes, V.R.; Keenan, S.N.; Bayliss, J.; Lloyd, E.M.; Meikle, P.J.; Grounds, M.D.; Watt, M.J. Dysferlin deficiency alters lipid metabolism and remodels the skeletal muscle lipidome in mice. J. Lipid Res. 2019, 60, 1350–1364. [Google Scholar] [CrossRef]
- Cea, L.A.; Bevilacqua, J.A.; Arriagada, C.; Cárdenas, A.M.; Bigot, A.; Mouly, V.; Sáez, J.C.; Caviedes, P. The absence of dysferlin induces the expression of functional connexin-based hemichannels in human myotubes. BMC Cell Biol. 2016, 17 (Suppl. 15), 127–136. [Google Scholar] [CrossRef] [Green Version]
- Cea, L.A.; Cisterna, B.A.; Puebla, C.; Frank, M.; Figueroa, X.F.; Cardozo, C.; Willecke, K.; Latorre, R.; Sáez, J.C. De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 16229–16234. [Google Scholar] [CrossRef] [Green Version]
- Cea, L.A.; Balboa, E.; Puebla, C.; Vargas, A.A.; Cisterna, B.A.; Escamilla, R.; Regueira, T.; Sáez, J.C. Dexamethasone-induced muscular atrophy is mediated by functional expression of connexin-based hemichannels. Biochim. Biophys. Acta 2016, 1862, 1891–1899. [Google Scholar] [CrossRef]
- Cea, L.A.; Balboa, E.; Vargas, A.A.; Puebla, C.; Brañes, M.C.; Escamilla, R.; Regueira, T.; Sáez, J.C. De novo expression of functional connexins 43 and 45 hemichannels increases sarcolemmal permeability of skeletal myofibers during endotoxemia. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2765–2773. [Google Scholar] [CrossRef]
- D’hondt, C.; Ponsaerts, R.; De Smedt, H.; Bultynck, G.; Himpens, B. Pannexins, distant relatives of the connexin family with specific cellular functions? Bioessays 2009, 31, 953–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine prevents renal alterations in diabetic rats. J. Diabetes Res. 2013, 2013, 593672. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.; Ezan, P.; Fernández, P.; Schmitt, J.; Sáez, J.C.; Giaume, C.; Koulakoff, A. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017, 65, 1607–1625. [Google Scholar] [CrossRef] [PubMed]
- Lostal, W.; Bartoli, M.; Bourg, N.; Roudaut, C.; Bentaib, A.; Miyake, K.; Guerchet, N.; Fougerousse, F.; McNeil, P.; Richard, I. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum. Mol. Genet. 2010, 19, 1897–1907. [Google Scholar] [CrossRef] [Green Version]
- Grounds, M.D.; Terrill, J.R.; Radley-Crabb, H.G.; Robertson, T.; Papadimitriou, J.; Spuler, S.; Shavlakadze, T. Lipid accumulation in dysferlin-deficient muscles. Am. J. Pathol. 2014, 184, 1668–1676. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Tunison, K.; Mitsche, M.A.; McDonald, J.G.; Garg, A. Insights into lipid accumulation in skeletal muscle in dysferlin-deficient mice. J. Lipid Res. 2019, 60, 2057–2073. [Google Scholar] [CrossRef] [PubMed]
- Fernández, G.; Arias, G.B.; Bevilacqua, J.A.; Castillo, M.; Caviedes, P.; Sáez, J.C.; Cea, L.A. Myofibers deficient in connexins 43 and 45 expression protect mice from skeletal muscle and systemic dysfunction promoted by a dysferlin mutation. Biochem Biophys Acta Mol. Basis Dis 2020, 1866, 165800. [Google Scholar] [CrossRef] [PubMed]
- Proulx, A.; Merrifield, P.A.; Naus, C.C. Blocking gap junctional intercellular communication in myoblasts inhibits myogenin and MRF4 expression. Dev. Genet. 1997, 20, 133–144. [Google Scholar] [CrossRef]
- Burr, A.R.; Molkentin, J.D. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015, 22, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- Farini, A.; Sitzia, C.; Navarro, C.; D’Antona, G.; Belicchi, M.; Parolini, D.; Del Fraro, G.; Razini, P.; Bottinelli, R.; Meregalli, M.; et al. Absence of T and B lymphocytes modulates dystrophic features in dysferlin deficient animal model. Exp. Cell Res. 2012, 318, 1160–1174. [Google Scholar] [CrossRef]
- Galassi, G.; Rowland, L.P.; Hays, A.P.; Hopkins, L.C.; Di Mauro, S. High serum levels of creatine kinase: Asymptomatic prelude to distal myopathy. Muscle Nerve 1987, 10, 346–350. [Google Scholar] [CrossRef]
- Haddix, S.G.; Lee, Y.I.; Kornegay, J.N.; Thompson, W.J. Cycles of myofiber degeneration and regeneration lead to remodeling of the neuromuscular junction in two mammalian models of Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0205926. [Google Scholar] [CrossRef]
- Ho, M.; Post, C.M.; Donahue, L.R.; Lidov, H.G.W.; Bronson, R.T.; Goolsby, H.; Watkins, S.C.; Cox, G.A.; Robert Brown, R.J. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum. Mol. Genet. 2004, 13, 1999–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, M.E.; Pinto-González, D.; Orson, F.M.; McKenzie, G.J.; Petry, G.R.; Barry, M.A. Role of endogenous endonucleases and tissue site in transfection and CpG-mediated immune activation after naked DNA injection. Hum. Gene 1999, 10, 2461–2480. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Das, A.; Chakrabarti, S.; Chakrabarti, O. Calcium dependent regulation of protein ubiquitination—Interplay between E3 ligases and calcium binding proteins. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Utokaparch, S.; Sharma, A.; Yu, C.; Abraham, T.; Borchers, C.; Bernatchez, P. Proteomic identification of dysferlin-interacting protein complexes in human vascular endothelium. Biochem. Biophys. Res. Commun. 2011, 415, 263–269. [Google Scholar]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Fulco, M.; et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, S.; Xie, L.-H.; John, S.A.; Subramaniam, S.; Lal, R. A Novel Role for Connexin Hemichannel in Oxidative Stress and Smoking-Induced Cell Injury. PLoS ONE 2007, 2, e712. [Google Scholar] [CrossRef] [Green Version]
- Aguiari, P.; Leo, S.; Zavan, B.; Vindigni, V.; Rimessi, A.; Bianchi, K.; Franzin, C.; Cortivo, R.; Rossato, M.; Vettor, R.; et al. High glucose induces adipogenic differentiation of muscle-derived stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 1226–1231. [Google Scholar] [CrossRef] [Green Version]
- Chi, J.; Li, L.; Liu, M.; Tan, J.; Tang, C.; Pan, Q.; Wang, D.; Zhang, Z. Pathogenic Connexin-31 Forms Constitutively Active Hemichannels to Promote Necrotic Cell Death. PLoS ONE 2012, 7, e32531. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, W.; Nie, Y.; Qaher, M.; Horton, H.E.; Yue, F.; Asakura, A.; Kuang, S. Loss of MyoD promotes fate transdifferentiation of myoblasts into Bbrown adipocytes. EBioMedicine 2017, 16, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.-H.; Hornsey, M.A.; Klinge, L.; Jørgensen, L.H.; Laval, S.H.; Charlton, R.; Barresi, R.; Straub, V.; Lochmüller, H.; Bushby, K. Attenuated muscle regeneration is a key factor in dysferlin-deficient muscular dystrophy. Hum. Mol. Genet. 2009, 18, 1976–1989. [Google Scholar] [CrossRef] [Green Version]
- Galvin, J.E.; Palamand, D.; Strider, J.; Milone, M.; Pestronk, A. The muscle protein dysferlin accumulates in the Alzheimer brain. Acta Neuropathol. 2006, 112, 665–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vuyst, E.; Decrock, E.; Cabooter, L.; Dubyak, G.R.; Naus, C.C.; Evans, W.H.; Leybaert, L. Intracellular calcium changes trigger connexin 32 hemichannel opening. Embo J. 2006, 25, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retamal, M.A.; Schalper, K.A.; Shoji, K.F.; Bennett, M.V.L.; Sáez, J.C. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc. Natl. Acad. Sci. USA 2007, 104, 8322–8327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, X.F.; Lillo, M.A.; Gaete, P.S.; Riquelme, M.A.; Sáez, J.C. Diffusion of nitric oxide across cell membranes of the vascular wall requires specific connexin-based channels. Neuropharmacology 2013, 75, 471–478. [Google Scholar] [CrossRef]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Sáez, P.J.; Sáez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines Released from Activated Microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Morozzi, G.; Beccafico, S.; Bianchi, R.; Riuzzi, F.; Bellezza, I.; Giambanco, I.; Arcuri, C.; Minelli, A.; Donato, R. Oxidative stress-induced S100B accumulation converts myoblasts into brown adipocytes via an NF-κB/YY1/miR-133 axis and NF-κB/YY1/BMP-7 axis. Cell Death Differ. 2017, 24, 2077–2088. [Google Scholar] [CrossRef]
- Gu, W.; Schneider, J.W.; Condorelli, G.; Kaushal, S.; Mahdavi, V.; Nadal-Ginard, B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell 1993, 72, 309–324. [Google Scholar] [CrossRef]
- Shao, D.; Lazar, M.A. Peroxisome proliferator activated receptor gamma, CCAAT/enhancer-binding protein alpha, and cell cycle status regulate the commitment to adipocyte differentiation. J. Biol. Chem. 1997, 272, 21473–21478. [Google Scholar] [CrossRef] [Green Version]
- Cisterna, B.A.; Vargas, A.A.; Puebla, C.; Fernández, P.; Escamilla, R.; Lagos, C.F.; Matus, M.F.; Vilos, C.; Cea, L.A.; Barnafi, E.; et al. Active acetylcholine receptors prevent the atrophy of skeletal muscles and favor reinnervation. Nat. Commun. 2020, 11, 1073. [Google Scholar] [CrossRef] [Green Version]
- Krajacic, P.; Pistilli, E.E.; Tanis, J.E.; Khurana, T.S.; Lamitina, S.T. FER-1/Dysferlin promotes cholinergic signaling at the neuromuscular junction in C. elegans and mice. Biol. Open 2013, 2, 1245–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostelnik, A.; Pohanka, M. Inhibition of acetylcholinesterase and butyrylcholinesterase by a plant secondary metabolite boldine. Biomed. Res. Int. 2018, 2018, 9634349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.J.; Cheng, Y.-W.; Fu, W.-M. Studies on neuromuscular blockade by boldine in the mouse phrenic-diafragm. Jpn. J. Pharm. 1998, 76, 207–211. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.; Carrasco-Pzo, C.; Speisky, H.C. Boldine and its antioxidant or health-promoting properties. Chem. Biol. Interact. 2006, 159, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Balboa, E.; Saavedra, F.; Cea, L.A.; Ramírez, V.; Escamilla, R.; Vargas, A.A.; Regueira, T.; Sáez, J.C. Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment on Skeletal Muscles. Int. J. Mol. Sci. 2020, 21, 4094. [Google Scholar] [CrossRef]
- Cisterna, B.A.; Vargas, A.A.; Puebla, C.; Sáez, J.C. Connexin hemichannels explain the ionic imbalance and lead to atrophy in denervated skeletal muscles. Biochim. Biophys. Acta 2016, 1862, 2168–2176. [Google Scholar] [CrossRef]
- Kurtenbach, S.; Kurtenbach, S.; Zoidl, G. Gap junction modulation and its implications for heart function. Front. Physiol. 2014, 5, 82. [Google Scholar] [CrossRef]
- Koopman, R.; Schaart, G.; Hesselink, M.K. Optimisation of Oil Red O Staining Permits Combination With Immunofluorescence and Automated Quantification of Lipids. Histochem. Cell Biol. 2001, 116, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, M.A.; Cea, L.A.; Vega, J.L.; Boric, M.P.; Monyer, H.; Bennett, M.V.; Frank, M.; Willecke, K.; Sáez, J.C. The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 2013, 75, 594–603. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cea, L.A.; Fernández, G.; Arias-Bravo, G.; Castillo-Ruiz, M.; Escamilla, R.; Brañes, M.C.; Sáez, J.C. Blockade of Hemichannels Normalizes the Differentiation Fate of Myoblasts and Features of Skeletal Muscles from Dysferlin-Deficient Mice. Int. J. Mol. Sci. 2020, 21, 6025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176025
Cea LA, Fernández G, Arias-Bravo G, Castillo-Ruiz M, Escamilla R, Brañes MC, Sáez JC. Blockade of Hemichannels Normalizes the Differentiation Fate of Myoblasts and Features of Skeletal Muscles from Dysferlin-Deficient Mice. International Journal of Molecular Sciences. 2020; 21(17):6025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176025
Chicago/Turabian StyleCea, Luis A., Gabriela Fernández, Guisselle Arias-Bravo, Mario Castillo-Ruiz, Rosalba Escamilla, María C. Brañes, and Juan C. Sáez. 2020. "Blockade of Hemichannels Normalizes the Differentiation Fate of Myoblasts and Features of Skeletal Muscles from Dysferlin-Deficient Mice" International Journal of Molecular Sciences 21, no. 17: 6025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176025